

ABSTRACT
Infected cell protein 0 (ICP0) of herpes simplex virus 1 (HSV-1) is a key regulator in both lytic and latent infections. In lytic infection, an important early event is the colocalization of ICP0 to nuclear domain 10 (ND10), the discrete nuclear bodies that impose restrictions on viral expression. ICP0 contains an E3 ubiquitin ligase that degrades promyelocytic leukemia protein (PML) and Sp100, two major components of ND10, and disperses ND10 to alleviate repression. We previously reported that the association between ICP0 and ND10 is a dynamic process that includes three steps: adhesion, fusion, and retention. ICP0 residues 245 to 474, defined as ND10 entry signal (ND10-ES), is a region required for the fusion step. Without ND10-ES, ICP0 adheres at the ND10 surface but fails to enter. In the present study, we focus on characterizing ND10-ES. Here we report the following. (i) Fusion of ICP0 with ND10 relies on specific sequences located within ND10-ES. Replacement of ND10-ES by the corresponding region from ORF61 of varicella-zoster virus did not rescue ND10 fusion. (ii) Three tandem ND10 fusion segments (ND10-FS1, ND10-FS2, and ND10-FS3), encompassing 200 amino acids within ND10-ES, redundantly facilitate fusion. Each of the three segments is sufficient to independently drive the fusion process, but none of the segments by themselves are necessary for ND10 fusion. Only when all three segments are deleted is fusion blocked. (iii) The SUMO interaction motif located within ND10-FS2 is not required for ND10 fusion but is required for the complete degradation of PML, suggesting that PML degradation and ND10 fusion are regulated by different molecular mechanisms.
IMPORTANCE ND10 nuclear bodies are part of the cell-intrinsic antiviral defenses that restrict viral gene expression upon virus infection. As a countermeasure, infected cell protein 0 (ICP0) of herpes simplex virus 1 (HSV-1) localizes to ND10s, degrades the ND10 organizer, and disperses ND10 components in order to alleviate repression. We studied the ICP0-ND10 association to delineate elements important for this dynamic interaction and to understand its role in viral replication and host defense. In this work, we show that ICP0 contains three redundant segments to ensure an effective mergence of ICP0 with ND10 nuclear bodies. This is the first study to systematically investigate ICP0 elements that are important for ICP0-ND10 fusion.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infected cell protein 0 (ICP0) is a multifunctional immediate early protein that plays a key role in both lytic and latent infections. The protein is essential at a low multiplicity of infection (MOI) in cultured cells (1). Its main functions are to counteract cell-intrinsic and innate defenses by targeting saturable cellular restrictive factors and consequently to enhance viral gene expression (1).
The 775-amino-acid ICP0 contains a RING (really interesting new gene) type E3 ubiquitin ligase in its second exon (2, 3), which ubiquitinates various cellular proteins for proteasomal degradation. Known ICP0 substrates include promyelocytic leukemia protein (PML) (4), speckled protein Sp100 (4), DNA-dependent protein kinase (DNA-PK) (5), centromeric protein A (CENP-A) (6), and interferon (IFN)-inducible protein 16 (IFI16) (7). Some of these ICP0 substrates are restrictive for HSV-1 infection, inasmuch as small interfering RNA (siRNA) knockdown of PML, Sp100, or IFI16, individually or in combination, enhances viral replication in the absence of ICP0 (8, 9).
A second tactic of ICP0 to overcome antiviral defenses is to regulate a diverse array of cell pathways via protein-protein interactions. For example, ICP0 interacts with corepressor of RE1-silencing transcription factor (CoREST). This interaction leads to the dissociation of histone deacetylases (HDACs) 1 and 2 from the REST/CoREST complex. Subsequently, the chromatin silencing imposed on the viral genome is released, and viral gene expression is enhanced (10–12). ICP0 also interacts with ubiquitin-specific protease 7 (USP7). Through this interaction, ICP0 and USP7 reciprocally modulate each other's stability (13, 14). Similar to ICP0 (4–7), USP7 also has a wide range of deubiquitination substrates involved in many critical checkpoints, such as cell cycle regulation, apoptosis, DNA repair, and gene regulation (15–18). Although the relationships among ICP0, USP7, and their substrates are not yet clear, it is justifiable to postulate that fine-tuning of the levels of these checkpoint proteins can have a pivotal role in the tug-of-war between HSV-1 and the host. Recently, SUMO (small ubiquitin-like modifier) interaction motifs (SIMs) have also been identified in ICP0 (19, 20), suggesting that ICP0 may regulate more cellular factors via SUMO-SIM interactions.
One important question that remains unclear is how ICP0 coordinates its concurrent interactions with multiple substrates and binding partners that belong to such a diverse array of cell pathways. For example, DNA-PK is involved in the DNA repair pathway, whereas USP7 is a part of the proteasome pathway. We hypothesize that cellular intersections connecting different pathways may serve as molecular platforms for ICP0 to effectively coordinate its multiple functions. A dynamic nuclear structure, nuclear domain 10 (ND10), which has been implicated in numerous cellular processes, including gene regulation (21, 22), apoptosis (23, 24), DNA repair (25), oncogenesis (26, 27), and antiviral defense (28), is a suitable candidate to bridge the cross talk and networking among different cell pathways.
ND10s, also known as promyelocytic leukemia nuclear bodies (PML-NBs), contain >150 proteins, many of which are regulatory factors and transient components recruited to ND10 only upon specific stimulation (28). ICP0 has an intimate interaction with ND10 early in HSV-1 infection. After entering the nucleus, viral DNA is found in the vicinity of ND10 (29). Components of ND10 act as host defenses to repress initial viral gene expression (8, 30). ICP0, upon its synthesis, is deployed to localize at ND10 (31) and targets two major ND10 constituents, PML and Sp100, for proteasomal degradation (4). Since PML is the main organizer protein for ND10 (26, 32, 33), the loss of PML leads to the dispersal of ND10 nuclear bodies. Subsequently, the restrictions imposed by ND10 are alleviated, and viral gene expression is enhanced. We focus on the delineation of the intimate relationship between ICP0 and ND10, with the goal of elucidating the spatial-temporal regulation of ICP0-ND10 interactions and understanding the role of ICP0-ND10 interactions in the coordination of ICP0 multifunctionality.
In a previous report, we demonstrated that the interaction between ICP0 and ND10s is a dynamic process that contains three sequential steps, adhesion, fusion, and retention, and specific ICP0 domains are involved in each of the three steps (34). These new findings have redefined the prior static description of ICP0-ND10 colocalization. The new concept of a dynamic ICP0-ND10 interaction embraces a hypothesis that ICP0 may interact with a distinctive set of ND10 components in each of the dynamic steps, which orchestrates spatial-temporal regulation on its approach to E3 ligase substrates within ND10 and on its acquiring cellular partners essential for viral replication.
To understand the spatial-temporal regulation of the ICP0-ND10 interaction, we need to dissect the protein network responsible for the dynamic association. A thorough characterization of ICP0 motifs involved in each step is the necessary groundwork to delineate protein-protein interactions. In the present study, we focus on the fusion event to determine the minimum sequence requirement for ICP0-ND10 fusion. We show that three discontinuous segments located in the central region of ICP0 redundantly facilitate the ND10 fusion process. Each segment by itself is sufficient to drive fusion, but only when all three segments are simultaneously deleted can the fusion of ICP0 to ND10 be blocked.
MATERIALS AND METHODS
Cells and viruses.
HEp2 cells and immortalized human embryonic lung (HEL) fibroblasts, generated by Thomas E. Shenk (Princeton University), were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS). Human osteosarcoma U2OS cells were grown in McCoy's 5A medium (Sigma) supplemented with 10% FBS. Recombinant viruses RHG101, RHG104, RHG110, and RHG120, which contain wild-type or mutant ICP0s, were previously described (34).
Construction of recombinant viruses.
