

ABSTRACT
Similar to H5N1 viruses, A(H7N9) influenza viruses have been associated with severe respiratory disease and fatal outcomes in humans. While high viral load, hypercytokinemia, and pulmonary endothelial cell involvement are known to be hallmarks of H5N1 virus infection, the pathogenic mechanism of the A(H7N9) virus in humans is largely unknown. In this study, we assessed the ability of A(H7N9) virus to infect, replicate, and elicit innate immune responses in both human bronchial epithelial cells and pulmonary microvascular endothelial cells, compared with the abilities of seasonal H3N2, avian H7N9, and H5N1 viruses. In epithelial cells, A(H7N9) virus replicated efficiently but did not elicit robust induction of cytokines like that observed for H5N1 virus. In pulmonary endothelial cells, A(H7N9) virus efficiently initiated infection; however, no released infectious virus was detected. The magnitudes of induction of host cytokine responses were comparable between A(H7N9) and H5N1 virus infection. Additionally, we utilized differentiated human primary bronchial and tracheal epithelial cells to investigate cellular tropism using transmission electron microscopy and the impact of temperature on virus replication. Interestingly, A(H7N9) virus budded from the surfaces of both ciliated and mucin-secretory cells. Furthermore, A(H7N9) virus replicated to a significantly higher titer at 37°C than at 33°C, with improved replication capacity at 33°C compared to that of H5N1 virus. These findings suggest that a high viral load from lung epithelial cells coupled with induction of host responses in endothelial cells may contribute to the severe pulmonary disease observed following H7N9 virus infection. Improved adaptation of A(H7N9) virus to human upper airway poses an important threat to public health.
IMPORTANCE A(H7N9) influenza viruses have caused over 450 documented human infections with a 30% fatality rate since early 2013. However, these novel viruses lack many molecular determinants previously identified with mammalian pathogenicity, necessitating a closer examination of how these viruses elicit host responses which could be detrimental. This study provides greater insight into the interaction of this virus with host lung epithelial cells and endothelial cells, which results in high viral load, epithelial cell death, and elevated immune response in the lungs, revealing the mechanism of pathogenesis and disease development among A(H7N9)-infected patients. In particular, we characterized the involvement of pulmonary endothelial cells, a cell type in the human lung accessible to influenza virus following damage of the epithelial monolayer, and its potential role in the development of severe pneumonia caused by A(H7N9) infection in humans.
INTRODUCTION
Human infection with avian influenza A(H7N9) viruses has been documented in 14 provinces and municipalities in China to date, with additional cases in Taiwan, Hong Kong, Malaysia, and Canada (1, 2). More than 450 laboratory-confirmed human cases of A(H7N9) virus infection have been reported, with a high fatality rate, approximately 30% (2). Additional seasonal waves of human infection with A(H7N9) virus will likely continue and pose an ongoing threat to public health.
A(H7N9) virus infection has resulted in severe clinical outcomes in patients, including hospitalization (99%), pneumonia or respiratory failure (90%), acute respiratory distress syndrome (ARDS) (34%), and admission to an intensive care unit (63%) (3–5). This is in contrast to prior human infections with H7 viruses, which have typically manifested as mild respiratory illness and/or conjunctivitis, with only infrequent reports of severe respiratory disease (6). Epidemiological studies have revealed that severe and fatal cases of A(H7N9) virus infection share several clinical features and laboratory findings with highly pathogenic avian influenza (HPAI) H5N1 virus infection, including high viral load and exacerbated cytokine production (3, 7, 8). Similar to H5N1, A(H7N9) viruses are capable of efficient replication in human bronchus and lung tissues and are detected at high titers throughout the respiratory tracts of experimentally infected mammalian models (9–12). Furthermore, hypercytokinemia has been reported among severe and fatal cases with both H5N1 and A(H7N9) viruses (13–15).
