

ABSTRACT
The life cycle of human papillomaviruses (HPVs) is dependent upon differentiation of the infected host epithelial cell as well as activation of the ataxia telangiectasia mutated (ATM) DNA repair pathway that in normal cells acts to repair double-strand DNA breaks. In normal cells, following DNA damage the acetyltransferase Tip60 must acetylate ATM proteins prior to their full activation by autophosphorylation. E6 proteins have been shown to induce the degradation of Tip60, suggesting that Tip60 action may not be required for activation of the ATM pathway in HPV-positive cells. We investigated what role, if any, Tip60 plays in regulating the differentiation-dependent HPV life cycle. Our study indicates that Tip60 levels and activity are increased in cells that stably maintain complete HPV genomes as episomes, while low levels are seen in cells that express only HPV E6 and E7 proteins. Knockdown of Tip60 with short hairpin RNAs in cells that maintain HPV episomes blocked ATM induction and differentiation-dependent genome amplification, demonstrating the critical role of Tip60 in the viral life cycle. The JAK/STAT transcription factor STAT-5 has previously been shown to regulate the phosphorylation of ATM. Our studies demonstrate that STAT-5 regulates Tip60 activation and this occurs in part by targeting glycogen synthase kinase 3β (GSK3β). Inhibition of either STAT-5, Tip60, or GSK3β blocked differentiation-dependent genome amplification. Taken together, our findings identify Tip60 to be an important regulator of HPV genome amplification whose activity during the viral life cycle is controlled by STAT-5 and the kinase GSK3β.
IMPORTANCE Human papillomaviruses (HPVs) are the etiological agents of cervical and other anogenital cancers. HPVs regulate their differentiation-dependent life cycle by activation of DNA damage pathways. This study demonstrates that HPVs regulate the ATM DNA damage pathway through the action of the acetyltransferase Tip60. Furthermore, the innate immune regulator STAT-5 and the kinase GSK3β mediate the activation of Tip60 in HPV-positive cells. This study identifies critical regulators of the HPV life cycle.
INTRODUCTION
Human papillomaviruses (HPVs) are double-strand DNA viruses that are the causative agents of cervical and other anogenital cancers (1). The high-risk HPV types, including HPV16, HPV18, HPV31, and HPV35, are sexually transmitted and play critical roles in the development of malignancy. HPVs infect cells in the basal layer of stratified epithelia and link the production of progeny virions to epithelial differentiation (2, 3). Upon infection of basal cells, HPVs establish their genomes as low-copy-number episomes at about 50 to 100 copies per cell. Following chromosomal replication and cell division, one of the daughter cells begins to differentiate as it migrates away from the basal layer. This leads to the activation of the late viral promoters and enhanced expression of viral replication proteins E1 and E2 (4–8). HPV proteins block cell cycle exit upon differentiation as well as activate the ataxia telangiectasia mutated (ATM) DNA damage to regulate late events in the viral life cycle.
The HPV genome encodes only a small number of proteins, and HPV depends on the action of host factors, such as polymerases and transcription factors, to mediate viral replication. Recently, activation of the ATM kinase (9, 10) has been shown to be necessary for HPV genome amplification (11). The phosphorylation of ATM triggers activation of various downstream targets, such as p53 and CHK2, along with the NBS1/BRCA1/SMC1 signaling pathway (12). In normal cells, ATM activation requires acetylation by the Tat interactive protein 60 (Tip60) acetyltransferase as well as recruitment to sites of double-strand breaks by the MRN complex (13, 14). Phospho-ATM (p-ATM) induces CHK2 and p53 phosphorylation, leading ultimately to arrest in S/G2 phase (15). These factors are constitutively activated in HPV-infected cells in the absence of obvious DNA damage, and the mechanisms underlying their activation remain unclear. Previous studies have shown that Tip60 is degraded by high-risk HPV E6 proteins, raising the question of how its function in ATM activation is circumvented in HPV-positive cells (16).
The histone acetyltransferase Tip60 is critical for activation of DNA repair but also plays roles in chromatin remodeling and histone acetylation (14). Tip60 acetylates the ε-amino groups of lysine residues on target proteins, including p53 (17), ATM (18), and histone H2A (19, 20). In normal cells, inhibition of Tip60 activity prevents acetylation of ATM, which is necessary for its autophosphorylation and activation (18). It has previously been shown that Tip60 acetylates ATM to facilitate its phosphorylation (21). Tip60 itself is activated by tyrosine phosphorylation through the action of the kinase glycogen synthase kinase 3β (GSK3β) (22, 23). Recent studies have shown that Epstein-Barr virus (EBV) activates Tip60 to promote viral replication (24) and that human T-cell lymphotropic virus type 1 utilizes Tip60 to enhance c-Myc transforming activity (25). In EBV infections, a virally encoded kinase, BGLF-4, rather than GSK3β mediates Tip60 activation (26). In contrast, Tip60 is degraded by human cytomegalovirus viral protein pUL27 (22) as well as the HPV E6 oncoprotein (16). These findings suggest that Tip60 function may be circumvented in HPV-positive cells and that ATM activation in these cells could be mediated through a mechanism independent of Tip60. In the present study, we investigated what role, if any, Tip60 has in regulating HPV replication. Our studies indicate that in cells with episomal HPV genomes, Tip60 levels are increased over those in normal cells or cells that express only E6 and E7 and its activity is necessary for HPV genome amplification. The activation of Tip60 in HPV-positive cells is shown to be mediated by STAT-5, a factor previously shown to activate the ATM pathway, acting in part through the kinase GSK3β.
MATERIALS AND METHODS
Cell culture.
Human foreskin keratinocytes (HFKs) were isolated from neonatal foreskins as previously described (27). To generate cell lines that maintain viral episomes, primary keratinocytes were transfected with recircularized viral genomes and selected for HPV-positive cells as previously described (27). HPV16, HPV18, and HPV31 are cell lines that stably maintain episomal copies of the corresponding viral genomes. HPV31E5KO is a cell line that expresses a genome with a knockout (KO) mutation in the E5 open reading frame (ORF). CIN612 cells are derived from a cervical intraepithelial neoplasia grade II (CIN II) biopsy specimen that stably maintains HPV31 episomes. E6- or E6E7-expressing cells were generated by infection of HFKs with recombinant retroviruses and selection, as previously described (28). All HFKs and HPV-positive cells were cultured in E medium supplemented with mouse epidermal growth factor (EGF; 5 ng/ml; Collaborative Biomedical Products, Bedford, MA) in the presence of mitomycin C-treated NIH 3T3 J2 fibroblast feeders (28). To induce differentiation, cells were cultured in keratinocyte basal medium (KBM) containing 1.5 mM CaCl2 but no other supplements for up to 96 h (27).