Deletions or mutations of ICP0 in recombinant viruses RHG111, RHG112, RHG113, RHG114, RHG115, RHG121, RHG122, RHG123, RHG124, RHG125, RHG126, and RHG130, as shown in Fig. 1B, panels 1 to 5, were constructed by a two-round PCR strategy. The pairs of forward and reverse primers designed for each mutant are complementary to each other (see Table S1 in the supplemental material). Each primer has 20 to 30 nucleotides upstream of the deletion junction directly connected to the 20 to 30 nucleotides downstream of the deletion junction. In the first round of PCR, forward primers were paired with a downstream primer, 5′-GCAGTCGACTTACCCGGGCCCACCCTGGCCGCG-3′, and reserve primers were paired with an upstream primer, 5′-GCAGGATCCGCGGTCTCGGGGGGGAGC-3′, to amplify the fragments downstream or upstream of the deletion or mutation junction, respectively. In the second round of PCR, both upstream and downstream PCR products were gel purified and mixed to serve as the template to be amplified with the upstream and downstream primers only. The second-round PCR products of the joined fragments contained the designated internal deletions or mutations. These deletion or mutation products were digested with XhoI and MluI to swap the fragment between the XhoI and MluI sites of plasmid pHG101, in which an mCherry-tagged full-length ICP0 cDNA is flanked with the 5′ and 3′ ICP0-flanking sequences from the HSV-1(F) genome (34). The plasmids obtained were named pHG111 to pHG130, corresponding to the virus names.
FIG 1.

Schematic diagrams of ICP0 mutants used in this study and their functions in ND10 fusion. (A) The ICP0 gene and its position in the HSV-1 genome. The key and icons are the same as those described previously (34). Line 1, the HSV-1 genome, with the terminal and internal repeats shown in patterned boxes; line 2, positions of the two copies of ICP0 in the terminal and internal repeats and expanded illustration of the ICP0 gene, with three exons intervened by two introns; line 3, ICP0 cDNA (775 amino acids) used for mutant construction, with positions of the RING finger domain (2, 3) and nuclear localization sequence (64) shown; line 4, schematic diagrams of recombinant viruses RHG101, RHG120, and RHG110, which were reported elsewhere (34); line 5, amino acid positions in the expanded region of residues 100 to 550. (B1 to B5) Recombinant viruses containing deletions and mutations in ICP0. The amino acid deletions and mutations are indicated in parentheses following the virus names. Symbols representing specific domains are shown in the key. Functions of individual ICP0 mutants in ND10 fusion are indicated to the right of each virus. +, ND10 fusion competent; −, ND10 fusion incompetent.
To replace ICP0 residues 244 to 475 with the corresponding region from varicella-zoster virus (VZV) ORF61, primers 5′-CTGGACGACGCAGACTACAGATCTGATATCGGGCCCTCCCGCGGCGCC-3′ and 5′-GGCGCCGCGGGAGGGCCCGATATCAGATCTGTAGTCTGCGTCGTCCAG-3′ were used for the two-round PCR strategy described above to generate BglII and EcoRV sites between the junction of amino acids 244 and 475 of ICP0. VZV ORF61 primers 5′-CGCGGATCCGATCTTCTGCCACCAAGC-3′ and 5′-ACGGATATCTGTATCTTTCCAGGTCCA-3′ were used to amplify ORF61 amino acids 105 to 335. The PCR template, plasmid pcDNA3.1(+)-VZV ORF61, was a generous gift from Ann Arvin (Stanford University). The PCR product containing ORF61 residues 105 to 335 was digested with BamHI and EcoRV and inserted into the BglII and EcoRV sites as described above. For the construction of RHG128 and RHG135, plasmids pHG113 and pHG123 were digested with NruI and MluI. The NruI-MluI fragments that contain the respective deletion of residues 343 to 391 (Δ343–391) and residues 343 to 441 (Δ343–441) were used to replace the wild-type version of the NruI-MluI fragment in plasmid pHG111, which already contains a deletion of ICP0 residues 242 to 291. The resulting plasmids were named pHG128, which contains a double deletion of residues 242 to 291 and residues 343 to 391, and pHG135, which contains a double deletion of residues 242 to 291 and residues 343 to 441.
For the construction of RHG136, we again adopted the two-round PCR strategy. We used pHG121 as the PCR template and the same primer pair as the one used for RHG114 construction. The product generated from the second-round PCR containing a double deletion of residues 242 to 341 and residues 393 to 441 was digested with XhoI and MluI to swap the wild-type XhoI-MluI fragment in pHG101. This generated plasmid pHG136.
For the construction of RHG137, plasmid pHG130, which contains I362G, V363A, and I364G (I362G/V363A/I364G) mutations, was digested with NruI and MluI. The NruI-MluI fragment containing the I362G/V363A/I364G mutations was used to replace the NruI-MluI fragment in plasmid pHG111. This generated an intermediate plasmid, pHG111-SLS4m, that contains a double mutation of Δ242–291 and I362G/V363A/I364G substitutions. We then used the two-round PCR strategy with pHG111-SLS4m as the PCR template and the same primer pair as the one used for RHG114 construction to generate a PCR product containing a triple mutation of Δ242–291 plus Δ393–441 and the I362G/V363A/I364G substitutions. This PCR product was digested with XhoI and MluI to swap the wild type XhoI-MluI fragment in pHG101. This generated plasmid pHG137.
The above-described internal deletions and mutations of ICP0 were then cloned into the KO5 plasmid as previously described (35). The series of KO5 constructs were then electroporated into the RR1 strain containing an ICP0-null bacterial artificial chromosome (BAC) (35). BAC DNAs extracted from positive colonies were transfected into U2OS cells to generate recombinant viruses. All viruses were plaque purified at least three times on U2OS cells. Viral DNAs were isolated, and the existence of ICP0 mutations in both terminal and internal repeats was verified by Southern blotting.
Confocal microscopy.
HEL cells grown on four-well glass slides (Thermo Fisher Scientific) were exposed to 10 PFU/cell of recombinant viruses for 1 h. The inocula were then removed, and the cells were incubated in growth medium. At the hours indicated in Results, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with phosphate-buffered saline (PBS) containing 5% horse serum and 1% bovine serum albumin. The cells were then reacted with primary antibodies at 4°C overnight, rinsed, and reacted with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit (Sigma) plus Texas Red-conjugated goat anti-mouse (Invitrogen) secondary antibodies. Images were taken with a Zeiss LSM780 confocal microscope at the Microscopy, Imaging and Cytometry Resources (MICR) Core facility at Wayne State University School of Medicine. Three-dimensional (3D) images were constructed by using Volocity 3D image analysis software.
Coimmunoprecipitation.
Infected HEp-2 cells cultured on 100-mm plates were lysed in 500 μl of lysis buffer (10 mM Tris-HCl [pH 8.0], 140 mM NaCl, 1.5 mM MgCl2, 1 mM dithiothreitol, and 0.5% NP-40 supplemented with a protease inhibitor cocktail [Sigma] and a phosphatase inhibitor cocktail [Sigma]). After brief sonication and centrifugation to remove the cell debris, the total cell lysates were cleared by incubation with 50 μl of a 50% slurry of protein A-Sepharose CL-4B beads (GE Healthcare, Pittsburgh, PA) at 4°C for 20 min. Upon the removal of the beads, the cleared lysates were reacted with a rabbit polyclonal antibody to USP7 (Bethyl Laboratories Inc.) overnight at 4°C. The lysate-antibody mixtures were then incubated with 50 μl of a 50% slurry of protein A-Sepharose CL-4B beads at 4°C for 2 h. The beads were then rinsed four times with lysis buffer and eluted with 1× Laemmli buffer at 95°C for 5 min. The eluates were electrophoretically separated on 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and subjected to Western blot analysis.
Western blot analysis.
HEL or HEp2 cells were mock infected or infected with 10 PFU/cell of recombinant viruses for 1 h. The inocula were then removed, and cells were incubated in DMEM supplemented with 10% newborn calf serum (Thermo Fisher Scientific). At the hours indicated, the cells were harvested, washed, and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, 0.25% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride). The total cell lysate was sonicated, electrophoretically separated on SDS-PAGE gels, and then transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore). The membrane was blocked with 1× Tris-buffered saline–Tween (TBST) (20 mM Tris [pH 7.5], 150 mM NaCl, 0.5% Tween 20) containing 5% nonfat dry milk and probed with primary antibodies, as indicated in Results. The membrane was then incubated with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Sigma) and visualized with ECL Western blotting detection reagent (GE Healthcare).