Acute lung injury is associated with altered permeability of alveolar epithelial and endothelial barriers, endothelial injury, and dysregulated inflammation (16). While the association of acute lung injury following human infection with A(H7N9) virus necessitates a greater understanding of the ability of this virus to cause severe disease, there are only limited studies examining the tropism of H7 subtype viruses for human lung tissues and the induction of host responses in these cells following virus infection (9, 12, 17–21). In this study, we characterized the infectivity, replication, and elicitation of cytokines and inflammatory mediators following A(H7N9) virus infection of human bronchial epithelial cells and pulmonary microvascular endothelial cells. In bronchial epithelial cells, A(H7N9) virus efficiently initiated infection and replication, inducing increased levels of proinflammatory cytokine expression and production, similar to the case with seasonal H3N2 and avian H7N9 viruses but lower than with an HPAI H5N1 virus. However, infection of A(H7N9) virus damaged the integrity of the epithelial monolayer through significantly higher levels of cell necrosis than with the seasonal H3N2 virus. In pulmonary microvascular endothelial cells, A(H7N9) virus resulted in efficient initiation of infection in the absence of productive virus replication. In comparison to seasonal H3N2 virus infection, A(H7N9) virus infection induced high levels of cytokine expression and production, similar to those observed with the HPAI H5N1 virus. In differentiated primary human bronchial/tracheal epithelial cells, A(H7N9) virus infected both ciliated and mucin-secretory cells; however, it did not replicate equally well at 33°C and 37°C, a feature associated with inefficient human transmission among other avian viruses. Our data suggest that the high viral load generated from epithelial cells and elevated cytokine production from infected endothelial cells may be responsible for the development of severe pneumonia observed in A(H7N9) virus-infected patients.
MATERIALS AND METHODS
Viruses.
The viruses used in this study are listed in Table 1. Influenza A viruses were grown in the allantoic cavities of 10-day-old embryonated hens' eggs (H5 and H7 viruses) or in Madin-Darby canine kidney (MDCK) cells, clarified by centrifugation, and immediately frozen at −80°C until use (22). Virus titers were determined by standard plaque assay (23). The identity of virus genes was confirmed by sequence analysis to verify that no inadvertent mutations were present during the generation of virus stocks. All experiments were conducted in biosafety level 3 facilities with enhancements required by the U.S. Department of Agriculture and the Select Agent Program (24), in accordance with U.S. federal and World Health Organization guidelines.
TABLE 1.
Viruses used in this study
| Virus | Abbreviation | Subtype | Patient symptomsa | Trypsin dependentb |
|---|---|---|---|---|
| A/Texas/50/12 | Texas/50 | H3N2 | Respiratory | Yes |
| A/Anhui/1/13 | Anhui/1 | H7N9 | Fatal respiratory | Yes |
| A/shv/Egypt/00215-NAMRU3/07 | shv/Egypt/07 | H7N9 | NA | Yes |
| A/Vietnam/1203/04 | VN/1203 | H5N1 | Fatal respiratory | No |
| A/Brisbane/59/07 | Brisbane/59 | H1N1 | Respiratory | Yes |
| A/Mexico/7218/12 | Mx/7218 | H7N3 | Respiratory | No |
| A/Netherlands/219/03 | NL/219 | H7N7 | Respiratory | No |
| A/Thailand/16/04 | Thai/16 | H5N1 | Respiratory | No |
Cell culture and viral infection.
Immortalized human bronchial epithelium cells (Calu-3) and lung microvascular endothelial cells (HULECs) were grown as previously described (19, 23). During infection, Calu-3 cells grown on transwells and HULECs grown on 12-well plates were infected with virus at multiplicities of infection (MOIs) of 0.01 or 1 (for replication kinetics) and 1 (for immunofluorescence staining, cytokine quantification, real-time PCR, and cell death enzyme-linked immunosorbent assay [ELISA]) for 1 h. After washing, cell type-specific serum free medium was added to all wells. A total of 300 μg/liter of N-p-tosyl-l-phenylalanine chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich, St. Louis, MO) was added to medium for HULECs to facilitate hemagglutinin (HA) cleavage.
EpiAirway cells (MatTek, Ashland, MA) are an in vitro model of differentiated human bronchial and tracheal epithelial (HTE) cells cultured on transwells under air-liquid interface (ALI) for 2 to 3 weeks. The cells were infected apically with virus at an MOI of 0.01 (replication) for 1 h and maintained under ALI conditions. For replication kinetics, 200 μl of medium was added to the apical compartment for 20 min at each time point and collected for viral titer determination.