Antibodies and Western blot analysis.
The antibodies used in this study were as follows: anti-involucrin and anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) (Santa Cruz, Santa Cruz, CA), anti-p53 and anti-Tip60 (Calbiochem, Gibbstown, NJ); anti-STAT-5, anti-phospho-STAT-5 (anti-p-STAT-5), anti-CHK2, anti-ATM, anti-phospho-CHK2 (anti-p-CHK2; Thr68), anti-p-ATM (Ser1981), anti-acetyl-p53, anti-phospho-GSK3, anti-GSK3β, anti-GSK3α (Cell Signaling, Danvers, MA), and anti-phospho-Tip60 (anti-p-Tip60) (Abcam, Cambridge, MA).
For Western blot analysis, keratinocytes were first separated from J2 feeders by treatment with phosphate-buffered saline containing 0.5 mM EDTA for 2 min at room temperature. The cells were then collected and lysed in radioimmunoprecipitation assay lysis buffer on ice for 30 min. The lysates were then separated by gel electrophoresis and transferred to a membrane as previously described (29). The membranes were developed using ECL prime or ECL reagents (Amersham, Pittsburgh, PA). Chemiluminescence signals were detected using Eastman Kodak X-ray films.
Southern/Northern blot analysis.
For Southern blot analysis, HPV-positive cells were isolated as described above and lysed in Southern lysis buffer (400 mM NaCl, 10 mM Tris-HCl, 10 mM EDTA). The lysates were then incubated at room temperature with 50 μg/ml RNase A (Sigma-Aldrich, St. Louis, MO) for 15 min, followed by addition of 50 μg/ml proteinase K (Sigma-Aldrich, St. Louis, MO) at 37°C overnight to remove residual RNA and proteins. Total DNA was then exacted by phenol-chloroform and then digested with XhoI at 37°C for 2 h. The DNA samples were then processed as previously described (30). For Northern blot analysis, the cells were lysed in STAT-60 (Tel-Test, Friendswood, TX) according to the manufacturer's protocol. The RNA samples were then processed as previously described (30).
Lentiviral virion production and transduction.
Mission short hairpin RNA (shRNA)-expressing lentiviral vectors were purchased from Sigma-Aldrich, St. Louis, MO. Four Tip60-specific shRNA constructs in a TRC1 plasmid backbone (named constructs shTip60-1 to shTip60-4) were individually transfected into 293T cells to produce lentiviral particles. The transfection mixture included 5 μg shRNA plasmid DNA, 1.66 μg vesicular stomatitis virus G glycoprotein plasmid DNA, and 3.37 μg Gag-Pol-Tat-Rev plasmid DNA along with polyethyleneimine (Polysciences, Warrington, PA) (normalized for a 10-cm dish). Culture medium was changed after 24 h, and lentivirus-containing supernatants were collected after an additional 48 h and filter sterilized using 0.45-μm-pore-size syringe filters (Pall, Ann Arbor, MI). The filtered particles were concentrated by the use of Ultra-15 centrifugal filter units (Millipore, Billerica, MA). CIN612 cells were incubated with 5 ml fresh medium including concentrated Tip60 shRNA or scrambled shRNA control lentivirus-containing supernatant in the presence of 4 μg/ml hexadimethrine bromide (Polybrene; Sigma-Aldrich, St. Louis, MO) overnight at 37°C. The culture medium was changed, and the transduced cells were cultured in fresh E medium for an additional 48 h before analysis.
RT-PCR.
The cells were isolated as described above, and total RNA was extracted using a Complete miniprep kit (Zymo). Five micrograms of RNA was then transcribed into cDNA using a SuperScript first-round synthesis system (Invitrogen, Carlsbad, CA). The reverse transcription (RT) products were mixed with LightCycler 480 SYBR green I master mix (Roche, Indianapolis, IN), and PCR was performed using a LightCycler 480 instrument. The primer pairs used in the study were designed to be specific for the following: Tip60 (forward primer, 5′-AAGGGCCAGTACATCCTCAC-3′; reverse primer, 5′-AGTGCAGACACTTGGAGTCG-3′), GSK3β (forward primer, 5′-GGAGAACTGGTCGCCATCAAG-3′; reverse primer, 5′-ACATTGGGTTCTCCTCGGACC-3′), and GAPDH (forward primer, 5′-GAGGACAGAGACCCAGCTGCC-3′; reverse primer, 5′-TGGAATTTGCCATGGGTG-3′). The results are normalized to those for GAPDH, and the figures are representative of observations from 3 independent experiments, unless indicated otherwise. Significance was determined using Student's t test, and a P value of <0.05 was considered significant.
RESULTS
Tip60 levels are increased in keratinocytes that stably maintain HPV31 episomes.
The differentiation-dependent amplification of HPV genomes is dependent upon the activation of the ATM DNA damage pathway, and in normal cells the activation of ATM requires acetylation by Tip60. Previous studies showed that HPV16 E6 could induce the rapid turnover of Tip60 proteins (16), yet at the same time the ATM pathway is constitutively activated in cells with HPV episomes (11). We therefore investigated if HPV proteins activate ATM autophosphorylation in a manner that is independent of Tip60 action. The levels of Tip60 proteins in HPV-positive keratinocytes that stably maintain either HPV31, HPV16, or HPV18 episomes were first examined by Western blotting and compared to the levels in normal human keratinocytes. We observed that Tip60 levels in keratinocytes that maintain HPV episomes were increased compared to the levels in normal keratinocytes (HFKs) (Fig. 1A). Importantly, the levels of the active phosphorylated form of Tip60 were also increased in HPV-positive cells compared to normal cells. In contrast, keratinocytes that expressed HPV31 E6 alone or E6 and E7 exhibited levels of Tip60 reduced from those seen in normal cells (Fig. 1B). This indicates that the retention of high levels of Tip60 is specific to cells with complete HPV genomes. We also examined cells that contained episomes with mutations in the E5 ORF and found similar or slightly lower levels of Tip60 compared to those in HFKs, suggesting that E5 or a combination of factors, such as E1 or E2, may contribute to activation. We conclude that while expression of E6 alone can reduce Tip60 levels, other proteins expressed from complete HPV genomes can overcome this activity, resulting in increased levels of Tip60.
FIG 1.