Antibodies.
Polyclonal anti-PML antibody and monoclonal antiactin antibody were purchased from Santa Cruz Biotechnology Inc. Monoclonal anti-mCherry antibody was purchased from Clontech. Polyclonal anti-USP7 antibody was purchased from Bethyl Laboratories Inc.
RESULTS
The functional ND10 entry signal encompasses 200 amino acids within the central region of ICP0.
Previously, we reported that the dynamic interaction between ICP0 and ND10 is composed of three sequential steps: adhesion, fusion, and retention (34). Upon synthesis, ICP0 adheres at the ND10 surface and then rapidly fuses with the ND10 body (34). The adhesion process is very transient. For wild-type ICP0, only fused molecules can be observed, which was previously defined as ICP0-ND10 colocalization (31, 36). In a preceding report, we showed that when the central region comprised of residues 245 to 474, namely, ND10 entry signal (ND10-ES) is deleted, the ND10 fusion process is blocked after adhesion, and ICP0 juxtaposes with ND10 for a short period of time (34). The present study is designed to characterize the molecular properties of ND10-ES. To identify the minimum sequence required for ND10 fusion, we conducted deletion mapping of the ICP0 central region and examined the positions of ICP0 mutants relative to those of ND10 nuclear bodies by high-resolution immunofluorescence confocal analysis. Throughout the study, we immunostained PML to represent the ND10 distribution in the nucleus. We adopted ICP0 lacking ND10-ES from recombinant virus RHG110 (Fig. 1A, line 4) (34) as the control for ICP0-ND10 juxtaposition (Fig. 2A, panels d to f, and 3A, panels d to f and h). We used wild-type ICP0 from virus RHG101 (Fig. 1A, line 4) (34) as the control for ICP0-ND10 colocalization (Fig. 2A, panels a to c). We also used ICP0 containing C116G/C156A substitutions in the RING domain (RING finger mutant [RFm]) from virus RHG120 (Fig. 1A, line 4) (34) as an additional control for ICP0-ND10 colocalization (Fig. 3A, panels a to c and g). ICP0 containing RFm lost its ability to degrade PML (35). Once it fuses with ND10, it is sequestered in the enzymatic transition state and is colocalized with ND10 throughout the infection (34, 35). The use of ICP0 RING finger mutant (RHG120) as a control for ICP0-ND10 colocalization can avoid the disappearance of the PML signal (ND10 marker) in cells infected with the wild-type virus. For each ICP0 mutant constructed in this study (Fig. 1B, panels 1 to 5), we compared ICP0 subnuclear distributions with both the ND10 juxtaposition control (RHG110) and the ND10 colocalization control (RHG101 or RHG120) to determine the relative positions of mutant ICP0 and ND10. Mutants that are juxtaposed with ND10 are defined as ND10 fusion incompetent, whereas those that are colocalized with ND10 are defined as ND10 fusion competent (Fig. 1).
FIG 2.

ICP0 localization in recombinant viruses that contain a 100- or 150-amino-acid deletion in the central region of ICP0. HEL cells were mock treated (left) or treated with 10 μM MG132 (right) before being infected with the indicated recombinant viruses. At 3 h postinfection, cells were fixed, permeabilized, and reacted with polyclonal anti-PML and monoclonal anti-mCherry antibodies. Representative images of infected cells were taken with a Zeiss LSM780 confocal microscope.
FIG 3.
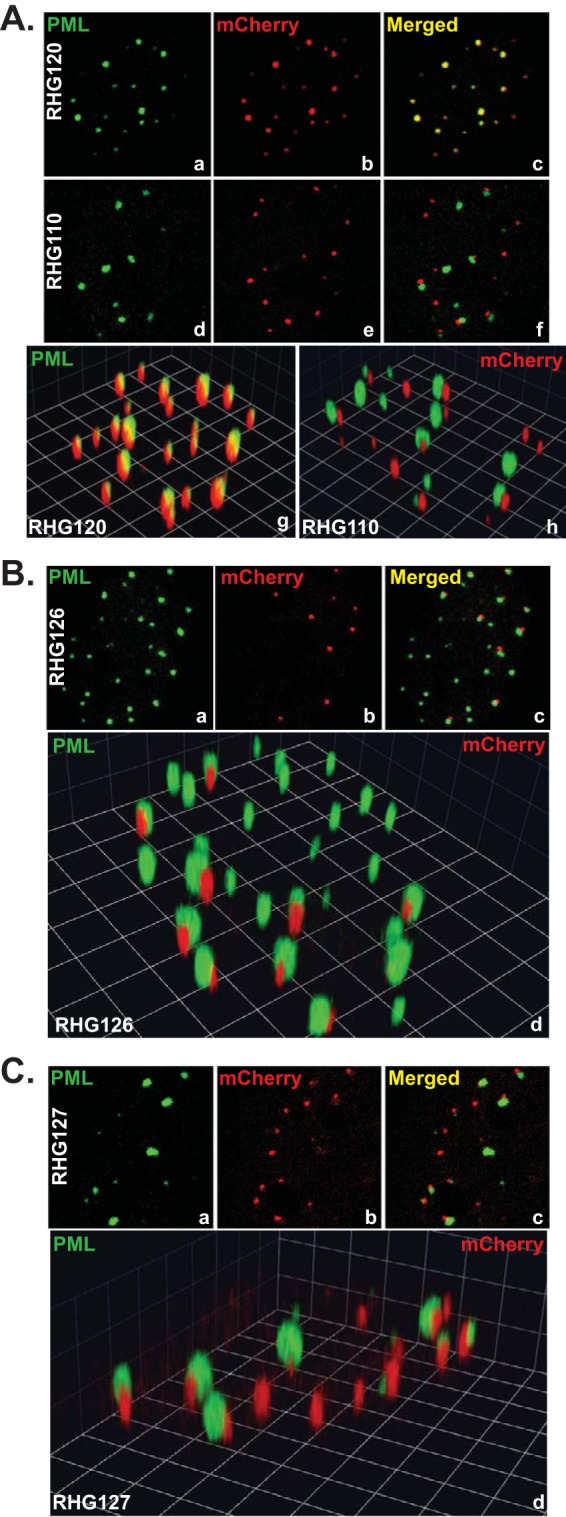
ICP0 localization of recombinant viruses RHG126 and RHG127. HEL cells were infected with RHG120 or RHG110 (A), RHG126 (B), or RHG127 (C) at 10 PFU/cell. At 3 h postinfection, cells were fixed, permeabilized, and reacted with polyclonal anti-PML and monoclonal anti-mCherry antibodies. Two- and three-dimensional images of representative infected cells were taken with a Zeiss LSM780 confocal microscope. The three-dimensional images were reconstructed with Volocity software.
We first constructed recombinant viruses RHG111 to RHG115, RHG121 to RHG123, and RHG124 and RHG125, in which ICP0 was internally deleted of 50, 100, and 150 amino acids at a time, respectively, to encompass the entire area of residues 242 to 441 (Fig. 1B, panels 1 to 3). To our surprise, ICP0 with a deletion of 50, 100, or 150 amino acids did not reveal any specific region that is necessary for ICP0 to fuse with ND10. As shown in Fig. 2 and in Fig. S1 in the supplemental material, in cells infected with viruses that contain a 50-amino-acid deletion (RHG111 to RHG115) (see Fig. S1a to S1o in the supplemental material), a 100-amino-acid deletion (RHG121 to RHG123) (Fig. 2B, panels a to i), or a 150-amino-acid deletion (RHG124 and RHG125) (Fig. 2B, panels j to o) in ICP0, the internally truncated ICP0 mutants were colocalized with ND10 and showed no apparent defects in ND10 fusion. Previously, we demonstrated that a blockage of the proteasomal pathway, by mutating the RING domain of ICP0 or by treating cells with the proteasome inhibitor MG132, leads to the sequestration of full-length ICP0 within ND10 bodies after ND10 fusion (34, 35). However, in the absence of ND10-ES, when ICP0 can only juxtapose at the ND10 surface without fusion, additional blockage of the proteasomal pathway leads to the diffusion of this particular mutant throughout the nucleus (34). Consistent with previously reported results, treatment with MG132 enhanced ICP0-ND10 colocalization for wild-type ICP0 in RHG101-infected cells (Fig. 2A, panels aa to cc) but caused ICP0 lacking ND10-ES to disperse in RHG110-infected cells (Fig. 2A, panels dd to ff). To confirm that ICP0 mutants with a 50-, 100-, or 150-amino-acid deletion were able to fuse with ND10, we examined their positions relative to that of ND10 in the presence of MG132. As shown in Fig. 2B, panels aa to oo and in Fig. S1aa to S1oo in the supplemental material, all deletion mutants in this series of experiments were colocalized with ND10 upon MG132 treatment, suggesting that ND10 fusion took place for these mutants and that they were sequestered within ND10 after fusion.