Immunofluorescence staining and microscopy.
To detect influenza A virus nucleoprotein (NP) antigen, Calu-3 cells grown on transwells and HULECs grown on collagen-coated 8-well chamber slides were infected with virus at an MOI of 1 for 1 h. At 8 h postinfection (p.i.), cells were fixed, permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 20 min, and incubated with mouse anti-NP monoclonal antibody A-3 (25), followed by fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Becton Dickinson Biosciences, San Diego, CA). Immunostained cells were counterstained with 4′,6-diamidino-2-phenyindole (DAPI) (Sigma-Aldrich) for nuclei and examined under a Zeiss Axioskop 2 fluorescence microscope. After counting of DAPI- and NP-positive cells in 4 independent 40× fields of view, the infection rate was determined as the number of NP-positive cells divided by the total cell number in each field.
Cytokine and chemokine quantification.
Calu-3 cells and HULECs were infected with virus at an MOI of 1. Supernatants were collected at 24 h p.i. and cytokine levels were examined using a customized BioPlex Pro assay according to the manufacturer's instructions (Bio-Rad, Hercules, CA). The customized cytokine panel included interleukin 1β (IL-1β), IL-6, IL-8, gamma interferon (IFN-γ), IP-10, MIP-1α, MIP-1β, RANTES, and tumor necrosis factor alpha (TNF-α). A minimum of three independent samples were collected and tested for each condition.
Cell death ELISA.
Calu-3 monolayers and HULECs were infected with virus at an MOI of 1. At 40 h p.i., cell supernatant and cell lysate were collected and examined using Cell Death Detection ELISAPLUS (Roche, Indianapolis, IN) for quantitative determination of cytoplasmic histone-associated DNA fragments according to the manufacturer's instructions. Mono- and oligonucleosomes from the supernatant (indicative of necrosis) or cytoplasm (indicative of apoptosis) were detected as absorbance at a wavelength of 405 nm.
Gene expression array.
Cells were infected with virus at an MOI of 1. Total RNA was extracted at 24 h p.i. from mock-infected or virus-infected Calu-3 cells or HULECs using the RNeasy minikit (Qiagen, Valencia, CA). The cDNA products were analyzed using the QuantiTect RT2 Profiler PCR array for 84 genes involved in human inflammatory cytokines and receptors (cytokine array; PAHS-011A) or 84 genes for the human NF-κB signaling pathway (NF-κB array; PAHS-025A) (Qiagen). The expression data from three independent treatments were analyzed through the vendor's web module (http://www.qiagen.com/us/products/genes%20and%20pathways/data-analysis-center-overview-page/). Genes with a >3-fold change and with statistical significance (P value of <0.05) compared to uninfected samples were normalized, calculated, and presented. Cluster 3.0/TreeView (Stanford University, CA) was used for hierarchical analysis of the array data.
Transmission electron microscopy (TEM).
Differentiated human EpiAirways cells (HTE cells) were infected with influenza viruses at an MOI of 1 from the apical surface. At 24 h p.i., cells were washed twice with PBS, fixed in 2.5% buffered glutaraldehyde for 1 h, and gamma irradiated (2 × 106 rads). Specimens were postfixed in 1% buffered osmium tetroxide, stained in 4% uranyl acetate, dehydrated, and embedded in epoxy resin. Finally, ultrathin sections were cut and examined with an FEI Tecnai Spirit electron microscope (FEI, Hillsboro, OR).
Statistical analysis.
Linear mixed models were used to estimate the means and differences for cytokine values and optical density (OD) values for cell apoptosis and necrosis among treatments using SAS software version 9.3 (SAS Institute Inc., Cary, NC). Logarithmic transformations of cytokine and OD values were performed to normalize the values as needed. The virus titers at 24 h p.i. were normalized by log transformation (log10), and comparison of means of the unmatched groups was performed using unpaired t test analysis. Nonparametric Mann-Whitney tests were used for statistical analysis of the area under the curve (AUC) for replication at 37°C and 33°C in human primary bronchial and tracheal epithelial cells. A P value of <0.05 was considered significant.