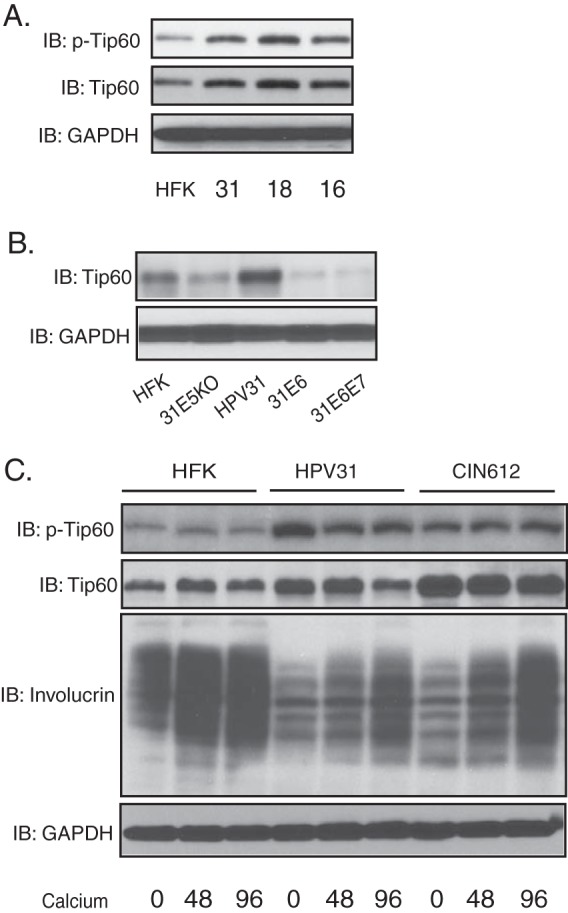
The levels of Tip60 are increased in HPV-positive cells that stably maintain episomes. (A) Western blot (immunoblot [IB]) analysis of Tip60, p-Tip60, and GAPDH levels in HFKs (lane HFK) and cells positive for HPV31 (lane 31), HPV18 (lane 18), and HPV16 (lane 16) grown in undifferentiated monolayer cultures. (B) Western blot analysis of Tip60 and GAPDH levels in HFKs and cells positive for HPV31 E5 KO (31E5KO), HPV31, HPV31 E6 (31E6), and HPV31 E6E7 (31E6E7) grown in monolayer cultures. (C) Western blot analysis of Tip60, p-Tip60, and GAPDH levels in HFKs, HPV31-positive keratinocytes, and HPV31-positive CIN612 cells differentiated in high-calcium medium for the times (in hours) indicated beneath the blots. All results are representative of observations from 3 independent experiments.
To determine how Tip60 levels were changed during the HPV life cycle, we examined the levels of Tip60 in HPV31-positive as well as normal keratinocytes upon differentiation in high-calcium medium. The addition of high-calcium medium to HPV-positive keratinocytes induces differentiation along with genome amplification within 48 to 96 h. As shown in Fig. 1C, the levels of Tip60 in HPV31-positive cells generated by transfection as well as CIN612 cells derived from an HPV31-positive biopsy specimen were similar to or slightly reduced from those in normal cells at late times of differentiation. Importantly, the active, phosphorylated forms of Tip60 were maintained at increased levels in HPV31-positive cells compared to those in HFKs throughout differentiation.
Tip60 knockdown by shRNAs blocks HPV31 genome amplification.
Since our studies indicate that the levels of p-Tip60 are maintained at high levels in cells with HPV episomes, it was important to determine if activated Tip60 played any role in the differentiation-dependent viral life cycle. To investigate if Tip60 was important for HPV genome amplification, protein levels were reduced using lentiviruses expressing shRNAs against Tip60. For this analysis, HPV31-positive CIN612 cells were infected with a series of recombinant lentiviruses expressing shRNAs against Tip60. At 72 h postransduction, cell lysates were harvested and assayed for Tip60 protein levels by Western blotting. Our data showed that two of the four Tip60-specific shRNAs, shTip60-1 and shTip60-4, significantly reduced the levels of Tip60 in CIN612 cells (Fig. 2A). For the subsequent experiments, lentivirus-containing supernatants from these two specific shRNAs were pooled to knock down Tip60 or examined individually. CIN612 cells transduced with shRNA-expressing lentiviruses were then induced to differentiate in high-calcium medium for an additional 72 h following infection. The cell lysates were assayed for Tip60 protein levels by Western blotting, and DNA was isolated for Southern blot analysis. The levels of Tip60 were unchanged upon differentiation in CIN612 cells transduced with nontargeting scrambled shRNA-expressing lentiviruses, whereas the levels were greatly reduced in cells transduced with lentiviruses expressing Tip60-specific shRNAs (Fig. 3). Importantly, Southern blot analysis showed that the loss of Tip60 impaired HPV31 genome amplification upon epithelial cell differentiation. Similar results were seen with each of two shRNAs when expressed individually (Fig. 2B). Finally, Northern blot analysis of these cells demonstrated a decrease in the levels of HPV late transcripts encoding E1^E4 and E5, which are expressed coordinately with amplification (Fig. 2C).
FIG 2.
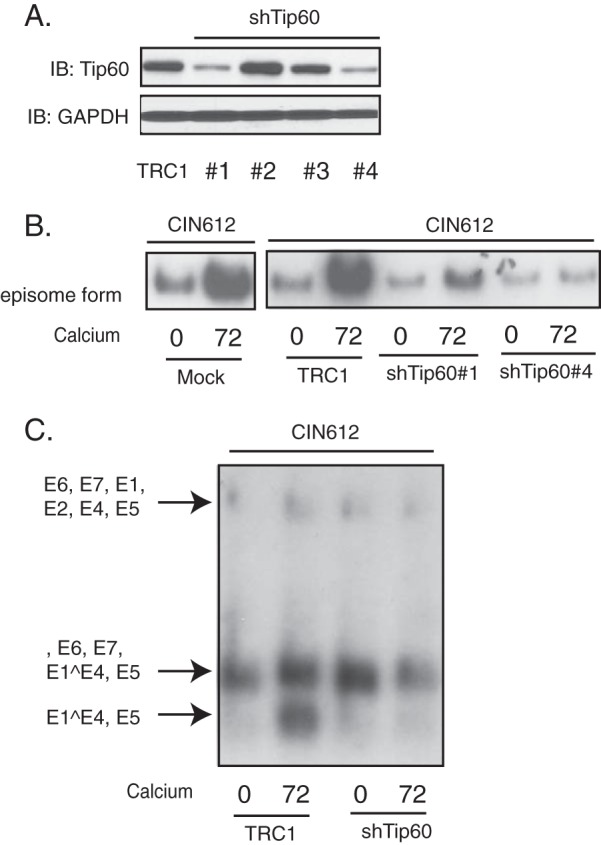
Suppression of Tip60 by shRNA knockdown blocks HPV genome amplification and late gene expression upon keratinocyte differentiation. HPV31-positive CIN612 cells were infected with lentiviruses expressing TRC1 scrambled shRNA or Tip60 shRNA and incubated for 72 h postinfection, followed by an additional 72 h of differentiation in high-calcium medium. (A) Western blot analysis of Tip60 and GAPDH proteins in monolayer CIN612 cells transduced with different shRNAs against Tip60. TRC1, TRC1 plasmid backbone; #1 to #4, shTip60-1 to shTip60-4, respectively. (B) Southern blot analysis for HPV31 episomes in CIN612 cells following infection with shRNA-expressing lentiviruses and differentiation in high-calcium medium for the times (in hours) indicated beneath the blots. (C) Northern blot analysis for HPV31 early and late gene expression in CIN612 cells following infection with shRNA-expressing lentiviruses and differentiation in high-calcium medium for the times (in hours) indicated beneath the blot. All results are representative of observations from 2 or more independent experiments.