We further constructed recombinant virus RHG126 (Fig. 1B, panel 4), in which ICP0 carries a 200-amino-acid deletion between residues 242 and 441. As shown in Fig. 3B, ICP0 lacking amino acids 242 to 441 was juxtaposed to ND10 and unable to fuse. This phenomenon recapitulated what we previously observed for ICP0 lacking the entire ND10-ES in RHG110-infected cells (34) (Fig. 2A, panels d to f, and 3A, panels d to f and h), with no substantial differences. Results from independently constructed virus RHG126 ruled out the possibility that incidental mutations in viral genes other than ICP0 cause ICP0 to juxtapose at ND10. The reproducibility between RHG110 and RHG126 also validated the importance of the ICP0 central region in ND10 fusion. Therefore, we conclude that ND10-ES encompasses 200 amino acids within the central region of ICP0, and smaller deletions fail to disrupt the function of ND10-ES.
Replacement of ND10-ES by the corresponding region from VZV ORF61 does not rescue ND10 fusion.
Two hypotheses can explain the failure in narrowing down the ND10-ES sequence to a domain smaller than 200 amino acids. In the first scenario, the region encompassing amino acids 242 to 441 constitutes a flexible linker to connect the N and C termini of ICP0. It is the specific length of this region, not the sequence per se, that is important for the overall structure of ICP0 and the fusion of ICP0 with ND10. In the second proposition, multiple discontinuous segments scattering throughout the 200 amino acids of the ICP0 central region redundantly facilitate the fusion of ICP0 with ND10. Smaller deletions disrupt only part of the redundant elements, while residual elements are still active in driving ND10 fusion.
To test the first hypothesis, we constructed a recombinant virus, RHG127 (Fig. 1B, panel 5), in which residues 245 to 474 of ICP0 were replaced with the corresponding region of ORF61 (amino acids 105 to 335) from varicella-zoster virus (VZV). In designing chimeric ICP0, we chose to replace the entire ND10-ES of residues 245 to 474, but not the smaller functional region of 200 amino acids, so that we could include a larger area from ORF61 and avoid losing important ORF61 sequences.
The localization of chimeric ICP0 was then examined. Confocal images showed that chimeric ICP0 was juxtaposed to ND10 nuclear bodies and unable to fuse with ND10 (Fig. 3C), just like ICP0 deleted of ND10-ES in RHG110-infected cells (Fig. 3A, panels d to f and h). The restoration of the distance between the N and C termini of ICP0 did not rescue fusion. Therefore, it is the specific sequence, not the length, of the ICP0 central region that is responsible for ND10 fusion.
Deletion of the ICP0 central region affects the E3 ligase activity located at the N terminus but not the USP7 binding ability located at the C terminus.
The dramatic change in the distributions of ICP0 lacking ND10-ES (RHG110) in the absence and presence of MG132 (Fig. 2A, panels d to f and dd to ff) (34) indicated a dynamic involvement of the proteasome in the ND10 adhesion process, which likely has biological relevance. It also argued against the possibility that ICP0 adhering at the ND10 surface is the simple result of random protein aggregation due to structural changes. To further validate this conception, we carefully analyzed the effects of the internal deletion on the overall functions of ICP0.
We first assessed the effects of an internal deletion of residues 245 to 474 on the USP7 binding activity located within C-terminal residues 594 to 633 (37). In control experiments, USP7 pulled down full-length ICP0 from virus RHG101 (Fig. 4A, lane 5), whereas it did not interact with C-terminally truncated ICP0 from virus RHG104 (34) (Fig. 4A, lane 6). As shown in Fig. 4A, lanes 7 and 8, both internally truncated ICP0 (RHG110) and VZV chimeric ICP0 (RHG127) maintained the ability to interact with USP7, suggesting that the C terminus of ICP0 is not substantially affected by the internal truncation or the sequence replacement from ORF61.
FIG 4.
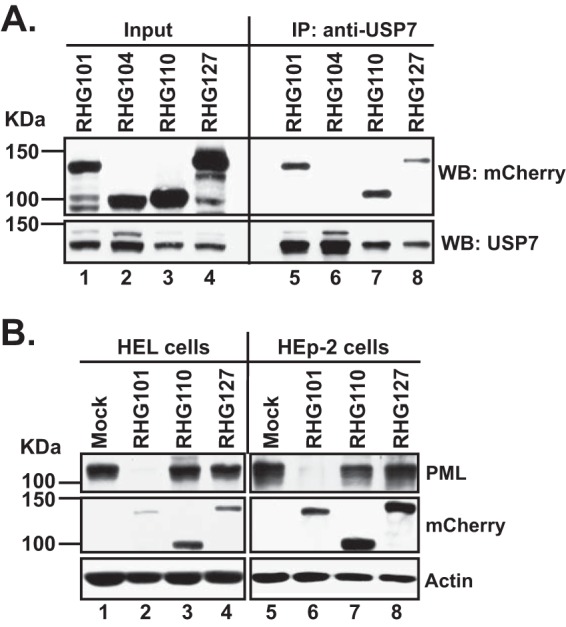
Effects of internal deletion on ICP0 functional domains located in its N and C termini. (A) ICP0 internal deletion does not affect its ability to interact with USP7. HEp2 cells were infected with recombinant viruses RHG101, RHG104, RHG110, and RHG127 at 5 PFU/cell for 14 h. Infected-cell lysates were then subjected to a coimmunoprecipitation (IP) assay with polyclonal anti-USP7 antibody. The immunoprecipitates were electrophoretically separated on SDS-PAGE gels and probed with the indicated antibodies by Western blotting (WB). (B) ICP0 internal deletion diminishes its E3 function in PML degradation. HEL and HEp2 cells were mock infected or infected with recombinant viruses RHG101, RHG110, and RHG127 at 10 PFU/cell. At 8 h postinfection, total cell lysates were probed with the indicated antibodies by Western blotting.
We also investigated the effects of the internal deletion on the E3 ubiquitin ligase activity located within the ICP0 N terminus. Previously, we reported that internally truncated ICP0 in RHG110 lost its ability to degrade PML (34). This was confirmed by the results shown in Fig. 4B. In both the HEL and HEp2 cell lines, the loss of residues 245 to 474 completely blocked PML degradation in infected cells (Fig. 4B, lanes 3, 4, 7, and 8). Various reports have shown that the E3 ligase activity of ICP0 is tightly regulated by multiple factors located in different domains of ICP0. For example, point mutations of D671A/E673A in the CoREST binding site can negatively affect PML degradation by ICP0 (35), and mutations in SLSs (SUMO interaction motif [SIM]-like sequences) of ICP0 also cooperatively influence the ability of ICP0 to degrade PML (20). Although, at this point, we cannot rule out the possibility that deletion of the ICP0 central region may change the RING structure, it is safe to postulate that even without structural changes in the RING finger, deletion of the ICP0 central region can have significant impacts on the E3 ligase activity of ICP0.
Simultaneous deletions of residues 242 to 291 and residues 343 to 441 abolish ND10 fusion.