RESULTS
A(H7N9) virus infects human lung epithelial and endothelial cells with comparable efficiencies.
To assess the ability of A(H7N9) viruses to infect multiple cell types of the human lung, we utilized Calu-3 cells and HULECs, representing human lung epithelial and endothelial cells, respectively. Polarized Calu-3 cells express receptors for both human viruses (α2,6-linked sialic acids [SA]) and avian viruses (α2,3-linked SA) in relatively equal proportions (23), while HULECs express a greater abundance of α2,3-linked SA (19). Calu-3 cells grown on transwell inserts or HULECs grown on chamber slides were inoculated with the seasonal H3N2 virus (Texas/50) virus, H7N9 virus (Anhui/1), a genetically related avian H7N9 virus (shv/Egypt/07), or the HPAI H5N1 virus (VN/1203) at an MOI of 1. At 8 h p.i., the intracellular expression of influenza virus nucleoprotein (NP) was evaluated (Fig. 1A). Consistent with previous studies, rates of infection of pulmonary endothelial cells with Texas/50 and VN/1203 viruses were significantly lower than for airway epithelial cells (P < 0.005) (Fig. 1B) (19). In contrast, Anhui/1 virus infected the two cell types equally well, reaching a 30% infection rate. In airway epithelial cells, the infection rate of Anhui/1 virus was comparable to that of VN/1203 virus but significantly higher than those observed for Texas/50 and shv/Egypt/07 viruses (P < 0.05). In pulmonary endothelial cells, the infection rate of Anhui/1 virus was significantly higher than those of all other examined viruses (P < 0.05). These results indicate that A(H7N9) viruses are able to infect human endothelial cells and epithelial cells with comparable efficiencies, in contrast to H3N2 and H5N1 viruses, which preferentially infect lung epithelial cells.
FIG 1.
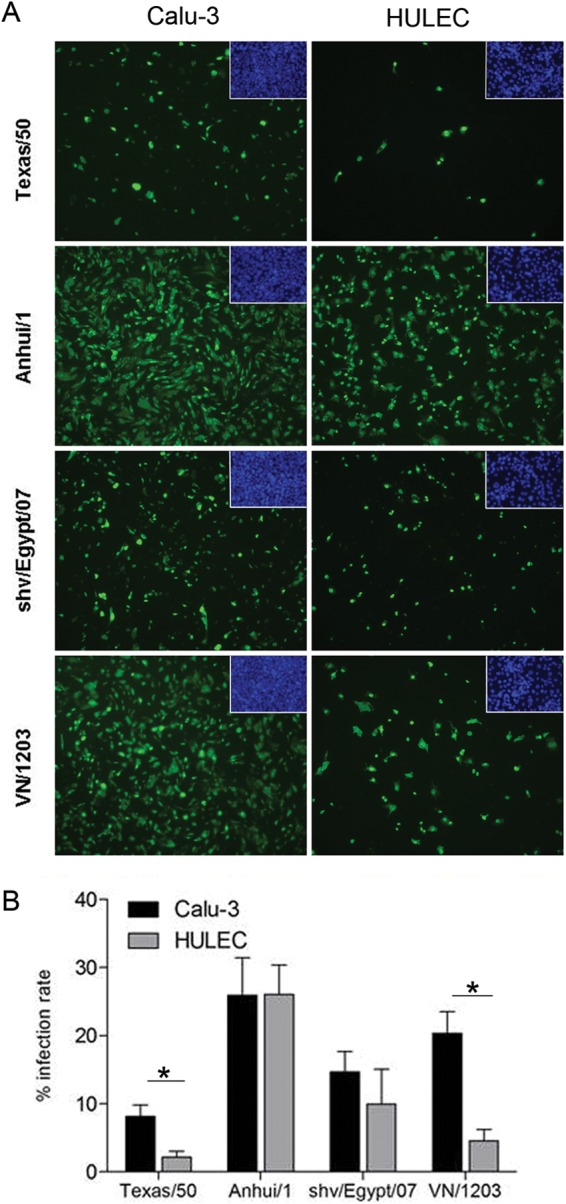
Infectivity of influenza viruses in human lung epithelial or endothelial cells. Calu-3 cells grown on transwell membranes or HULECs grown on chamber slides were infected with influenza virus at an MOI of 1 for 1 h. Cells were fixed and stained for influenza virus NP (green) or DAPI (insets, blue) at 8 h p.i. (A) Immunofluorescent staining of virus-infected cells. The confluent cell monolayers are shown as insets at the top right corners. (B) Assessment of infection rate in virus-infected cells. NP-positive cells and total cells were counted at a higher magnification (×400) for calculation of infection rate (number of NP-positive cells/number of total cells). Values represent the means of four independent areas, with the standard deviations indicated. An asterisk indicates a statistically significant difference between groups (P < 0.05).