FIG 3.
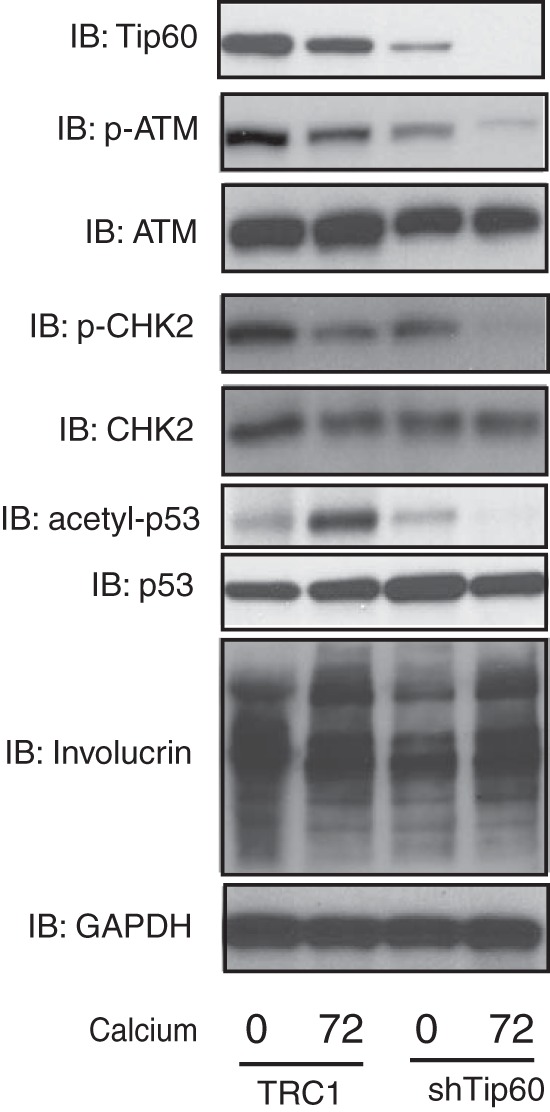
Knockdown of Tip60 suppresses activation of the ATM DNA damage response. HPV31-positive CIN612 cells were transduced with lentivirus expressing shRNAs against Tip60 (shTip60-1) as described in legend to Fig. 2. The transduced cells were assayed by Western blotting for Tip60, p-ATM, ATM, p-CHK2, CHK2, acetyl-p53, p53, and GAPDH protein levels upon differentiation in high-calcium medium for the times (in hours) indicated beneath the blots. All results are representative of observations from 2 or more independent experiments.
It was next important to examine how Tip60 participates in HPV genome amplification, and we investigated whether there was any alteration of the ATM DNA damage response in the Tip60-knockdown cells. Previous studies have shown that HPV-positive cells maintain high levels of phospho-ATM and phospho-CHK2 compared to those in HFKs (11). As shown in Fig. 3, the levels of phosphorylated ATM, but not the levels of total ATM, were significantly reduced in the cells expressing Tip60-knockdown shRNAs. Similar effects were seen with total and phosphorylated CHK2. Another target of Tip60 is p53 (31), and we found that the levels of acetylated p53 increased upon differentiation of HPV31-positive cells. p53 is a degradation target for high-risk HPV E6 proteins, including HPV31 E6; however, significant levels of p53 are retained in cells that stably maintain HPV31 episomes, where the level of E6 expression may be low (S. Hong et al., unpublished data) (Fig. 3). Knockdown of Tip60 was found to reduce the levels of acetylated p53 in HPV-positive cells upon differentiation (Fig. 3). We also confirmed that the levels of the differentiation marker involucrin increased in Tip60-knockdown cells, similar to the findings for the controls. These observations indicate that Tip60 is critical for activation of the DNA damage response as well as HPV genome amplification.
Tip60 activation is dependent on STAT-5.
It was next important to identify how HPV regulates Tip60. STAT-5 is a factor previously identified to be important in the HPV-induced activation of the DNA damage response (30), and we examined if it had any role in regulating Tip60 levels. For this analysis, we first examined the effects of the STAT-5 inhibitor pimozide, which specifically inhibits the phosphorylation of STAT-5 (30), on the levels of Tip60 in HPV-positive cells. The inhibitor has previously been shown not to affect keratinocyte differentiation or proliferation at the concentrations used (30). RT-PCR studies showed that the presence of pimozide did not change Tip60 mRNA levels, indicating that STAT-5 did not act to increase Tip60 transcription (Fig. 4A). As shown in Fig. 4B, the addition of pimozide greatly reduced the levels of phospho-Tip60 but not the total Tip60 levels, demonstrating that STAT-5 targets Tip60 activation.
FIG 4.