We then tested the hypothesis that segments scattered across the region spanning residues 242 to 441 redundantly drive ICP0 to fuse with ND10. We constructed recombinant viruses RHG135 and RHG136, in which a total of 150 amino acids were deleted in a discontinuous fashion (Fig. 1B, panel 5). RHG135 was constructed on the basis of RHG123 (lacking residues 343 to 441 in ICP0) with a further deletion of ICP0 residues 242 to 291. RHG136 was built on the basis of RHG121 (lacking residues 242 to 341 in ICP0) with an additional deletion of ICP0 residues 393 to 441. We found that ICP0 lacking both regions spanning residues 242 to 291 and residues 343 to 441 (RHG135) juxtaposed to ND10 but failed to fuse (Fig. 5A, panels a to d), whereas ICP0 with deletions of residues 242 to 341 and 393 to 441 (RHG136) maintained the ability to fuse and colocalize with ND10 (Fig. 5A, panels e to h). In these immunofluorescence studies, we again used full-length ICP0 containing the C116G/C156A mutations in the RING finger (RHG120) as the control for ICP0-ND10 colocalization (Fig. 5A, panels l to n) and ICP0 lacking ND10-ES (RHG110) as the control for ICP0-ND10 juxtaposition (Fig. 5A, panels i to k). All confocal microscopic pictures for the same batch of experiments were taken under the same confocal parameters calibrated with both RHG120 and RHG110.
FIG 5.

Residues 242 to 341 and residues 343 to 441 in the central region of ICP0 are responsible for ND10 fusion. (A to C) HEL cells were infected with RHG135 and RHG136 at 10 PFU/cell. RHG110 and RHG120 served as controls for ND10 juxtaposition and ND10 colocalization to calibrate the parameters for confocal imaging. (A) At 1.5 h postinfection, infected cells were fixed, stained, and visualized as described above. (B) At 1.5 and 3 h postinfection, cells expressing ICP0 (left) and cells containing ICP0 exclusively in punctate structures (right) were tabulated. (C) At 1.5 and 3 h postinfection, relative positions of ICP0 and ND10 dots (juxtaposition or colocalization) were tabulated. (D) HEL cells were infected with RHG128 at 10 PFU/cell for 1.5 h and then fixed, stained, and visualized as described above.
We then tabulated the infected cells containing discrete dotted structures to analyze the statistical differences between RHG135 and RHG136. In Fig. 5B, left, we show that at 1.5 h postinfection, of ∼200 cells tabulated, 47% of cells infected with RHG110, 50% of cells infected with RHG135, and 42% of cells infected with RHG136 expressed ICP0 to a detectable level. At 3 h postinfection, the percentage of cells expressing ICP0 was slightly increased for all three viruses, indicating that initial viral loads were at equivalent levels for the control and experiment viruses.
We previously reported that ICP0 lacking ND10-ES (RHG110) transiently adhered at the ND10 surface in juxtaposition. On a fixed slide, only about one-third of the RHG110-infected cells contain ICP0 lacking ND10-ES in punctate structures at 1.5 h postinfection, and this number diminishes with prolonged infection (34). Reproducible results for RHG110 were again found, as shown in Fig. 5B, right. This pattern of ICP0 distribution was recapitulated in RHG135 infection (Fig. 5B, right). However, in RHG136 infection, more cells had punctate structures at 1.5 h postinfection, and the drop at 3 h postinfection was steeper.
We further examined the positions of dotted ICP0 relative to that of ND10 with high-magnification objectives. We tabulated ICP0 and ND10 dots in two groups of 20 infected cells that contained ICP0 exclusively in discrete dots with no detectable nucleoplasmic staining. On average, there were 15 to 20 ND10 bodies per HEL cell. Therefore, ∼300 to 400 nuclear dots stained by anti-PML antibody were examined for each 20-cell group. In cells infected by RHG135, at 1.5 and 3 h postinfection, averages of 89.2% and 85.6% of the ICP0 dots were juxtaposed to ND10, while 9.7% and 13.2% of the ICP0 dots were colocalized with ND10, respectively (Fig. 5C). Reversed phenomena were observed for cells infected by RHG136. At 1.5 and 3 h postinfection, averages of 11.9% and 12.6% of the ICP0 dots were juxtaposed to ND10, while 87.8% and 86.5% of the ICP0 dots were colocalized with ND10, respectively (Fig. 5C). In both RHG135 and RHG136 infections, ∼1% of the ICP0 dots were aggregates without an apparent association with ND10. From these data, we conclude that deletion of both regions spanning residues 242 to 291 and residues 343 to 441 abolished ND10 fusion, causing ICP0 to juxtapose at the ND10 surface but fail to fuse with the ND10 body.
Three redundant segments within the region spanning amino acids 242 to 441 independently facilitate the fusion of ICP0 with ND10.
To clearly demonstrate ICP0 segments that are responsible for ND10 fusion, we compiled all the results for ND10 fusion analysis shown in Fig. 1 along with the schematic diagrams of ICP0 mutants. Comparing ICP0 from RHG123, which is ND10 fusion competent (Fig. 1B, panel 2), with ICP0 from RHG135, which is ND10 fusion incompetent (Fig. 1B, panel 5), deletion of residues 343 to 441 alone (RHG123) did not affect ND10 fusion, but double deletion of both regions spanning residues 242 to 291 and residues 343 to 441 (RHG135) blocked ICP0 from fusing with ND10. This led us to conclude that the sequence between residues 242 and 291 by itself is sufficient for ND10 fusion. We define this sequence as ND10 fusion segment 1 (ND10-FS1).
Comparing ICP0 from RHG136, which is ND10 fusion competent (Fig. 1B, panel 5), with ICP0 from RHG126, which is ND10 fusion incompetent (Fig. 1B, panel 4), the presence of ICP0 residues 343 to 391 in RHG136 is sufficient for ND10 fusion to take place. We therefore define the fragment of residues 343 to 391 as ND10-FS2.
Comparing ICP0 from RHG124, which is ND10 fusion competent (Fig. 1B, panel 3), with ICP0 from RHG126, which is ND10 fusion incompetent (Fig. 1B, panel 4), we found that deletion of continuous residues 242 to 391 (RHG124) did not completely remove the sequences required for ND10 fusion but that the additional deletion of residues 393 to 441 (RHG126) did. This suggested that the sequence between residues 393 and 441 contains another segment sufficient for ICP0 to fuse with ND10. To confirm this result, we constructed another recombinant virus, RHG128 (Fig. 1B, panel 5), in which ICP0 residues 242 to 291 (ND10-FS1) and residues 343 to 391 (ND10-FS2) were deleted simultaneously. As shown in Fig. 5D, panels a to d, and 6B, in RHG128-infected cells, in the absence of both ND10-FS1 and ND10-FS2, 95.0% of the ICP0 dots were colocalized to ND10 while 4.3% of the ICP0 dots were juxtaposed to ND10. Results for RHG135-infected cells, in which 11.8% of the ICP0 dots were colocalized to ND10 while 86.4% of the ICP0 dots were juxtaposed to ND10, were consistent with the data shown in Fig. 5C. These results indicate that RHG128 is ND10 fusion competent but RHG135 is ND10 fusion incompetent (Fig. 1B, panel 5). The only difference between RHG128 and RHG135 is the presence of ICP0 residues 393 to 441 in RHG128 (Fig. 1B, panel 5). The congruent results from the RHG124/RHG126 comparison and the RHG128/RHG135 comparison led us to the conclusion that ICP0 residues 393 to 441 are also sufficient to drive ICP0 to fuse with ND10. We define this third fusion segment as ND10-FS3.
FIG 6.
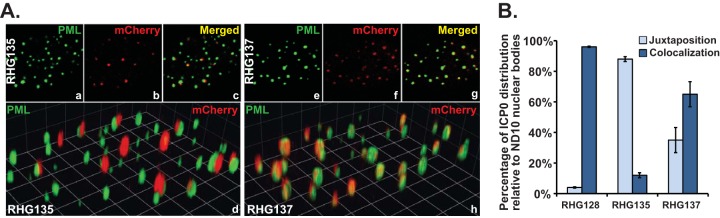
Involvement of ICP0 SLS4 in the ND10 fusion process. (A) ICP0 localization of recombinant viruses RHG135 and RHG137. HEL cells were infected with RHG135 and RHG137 at 10 PFU/cell. At 1.5 h postinfection, infected cells were fixed, stained, and visualized as described above. (B) Relative positions of ICP0 and ND10 dots (juxtaposition or colocalization) were tabulated as described in the text.