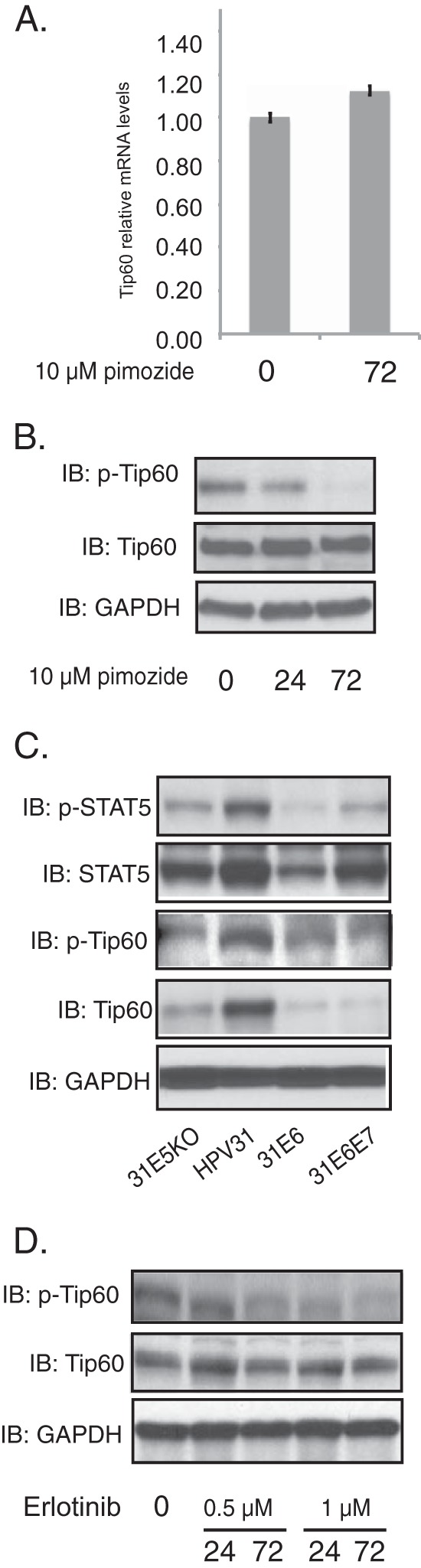
Inhibition of STAT-5 suppresses Tip60-dependent activation of the ATM DNA damage response. (A) RT-PCR analysis of Tip60 mRNA levels in CIN612 cells in the presence or absence of pimozide (10 μM) for the times (in hours) indicated beneath the bars. (B) Western blot analysis of Tip60, p-Tip60, and GAPDH protein levels of CIN612 cells for the times (in hours) indicated beneath the blots. (C) Western blot analysis for p-STAT-5, STAT-5, p-Tip60, Tip60, and GAPDH levels in cells positive for HPV31, HPV31 E5 KO, HPV31 E6, and HPV31 E6E7 grown in monolayer cultures. (D) Western blot analysis for p-Tip60, Tip60, and GAPDH levels in CIN612 cells treated with erlotinib at different concentrations for the various times (in hours) indicated beneath the blots. All results are representative of observations from 3 independent experiments.
Our studies demonstrate that the levels of p-Tip60 and p-STAT-5 are also significantly increased in cells that contain complete HPV genomes but are reduced in cells that express only E6 (Fig. 4C). Previous studies indicated that expression of E7 alone increased the levels of p-STAT-5 by approximately 2-fold. Our current analysis confirms that a moderate increase occurs due to E7 but also indicates that other viral proteins are likely the major mediators of p-STAT-5 activation. This is consistent with our observation that cells that maintain complete HPV genomes with stop codons in E5 exhibit decreased levels of p-STAT-5 (Fig. 4C). Since significantly higher levels of p-STAT-5 are observed in cells harboring complete viral genomes than cells expressing E6 and E7, it appears that E5, E2, or E1, alone or in combination with E7, is the major regulator of STAT-5 activation observed in cells with complete viral genomes.
HPV E5 proteins have been shown to interact with the EGF receptor (EGFR) and that this contributes to the transformation of HPV-infected cells (26, 32). We therefore tested whether Tip60 activation could be regulated through EGFR signaling by treating HPV-positive cells with the EGFR inhibitor erlotinib at two concentrations for various times (Fig. 4D). The treated cells were then lysed and assayed for p-Tip60, Tip60, and GAPDH by Western blot analysis. Our studies showed that the levels of p-Tip60 were reduced within 24 h when the cells were treated with 1 μM erlotinib, and this further decreased after 72 h of treatment. This indicates that the EGFR pathway, which is targeted by E5, is important for Tip60 phosphorylation.
STAT-5 acts on GSK3β to regulate Tip60 activation.
We next sought to determine what intermediate factors are involved in STAT-5-dependent Tip60 activation in HPV-positive cells. One factor that has been suggested to be an upstream regulator of Tip60 is glycogen synthase kinase 3β (GSK3β) (23, 33). Western blot analysis of HPV-positive cells demonstrated that the levels of GSK3β in HPV-positive cells were increased by approximately 2-fold compared to those in HFKs, and these levels were maintained at elevated levels throughout differentiation. In contrast, there was no change in GSK3α levels in HPV-positive cells (Fig. 5A). To determine whether GSK3β plays any role in HPV genome amplification, we used the GSK3β inhibitor CT98014 (34). CT98014 has been shown to be a specific inhibitor of GSK3β (35, 36). CIN612 cells were treated with CT98014 or vehicle, and we found that treatment with the GSK3β inhibitor significantly decreased HPV31 genome amplification upon differentiation (Fig. 5C). Importantly, the addition of CT98014 did not alter the rate of proliferation of treated cells. While CT98014 treatment reduced the levels of GSK3β, it had no effect on the levels of total GSK3α. In addition, inhibition of GSK3β resulted in a decrease in the levels of phospho-Tip60 and phospho-ATM but not the levels of total Tip60 and ATM (Fig. 5D). This indicates that GSK3β contributes to regulating Tip60 activity in HPV-positive cells.
FIG 5.
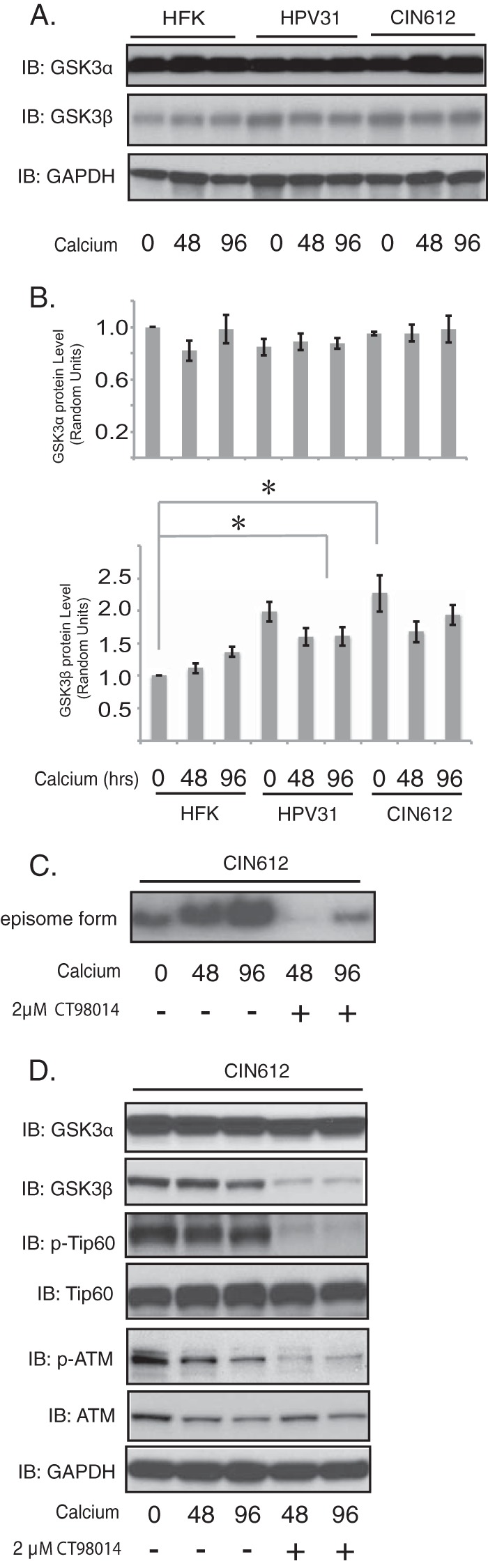
Inhibition of GSK3β activity by CT98014 blocks HPV genome amplification upon keratinocyte differentiation. (A) Western blot analysis of GSK3α, GSK3β, and GAPDH levels in HFKs, HPV31-positive keratinocytes, and HPV31-positive CIN612 cells differentiated in high-calcium medium for the times (in hours) indicated beneath the blots. (B) Relative levels of expression of target proteins normalized to the levels of GAPDH expression from the Western analysis whose results are shown in panel A. The statistical analysis was performed by 2-tailed t test. Data are means ± standard errors. *, P < 0.05. The band intensities were determined by ImageJ (64-bit) software. (C) Southern blot analysis for HPV31 episomes in CIN612 cells untreated or treated with CT98014 (2 μM) following differentiation in high-calcium medium for the times (in hours) indicated beneath the blots. (D) Western blot analysis of the GSK3α, GSK3β, p-Tip60, Tip60, p-ATM, ATM, and GAPDH proteins of CIN612 cells following differentiation induced in high-calcium medium in the presence or absence of CT98014 (2 μM) for the times (in hours) indicated beneath the blots. All results are representative of observations from 3 independent experiments.
Finally, we tested whether the levels of GSK3β were altered by STAT-5 inhibition in HPV-positive cells. CIN612 cells were induced to differentiate in high-calcium medium in the presence of pimozide, and cell extracts were isolated and examined for the levels of GSK3β and GSK3α. Suppression of STAT-5 activation resulted in reductions in the levels of GSK3β, but no change in GSK3α levels was observed (Fig. 6A). The levels of GSK3β mRNAs were also examined following pimozide treatment and were found to be decreased, suggesting that STAT-5 targets its expression (Fig. 6B). We conclude that p-STAT-5 acts in part through GSK3β to regulate p-Tip60 levels and to activate the ATM DNA damage response.
FIG 6.
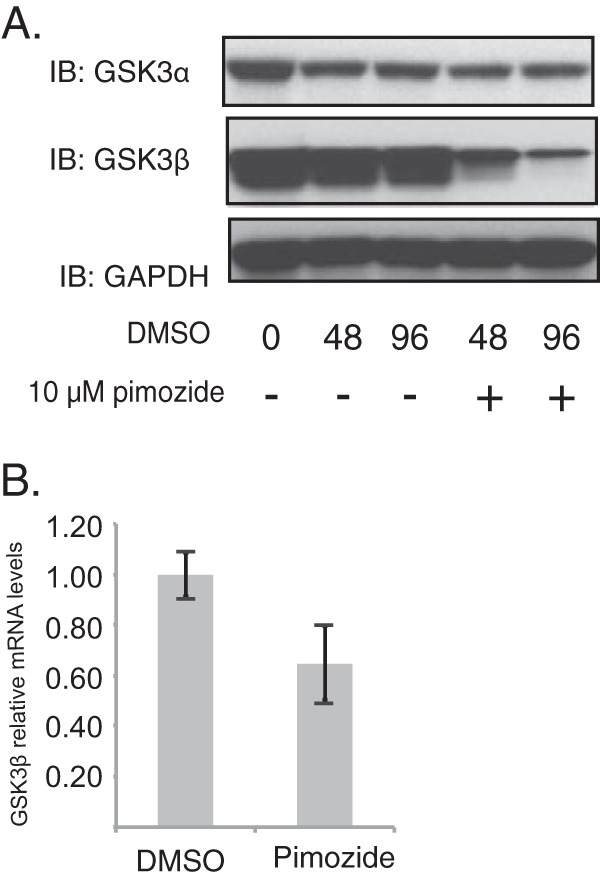
Loss of STAT-5 reduces GSK3β activation. (A) Western blot analysis of the GSK3α, GSK3β, and GAPDH proteins of CIN612 cells following differentiation induced in high-calcium medium in the presence or absence of pimozide (10 μM) for the times (in hours) indicated beneath the blots. (B) RT-PCR analysis of GSK3β mRNA levels in CIN612 cells in the presence or absence of pimozide (10 μM). The difference in the results between assays performed in the presence and the absence of pimozide was statistically significant (P < 0.05). All results are representative of observations from 3 independent experiments. DMSO, dimethyl sulfoxide.
DISCUSSION
In normal cells, Tip60 must acetylate the ATM kinase before it can undergo autophosphorylation and activation (14). The ATM pathway is constitutively activated in cells that stably maintain HPV episomes in the absence of external DNA-damaging agents, and activation of this pathway is necessary for the differentiation-dependent amplification of viral genomes. Previous studies demonstrated that high-risk HPV E6 proteins induce the rapid turnover of Tip60 (16), suggesting that HPV may circumvent the requirement for Tip60-directed acetylation for activation of the ATM pathway. Our studies demonstrate that Tip60 levels are increased in cells that stably maintain complete HPV episomes and that Tip60 activity is required for differentiation-dependent viral genome amplification. Furthermore, this increase in the levels of phosphorylated Tip60 is dependent on the action of the innate immune regulator STAT-5.