Taking all the deletion mapping data together, we identified three segments in ICP0 that redundantly facilitate the ICP0-ND10 fusion process, as illustrated in Fig. 8. ND10-FS2 and ND10-FS3 are located next to each other, while ND10-FS1 is segregated from the other two. Each of the three segments ND10-FS1, ND10-FS2, and ND10-FS3 is sufficient but not necessary for ND10 fusion. The simultaneous removal of all three ND10-FSs can completely abolish ND10 fusion with ICP0.
FIG 8.
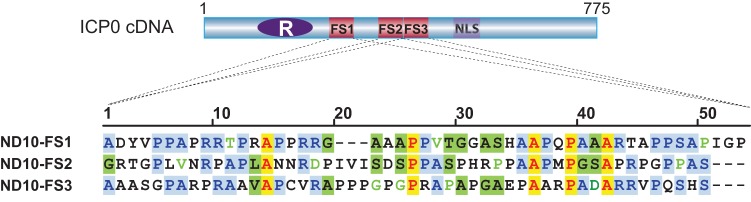
Alignment of the three ND10-FS sequences. The positions of ND10-FS1, ND10-FS2, and ND10-FS3 in ICP0 cDNA are illustrated. Sequence alignment was conducted by using the AlignX program of Vector NTI software. In the aligned sequences, identical amino acids are illustrated by red letters highlighted in yellow, conservative amino acids are highlighted in light blue, amino acids with strong similarity are highlighted in green, and amino acids with weak similarity are illustrated by light green letters.
SLS4 located within ND10-FS2 is not required for ND10 fusion.
Previously, Boutell et al. reported seven SLSs scattered throughout ICP0 (19). Among them, SLS4 interacts with SUMO2/3 and functions to promote ubiquitination by ICP0 in in vitro polyubiquitination assays (19). SLS4 is located at hydrophobic residues 362 to 364 of ICP0, which coincides with ND10-FS2 (residues 343 to 391). To examine whether this particular SUMO interaction within ND10-FS2 plays a role in ND10 fusion, we constructed a recombinant virus, RHG137, in which ND10-FS1 and ND10-FS3 were deleted from ICP0 but ND10-FS2 was retained with the SIM mutated by the I362G/V363A/I364G substitutions (Fig. 1B, panel 5). In RHG137-infected cells, 65% of the ICP0 dots were colocalized at ND10 (Fig. 6A, panels e to h, and B), different from either RHG128- or RHG135-infected cells (Fig. 6B). Since the majority of the ICP0 triple mutant in RHG137-infected cells fused to ND10 in the presence of ND10-FS2 lacking SLS4, we conclude that a SUMO interaction is not required for ND10 fusion but plays an auxiliary role to facilitate effective fusion.
ND10 fusion and PML degradation are regulated by independent mechanisms.
We then investigated the relationship between ICP0-ND10 fusion and PML degradation by ICP0. In Fig. 4B, we show that the central region of ICP0 is essential for ICP0 to degrade PML. To understand how the sequence spanning residues 245 to 474 affects E3 ligase activity, we examined PML degradation in cells infected by an array of viruses containing 50-amino-acid deletions, RHG111 to RHG115. As shown in Fig. 7A, lane 2, RHG101 that contains wild-type ICP0 can completely degrade PML at 8 h postinfection. In contrast, RHG113, which contains a deletion of residues 343 to 391, is the only recombinant virus with defective PML degradation (Fig. 7A, lane 5), whereas ICP0s containing other 50-amino-acid deletions degraded PML to the same extent as did wild-type ICP0 (Fig. 7A, lanes 3, 4, 6, and 7). Since deletions of ND10-FS2 did not affect ND10 fusion (see Fig. S1g to S1i in the supplemental material) but impaired PML degradation (Fig. 7A, lane 5), we conclude that ND10 fusion is not sufficient for effective PML degradation.
FIG 7.
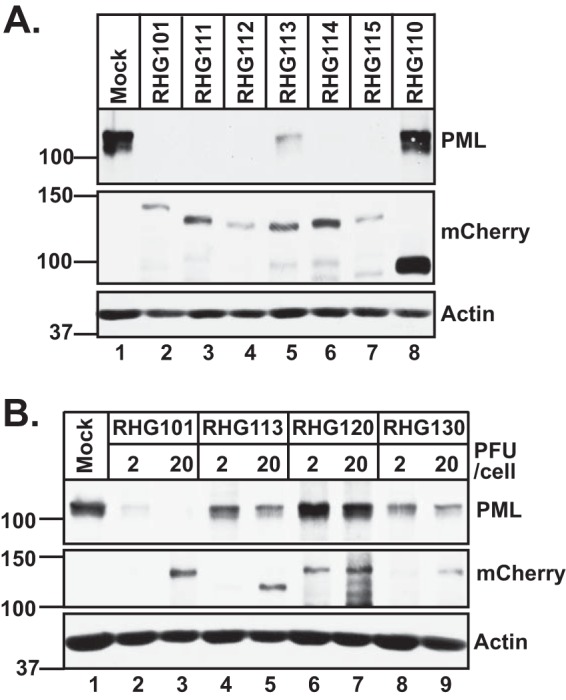
Involvement of SLS4 in PML degradation by ICP0. HEL cells were infected with the indicated viruses at 10 PFU/cell (A) or at 2 and 20 PFU/cell (B). At 8 h postinfection, total cell lysates were probed with the indicated antibodies by Western blotting.
To investigate whether SLS4 located in ND10-FS2 is responsible for defective PML degradation in RHG113-infected cells (Fig. 7A, lane 5), we constructed recombinant virus RHG130, which contains only the SIM mutations I362G/V363A/I364G in SLS4 (Fig. 1B, panel 5). Figure 7B shows that at either 2 or 20 PFU/cell, both ICP0 lacking ND10-FS2 (RHG113) and ICP0 containing I362G/V363A/I364G substitutions (RHG130) were unable to completely degrade PML, suggesting that defective PML degradation is caused by mutations in SLS4 located within residues 343 to 391, consistent with data from a previous report by Everett et al. (20). The fact that increasing the MOI from 2 PFU/cell to 20 PFU/cell did not substantially enhance PML degradation (Fig. 7B, lanes 4, 5, 8, and 9) suggests that degradation of a subpopulation of PML requires SLS4, whereas another subset of PML can be degraded in the absence of SLS4. The molecular mechanisms regulating PML degradation by ICP0 are fundamentally different from those that modulate ND10 fusion.
DISCUSSION
ND10 nuclear bodies are part of cell antiviral defense systems. Interferon (IFN) treatment increases the expressions of PML and Sp100 as well as the size and number of ND10 structures (38, 39). Many DNA viruses modify or disrupt ND10 as a counteraction to subjugate cell machineries and enhance viral replication (40–42). ICP0 of HSV-1 interacts with ND10 and degrades PML and Sp100 to alleviate the restrictions imposed by ND10 (8). In a previous study, we reported that ICP0 interacts with ND10 in three sequential steps: adhesion, fusion, and retention (34). It is appealing to hypothesize that at each ND10 interaction step, ICP0 binds to a distinctive set of ND10 components to orchestrate efficient viral replication.
A few lines of evidence suggest that some ND10 components may participate in HSV-1 replication. (i) HSV-1 transcription-replication compartments initiate near ND10 loci (29, 43). (ii) There are many transcriptional and posttranscriptional regulators in ND10, such as CBP, p53, mTOR, and eIF4E (21, 22, 44). Conceivably, the higher local concentration of regulatory factors in ND10 makes it easier for HSV-1 to adopt some of them to modulate its gene expression. (iii) Cellular proteins such as CoREST and CLOCK are evenly distributed throughout uninfected nuclei but are recruited to aggregate at ND10 during HSV-1 infection (45, 46), suggesting that they may be needed to establish HSV-1 replication.