The knockdown of Tip60 in HPV-positive cells blocks ATM activation as well as genome amplification upon differentiation and alters the activity of many of its downstream targets. While the levels of phospho-ATM and phospho-CHK2 are dramatically reduced in Tip60-knockdown cells, there is no effect on the levels of total ATM and CHK2. The knockdown of Tip60 also prevents p53 acetylation, which may play a role in the DNA damage response in HPV-positive cells. In addition, Tip60 controls the acetylation of histone H2AX, which plays important roles in DNA repair (37) and has been shown to localize to HPV genomes during amplification (38). We conclude that Tip60 is critical for the induction of HPV genome amplification and it acts through a number of different factors to regulate DNA damage responses. This model is presented in Fig. 7.
FIG 7.
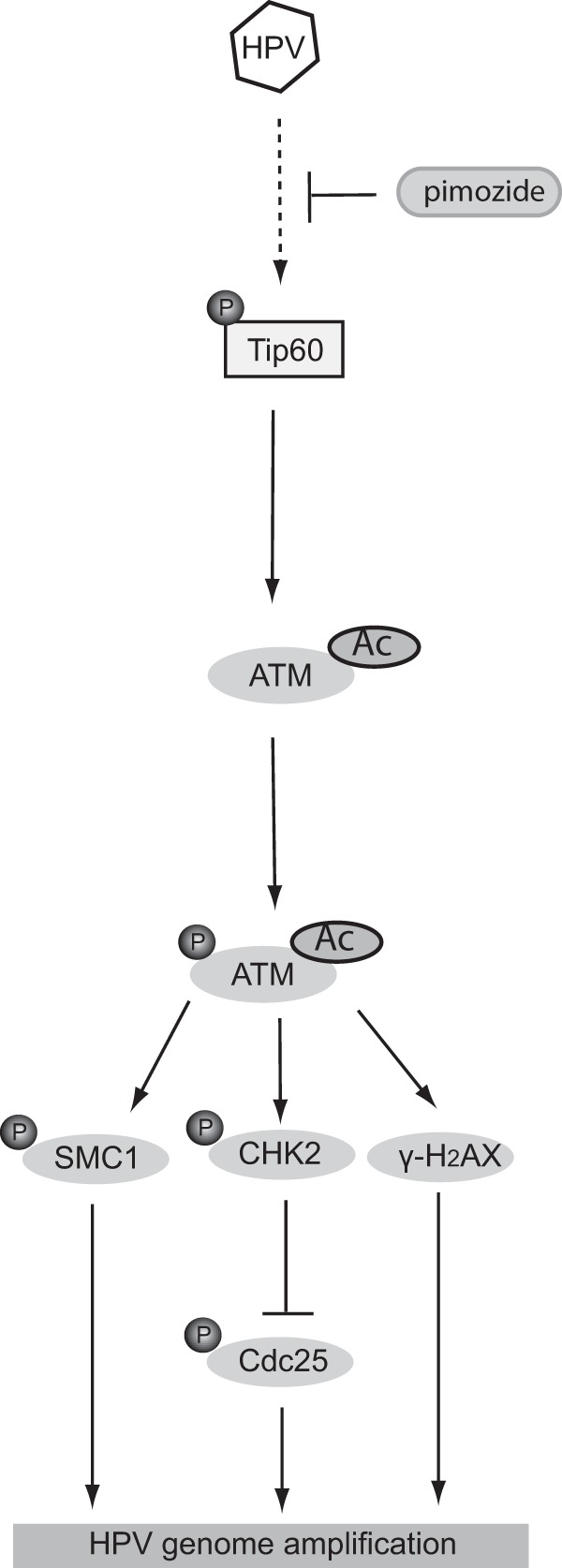
Model of how Tip60 activation by HPV could lead to ATM activation and amplification upon differentiation. HPV proteins work in combination to suppress E6 expression and/or function, leading to Tip60 activation. Tip60 activation can be suppressed by pimozide treatment, leading to inhibition of amplification. Activated Tip60 acetylates (Ac) the ATM kinase, resulting in ATM autophosphorylation. The activated ATM then induces the phosphorylation (P) of SMC1, CHK2, and γ-H2AX, which contribute to HPV genome amplification.
The innate immune regulator STAT-5 is an important regulator of the ATM pathway in HPV-positive cells (30). STAT-5 is constitutively phosphorylated in HPV-positive cells, and abrogation of its activity through the use of chemical inhibitors blocks ATM activation and genome amplification. Our studies demonstrate the importance of STAT-5 in the regulation of Tip60 phosphorylation, as inhibition of STAT-5 activation blocked Tip60 activation. While STAT-5 is a transcription factor, it does not control the levels of Tip60 transcripts, which remained unchanged. Since the levels of total Tip60 were moderately increased in HPV-positive cells, we suspect that HPV proteins may alter Tip60 protein turnover to increase its levels in cells with complete HPV genomes. Previous studies have indicated that the phosphorylation of STAT-5 can be induced at low levels by E7 (30); however, our ongoing studies indicate that other viral proteins act cooperatively to significantly enhance the levels above those seen with E7 alone. These other proteins include E5 and, possibly, E1, which has also been shown to activate the ATM DNA damage response in transient assays (39–41).
Our studies further indicate that the phosphorylation of Tip60 is controlled by the kinase GSK3β (23, 33) and that treatment with a GSK inhibitor blocks Tip60 activation as well as the phosphorylation of ATM. In addition, treatment with the GSK inhibitor results in reduced levels of viral episomes and impaired genome amplification. Importantly, our studies link STAT-5 activation with control of the levels and activities of GSK3β but not those of GSK3α. The levels of GSK3β are only moderately increased in HPV-positive cells, indicating that other factors, in addition to STAT-5, control its basal level of expression.
We further demonstrate that when HPV31 E6 is expressed by itself or in combination with E7, as in HPV-transformed cells, Tip60 levels are significantly reduced from the levels seen in normal cells. In contrast, when E6 is expressed in the context of complete viral genomes during the replicative cycle of the virus, an increase in the levels of total and phosphorylated Tip60 is observed. Similar effects were seen with genomes from HPV16 or HPV18. This increase in Tip60 levels may be due in part to the decreased expression of E6 from complete genomes, where early expression is repressed by the E2 protein. Upon integration of HPV genomes, which occurs in many cancers, expression of all HPV early genes except E6 and E7 is disrupted and degradation of Tip60 is prominent. It is also possible that other viral proteins, such as E1, E2, or E5, alone or in combination, could interfere with the E6-mediated degradation of Tip60. These models are supported by our observation that cells that stably maintain genomes with premature stop codon mutations in E5 showed reduced levels of Tip60, indicating that E5 contributes to the modulation of E6-mediated effects on Tip60. The addition of inhibitors of EGFR, a target of the E5 protein, also reduced Tip60 phosphorylation. Preliminary studies examining the transient expression of E2, E1, or E5 alone with retroviral vectors failed to activate Tip60, suggesting that a combination of factors may be responsible, and investigation of this mechanism will be a focus of future studies.