More than 150 proteins were identified as ND10 partners in a recent database search (47). Many of these factors are recruited to ND10 only upon specific stimulations (28). It has been shown that within each ND10 body, PML and Sp100 are located in different patches (48), indicating that ND10 components are not in a uniform distribution inside ND10. The immense dynamics of ND10 inevitably cause the diversity in ND10 nuclear bodies (49). Subsets of ND10s have been demonstrated to be associated with different cell functions. For example, active transcription sites are found to be associated with only a portion and not all of the ND10 bodies (50), as are the telomere loci (51). With the technical limitation in the numbers of nucleic acids and proteins that can be simultaneously probed in situ, it remains unclear how subsets of ND10s, such as transcription-associated ND10 and telomere-associated ND10, relate to each other.
The dynamic interaction between ICP0 and ND10 raises the potential that through stepwise interactions between ICP0 and ND10, ICP0 can place both spatial and temporal regulations on its approach to ICP0 substrates and binding partners. In our studies, we observed that ICP0 mutants that fail to enter ND10 (as in RHG110, RHG126, RHG127, and RHG135 infections) juxtapose to a fraction of, but not all, ND10 bodies (Fig. 3A, panel h, B, and C and 5A, panel d). It would be interesting to find out whether ICP0 approaches a specific subset of ND10 nuclear bodies before it reaches out to all other ND10s and how the spatial-temporal regulation of ICP0 interacting with ND10 plays a role in HSV-1 replication. To answer these questions, we need to have a better understanding of ND10 dynamics and diversity in cell biology and also a further elucidation of the ICP0-ND10 interaction network during HSV-1 infection.
In this study, we have focused on the ND10 fusion process and identified three tandem segments (ND10-FS1, ND10-FS2, and ND10-FS3) located within the central region of ICP0 that are important for ICP0-ND10 fusion. Each of the three segments by itself is sufficient to drive ND10 fusion, but only when all three segments are simultaneously deleted can fusion be completely blocked. The existence of redundant elements that are each sufficient suggests the importance of ND10 fusion in HSV-1 infection. Redundant and cooperative interactions may also be the reason why ND10 fusion occurs at a massive speed. Although, with current technology, it is difficult to determine whether the three segments work synergistically to expedite the fusion process, the fact that ICP0-ND10 juxtaposition has never been observed for wild-type ICP0 indicates that the speed and force of ND10 fusion are astronomical. The experiments in this study were carried out with an MOI of 10 or higher so that ICP0 could reach a detectable level very early during infection (e.g., 1.5 h). The three segments seem to work equally well and are redundant at such a high MOI. It is not clear whether the three segments will have a different efficiency in driving ND10 fusion at a low MOI.
Sequence alignment showed that the three fusion segments shared 52.8% consensus positions and 9.4% identity positions in the Vector NTI AlignX program (Fig. 8). One conserved feature of all three segments is the unusually high proline content. Proline accounts for 6.3% of the total amino acids in the human proteome (52). The overall proline content for ICP0 is 13%. However, in the three ND10-FSs, proline constitutes 28% of the total of 150 amino acids. One characteristic property of the proline-rich domain is its structural flexibility (53). Since the known ICP0 functions, E3 ligase activity and protein interactions, are located mostly at the N and C termini (2, 35, 37, 54–56), a flexible central region may increase the versatility of ICP0 in its interactions with various substrates and binding partners by the N and C termini.
Proline-rich sequences tend to form a polyproline-II (PPII) helix, a structure different from the α helix and β sheet (57). The main function of PPII is to mediate protein-protein interactions (53, 57). A wide range of protein motifs are known to interact with PPII, including the SH3 domain (58, 59), the WW domain (60), and major histocompatibility complex class II (MHCII)-peptide epitopes (61). PPII has an extended structure and tends to locate on the surface of a protein, which bears less specificity and higher flexibility when interacting with its binding partners (53, 57, 62). This property favors transient interactions in dynamic complexes and suits ICP0 well when it comes to interactions with the diverse and ever-changing components of ND10. Identification of cellular partners that interact with the proline-rich ND10-FSs will help us understand the spatial-temporal regulation of the ICP0-ND10 association.
We found that the SUMO interaction with SLS4 is an important regulatory event in the E3 ubiquitin ligase activity of ICP0, consistent with what has been reported previously (19, 20). However, a SUMO interaction with SLS4 is not required for ND10 fusion, but it acts as an auxiliary element to enhance fusion. Conceivably, the SUMO-SLS4 interaction may stabilize the association between ICP0 PPII and the PPII-interacting protein(s). However, ND10-FS2 is the only fusion segment that contains a SUMO interaction domain. Again, although the three segments were equally functional in the high-MOI infection conducted in this study, the additional SUMO interaction in ND10-FS2 may have a more important role in a low-MOI infection.
In ND10 assembly, PML tends to form the outer shell, whereas PML binding partners are usually inside (49). Cuchet-Lourenco et al. recently reported an interaction between ICP0 and overexpressed PML isoform I in the absence of endogenous PML (63). Those researchers also identified ICP0 residues 241 to 388, which overlap both ND10-FS1 and ND10-FS2, as the PML isoform I interaction domain in a yeast two-hybrid assay (63). Although a direct interaction between ICP0 and endogenous PML has not yet been demonstrated, and electron microscopy images did not show an apparent clustering of ICP0 with PML (36), it is still tempting to presume that a very transient interaction between PML isoform I and ND10-FSs at the ND10 surface is important for ND10 fusion. Whether PML interacts with PPII or whether additional protein-protein interactions coordinate with the PML-ICP0 interaction remains to be investigated. Moreover, how this spatial effect of ND10 fusion regulates PML degradation and viral replication is also a key question to be asked in understanding HSV-1 infection and pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
These studies were supported by a Wayne State University startup fund awarded to Haidong Gu.
We thank Ann Arvin for the VZV ORF61 plasmid. We thank Mark Vanberkum and Bernard Roizman for candid suggestions in preparing the manuscript. We thank the Microscopy, Imaging and Cytometry Resources (MICR) Core facility at Wayne State University for technical support on confocal imaging.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03658-14.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Boutell C, Sadis S, Everett RD. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol 76:841–850. doi: 10.1128/JVI.76.2.841-850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagglund R, Van Sant C, Lopez P, Roizman B. 2002. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc Natl Acad Sci U S A 99:631–636. doi: 10.1073/pnas.022531599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chelbi-Alix MK, de The H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- 5.Parkinson J, Lees-Miller SP, Everett RD. 1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 73:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lomonte P, Sullivan KF, Everett RD. 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J Biol Chem 276:5829–5835. doi: 10.1074/jbc.M008547200. [DOI] [PubMed] [Google Scholar]
- 7.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82:2661–2672. doi: 10.1128/JVI.02308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM. 2013. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110:E4492–E4501. doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A 102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A 104:17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferenczy MW, Ranayhossaini DJ, Deluca NA. 2011. Activities of ICP0 involved in the reversal of silencing of quiescent herpes simplex virus 1. J Virol 85:4993–5002. doi: 10.1128/JVI.02265-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boutell C, Canning M, Orr A, Everett RD. 2005. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J Virol 79:12342–12354. doi: 10.1128/JVI.79.19.12342-12354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalamvoki M, Gu H, Roizman B. 2012. Overexpression of the ubiquitin-specific protease 7 resulting from transfection or mutations in the ICP0 binding site accelerates rather than depresses herpes simplex virus 1 gene expression. J Virol 86:12871–12878. doi: 10.1128/JVI.01981-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. 2002. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 16.Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. 2008. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455:813–817. doi: 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T, Guan J, Li S, Zhang X, Zheng X. 2014. HSCARG downregulates NF-kappaB signaling by interacting with USP7 and inhibiting NEMO ubiquitination. Cell Death Dis 5:e1229. doi: 10.1038/cddis.2014.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Y, Koppen A, Rakhshandehroo M, Tasdelen I, van de Graaf SF, van Loosdregt J, van Beekum O, Hamers N, van Leenen D, Berkers CR, Berger R, Holstege FC, Coffer PJ, Brenkman AB, Ovaa H, Kalkhoven E. 2013. Early adipogenesis is regulated through USP7-mediated deubiquitination of the histone acetyltransferase TIP60. Nat Commun 4:2656. doi: 10.1038/ncomms3656. [DOI] [PubMed] [Google Scholar]