Overall, our studies indicate that HPV proteins activate STAT-5, resulting in activation of Tip60 and induction of the ATM DNA damage pathway. Furthermore, STAT-5 acts through GSK3β to regulate Tip60 activation. These observations link the innate immune regulator STAT-5 with the activation of Tip60 and the ATM DNA damage pathway to facilitate differentiation-dependent genome replication.
ACKNOWLEDGMENTS
We thank Jim DeCaprio, Renfeng Li, and Dianne Hayword for valuable reagents as well as Erika Langsfeld for helpful comments on the manuscript.
This work was supported by grants from NCI to L.A.L. (CA 59655 and CA 142861).
REFERENCES
- 1.zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Moody CA, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 3.Bodily J, Laimins LA. 2011. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol 19:33–39. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliveira JG, Colf LA, McBride AA. 2006. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc Natl Acad Sci U S A 103:1047–1052. doi: 10.1073/pnas.0507624103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poddar A, Reed SC, McPhillips MG, Spindler JE, McBride AA. 2009. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J Virol 83:640–650. doi: 10.1128/JVI.01936-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang CM, Ustav M, Stenlund A, Ho TF, Broker TR, Chow LT. 1992. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc Natl Acad Sci U S A 89:5799–5803. doi: 10.1073/pnas.89.13.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ustav M, Stenlund A. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J 10:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romanczuk H, Howley PM. 1992. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc Natl Acad Sci U S A 89:3159–3163. doi: 10.1073/pnas.89.7.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cremona CA, Behrens A. 2014. ATM signalling and cancer. Oncogene 33:3351–3360. doi: 10.1038/onc.2013.275. [DOI] [PubMed] [Google Scholar]
- 10.Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- 11.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong S, Laimins LA. 2013. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol 8:1547–1557. doi: 10.2217/fmb.13.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JH, Paull TT. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 14.Sun Y, Jiang X, Price BD. 2010. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle 9:930–936. doi: 10.4161/cc.9.5.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reinhardt HC, Yaffe MB. 2009. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 21:245–255. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jha S, Vande Pol S, Banerjee NS, Dutta AB, Chow LT, Dutta A. 2010. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol Cell 38:700–711. doi: 10.1016/j.molcel.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang Y, Luo J, Zhang W, Gu W. 2006. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 18.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. 2005. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR III, Abmayr SM, Washburn MP, Workman JL. 2004. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 20.Squatrito M, Gorrini C, Amati B. 2006. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 16:433–442. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Kaidi A, Jackson SP. 2013. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 498:70–74. doi: 10.1038/nature12201. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Reitsma JM, Savaryn JP, Faust K, Sato H, Halligan BD, Terhune SS. 2011. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe 9:103–114. doi: 10.1016/j.chom.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Liu Q, Ruan K, Wang Z, Zhang CS, Chien KY, Wu J, Li Q, Han J, Lin SC. 2012. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336:477–481. doi: 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, Desai P, Ambinder RF, Hayward GS, Qian J, Zhu H, Hayward SD. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10:390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, Motloch P, Takemoto S, Taguchi H, Cole MD, Luscher B, Dittrich O, Tagami H, Nakatani Y, McGee M, Girard AM, Gaughan L, Robson CN, Monnat RJ Jr, Harrod R. 2005. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol Cell Biol 25:6178–6198. doi: 10.1128/MCB.25.14.6178-6198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straight SW, Hinkle PM, Jewers RJ, McCance DJ. 1993. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol 67:4521–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fehrmann F, Laimins LA. 2005. Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol Biol 292:317–330. doi: 10.1385/1-59259-848-X:317. [DOI] [PubMed] [Google Scholar]
- 28.Hong S, Mehta KP, Laimins LA. 2011. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol 85:9486–9494. doi: 10.1128/JVI.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong S, Brass A, Seman M, Haag F, Koch-Nolte F, Dubyak GR. 2007. Lipopolysaccharide, IFN-gamma, and IFN-beta induce expression of the thiol-sensitive ART21 Ecto-ADP-ribosyltransferase in murine macrophages. J Immunol 179:6215–6227. doi: 10.4049/jimmunol.179.9.6215. [DOI] [PubMed] [Google Scholar]
- 30.Hong S, Laimins LA. 2013. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog 9:e1003295. doi: 10.1371/journal.ppat.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Legube G, Linares LK, Tyteca S, Caron C, Scheffner M, Chevillard-Briet M, Trouche D. 2004. Role of the histone acetyl transferase Tip60 in the p53 pathway. J Biol Chem 279:44825–44833. doi: 10.1074/jbc.M407478200. [DOI] [PubMed] [Google Scholar]
- 32.Pim D, Collins M, Banks L. 1992. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 7:27–32. [PubMed] [Google Scholar]
- 33.Charvet C, Wissler M, Brauns-Schubert P, Wang SJ, Tang Y, Sigloch FC, Mellert H, Brandenburg M, Lindner SE, Breit B, Green DR, McMahon SB, Borner C, Gu W, Maurer U. 2011. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol Cell 42:584–596. doi: 10.1016/j.molcel.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selenica ML, Jensen HS, Larsen AK, Pedersen ML, Helboe L, Leist M, Lotharius J. 2007. Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br J Pharmacol 152:959–979. doi: 10.1038/sj.bjp.0707471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, Ma ST, Reeder JW, Samuels I, Slabiak T, Wagman AS, Hammond ME, Harrison SD. 2003. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- 36.Tan JY, Sriram G, Rufaihah AJ, Neoh KG, Cao T. 2013. Efficient derivation of lateral plate and paraxial mesoderm subtypes from human embryonic stem cells through GSKi-mediated differentiation. Stem Cells Dev 22:1893–1906. doi: 10.1089/scd.2012.0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lowndes NF, Toh GW. 2005. DNA repair: the importance of phosphorylating histone H2AX. Curr Biol 15:R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 38.Gillespie KA, Mehta KP, Laimins LA, Moody CA. 2012. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol 86:9520–9526. doi: 10.1128/JVI.00247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadaja M, Isok-Paas H, Laos T, Ustav E, Ustav M. 2009. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog 5:e1000397. doi: 10.1371/journal.ppat.1000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J Virol 85:8996–9012. doi: 10.1128/JVI.00542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol 85:8981–8995. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]