- 19.Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog 7:e1002245. doi: 10.1371/journal.ppat.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett RD, Boutell C, Pheasant K, Cuchet-Lourenco D, Orr A. 2014. Sequences related to SUMO interaction motifs in herpes simplex virus 1 protein ICP0 act cooperatively to stimulate virus infection. J Virol 88:2763–2774. doi: 10.1128/JVI.03417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong S, Salomoni P, Pandolfi PP. 2000. The transcriptional role of PML and the nuclear body. Nat Cell Biol 2:E85–E90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- 22.Cohen N, Sharma M, Kentsis A, Perez JM, Strudwick S, Borden KL. 2001. PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. EMBO J 20:4547–4559. doi: 10.1093/emboj/20.16.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, Grosveld F, Pandolfi PP. 1998. Role of PML in cell growth and the retinoic acid pathway. Science 279:1547–1551. doi: 10.1126/science.279.5356.1547. [DOI] [PubMed] [Google Scholar]
- 24.Bernardi R, Pandolfi PP. 2003. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 22:9048–9057. doi: 10.1038/sj.onc.1207106. [DOI] [PubMed] [Google Scholar]
- 25.Carbone R, Pearson M, Minucci S, Pelicci PG. 2002. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene 21:1633–1640. doi: 10.1038/sj.onc.1205227. [DOI] [PubMed] [Google Scholar]
- 26.Salomoni P, Pandolfi PP. 2002. The role of PML in tumor suppression. Cell 108:165–170. doi: 10.1016/S0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 27.Zhou W, Bao S. 2014. PML-mediated signaling and its role in cancer stem cells. Oncogene 33:1475–1484. doi: 10.1038/onc.2013.111. [DOI] [PubMed] [Google Scholar]
- 28.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 29.Maul GG, Ishov AM, Everett RD. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- 30.Lukashchuk V, Everett RD. 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J Virol 84:4026–4040. doi: 10.1128/JVI.02597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maul GG, Everett RD. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J Gen Virol 75(Part 6):1223–1233. [DOI] [PubMed] [Google Scholar]
- 32.Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honore N, Doubeikovsky A, Duprez E, Pandolfi PP, Puvion E, Freemont P, de The H. 2001. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J Exp Med 193:1361–1371. doi: 10.1084/jem.193.12.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147:221–234. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu H, Zheng Y, Roizman B. 2013. The interaction of herpes simplex virus ICP0 with ND10 bodies: a sequential process of adhesion, fusion, and retention. J Virol 87:10244–10254. doi: 10.1128/JVI.01487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu H, Roizman B. 2009. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J Virol 83:181–187. doi: 10.1128/JVI.01940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopez P, Jacob R, Roizman B. 2002. Overexpression of promyelocytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J Virol 76:9355–9367. doi: 10.1128/JVI.76.18.9355-9367.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meredith M, Orr A, Elliott M, Everett R. 1995. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 209:174–187. doi: 10.1006/viro.1995.1241. [DOI] [PubMed] [Google Scholar]
- 38.Chelbi-Alix MK, Pelicano L, Quignon F, Koken MH, Venturini L, Stadler M, Pavlovic J, Degos L, de The H. 1995. Induction of the PML protein by interferons in normal and APL cells. Leukemia 9:2027–2033. [PubMed] [Google Scholar]
- 39.Stadler M, Chelbi-Alix MK, Koken MH, Venturini L, Lee C, Saib A, Quignon F, Pelicano L, Guillemin MC, Schindler C, DeThe H. 1995. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 11:2565–2573. [PubMed] [Google Scholar]
- 40.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Oliver SL, Sommer M, Rajamani J, Reichelt M, Arvin AM. 2011. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog 7:e1002157. doi: 10.1371/journal.ppat.1002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leppard KN, Emmott E, Cortese MS, Rich T. 2009. Adenovirus type 5 E4 Orf3 protein targets promyelocytic leukaemia (PML) protein nuclear domains for disruption via a sequence in PML isoform II that is predicted as a protein interaction site by bioinformatic analysis. J Gen Virol 90:95–104. doi: 10.1099/vir.0.005512-0. [DOI] [PubMed] [Google Scholar]
- 43.Everett RD, Sourvinos G, Leiper C, Clements JB, Orr A. 2004. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J Virol 78:1903–1917. doi: 10.1128/JVI.78.4.1903-1917.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. 2006. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 442:779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- 45.Gu H, Roizman B. 2009. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J Virol 83:4376–4385. doi: 10.1128/JVI.02515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A 107:17721–17726. doi: 10.1073/pnas.1012991107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Damme E, Laukens K, Dang TH, Van Ostade X. 2010. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci 6:51–67. doi: 10.7150/ijbs.6.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lang M, Jegou T, Chung I, Richter K, Munch S, Udvarhelyi A, Cremer C, Hemmerich P, Engelhardt J, Hell SW, Rippe K. 2010. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J Cell Sci 123:392–400. doi: 10.1242/jcs.053496. [DOI] [PubMed] [Google Scholar]
- 49.Lallemand-Breitenbach V, de The H. 2010. PML nuclear bodies. Cold Spring Harb Perspect Biol 2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiesslich A, von Mikecz A, Hemmerich P. 2002. Cell cycle-dependent association of PML bodies with sites of active transcription in nuclei of mammalian cells. J Struct Biol 140:167–179. doi: 10.1016/S1047-8477(02)00571-3. [DOI] [PubMed] [Google Scholar]
- 51.Chung I, Osterwald S, Deeg KI, Rippe K. 2012. PML body meets telomere: the beginning of an ALTernate ending? Nucleus 3:263–275. doi: 10.4161/nucl.20326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan AA, Rubenstein E. 2013. Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One 8:e53785. doi: 10.1371/journal.pone.0053785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rath A, Davidson AR, Deber CM. 2005. The structure of “unstructured” regions in peptides and proteins: role of the polyproline II helix in protein folding and recognition. Biopolymers 80:179–185. doi: 10.1002/bip.20227. [DOI] [PubMed] [Google Scholar]
- 54.Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J Virol 71:1019–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chaurushiya MS, Lilley CE, Aslanian A, Meisenhelder J, Scott DC, Landry S, Ticau S, Boutell C, Yates JR III, Schulman BA, Hunter T, Weitzman MD. 2012. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol Cell 46:79–90. doi: 10.1016/j.molcel.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Sant C, Kawaguchi Y, Roizman B. 1999. A single amino acid substitution in the cyclin D binding domain of the infected cell protein no. 0 abrogates the neuroinvasiveness of herpes simplex virus without affecting its ability to replicate. Proc Natl Acad Sci U S A 96:8184–8189. doi: 10.1073/pnas.96.14.8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adzhubei AA, Sternberg MJ, Makarov AA. 2013. Polyproline-II helix in proteins: structure and function. J Mol Biol 425:2100–2132. doi: 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 58.Ren R, Mayer BJ, Cicchetti P, Baltimore D. 1993. Identification of a ten-amino acid proline-rich SH3 binding site. Science 259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 59.Alexandropoulos K, Cheng G, Baltimore D. 1995. Proline-rich sequences that bind to Src homology 3 domains with individual specificities. Proc Natl Acad Sci U S A 92:3110–3114. doi: 10.1073/pnas.92.8.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen HI, Sudol M. 1995. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci U S A 92:7819–7823. doi: 10.1073/pnas.92.17.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. 1996. Crystallographic analysis of endogenous peptides associated with HLA-DR1 suggests a common, polyproline II-like conformation for bound peptides. Proc Natl Acad Sci U S A 93:734–738. doi: 10.1073/pnas.93.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siligardi G, Drake AF. 1995. The importance of extended conformations and, in particular, the PII conformation for the molecular recognition of peptides. Biopolymers 37:281–292. doi: 10.1002/bip.360370406. [DOI] [PubMed] [Google Scholar]
- 63.Cuchet-Lourenco D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J Virol 86:11209–11222. doi: 10.1128/JVI.01145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mullen MA, Ciufo DM, Hayward GS. 1994. Mapping of intracellular localization domains and evidence for colocalization interactions between the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. J Virol 68:3250–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.