A(H7N9) virus induces differential cytokine production in Calu-3 cells.
Hyperinduction of host proinflammatory responses has been detected following H5N1 virus infection of multiple cell types derived from the human respiratory tract (23, 26). However, H7 subtype viruses have generally exhibited a delayed and weakened induction of proinflammatory mediators following infection in these cell types (17, 20). As shown previously, A(H7N9) viruses replicated efficiently in Calu-3 cells (10) (Fig. 2), but their ability to elicit host proinflammatory responses in this cell type has not been previously examined. To determine if induction of host responses contributes to the severity of A(H7N9) virus infection, Calu-3 cells were infected with different subtype viruses and the production of proinflammatory cytokines and chemokines at 24 h p.i. was measured (Fig. 2). In general, all tested viruses replicated to similar levels and elicited significantly elevated cytokine and chemokine production in infected Calu-3 cells compared with that in uninfected cells (P < 0.05), with the highest levels induced by HPAI H5N1 virus. Similar to previous studies of H7 subtype viruses (17), Calu-3 cells infected with Anhui/1 virus elicited attenuated levels of numerous cytokines and chemokines compared with VN/1203 virus. Notably, levels of IP-10, IFN-γ, IL-6, TNF-α, and MIP-1β were significantly reduced during Anhui/1 virus infection compared with those in VN/1203 virus infection (P < 0.05). However, cells infected with Anhui/1 virus produced levels of RANTES and IL-8 that were comparable to those produced by VN/1203-infected cells, which were significantly higher than levels in Texas/50- and shv/Egypt/07-infected cells (P < 0.05). Anhui/1 virus induced significantly higher levels of IL-8, TNF-α, IFN-γ, and RANTES (P < 0.05) than did the seasonal Texas/50 virus. Previous work has shown that A(H7N9) viruses share high sequence similarity of surface glycoprotein genes (HA and neuraminidase [NA]) with avian H7N9 viruses, with internal genes derived from avian H9N2 viruses (27). Despite this genetic relatedness, significantly different cytokine production profiles were observed. Anhui/1 virus replicated to a higher titer and induced a significantly higher production of IL-6, IL-8, TNF-α, IFN-γ, MIP-1β, and RANTES than did shv/Egypt/07 virus. Our data indicate that A(H7N9) virus elicited cytokine production intermediate between those observed for H5N1 and H3N2 subtype viruses and higher than that elicited by the avian H7N9 virus.
FIG 2.

Cytokine production following influenza virus infection in human lung epithelial cells. Calu-3 cells were infected with influenza virus at an MOI of 1. Viral titer was determined by standard plaque assay at 24 h p.i. Supernatants were collected at 24 h p.i., and levels of each analyte were quantified by BioPlex. Values represent the means of triplicate independent cultures per virus plus standard deviations. Levels of each analyte in uninfected cells are indicated with a dashed line. An asterisk indicates a statistically significant difference between groups (P < 0.05).
A(H7N9) virus induces differential cytokine gene expression in Calu-3 cells.
To examine more broadly the induction of host innate immune responses in lung epithelial cells following A(H7N9) virus infection, the expression of a panel of genes involved with host inflammatory responses was examined using a cytokine array. Calu-3 cells were infected with Anhui/1, Texas/50, or VN/1203 virus at an MOI of 1, and total RNA was extracted at 24 h p.i. and analyzed by real-time PCR. Genes significantly upregulated (P < 0.05), with more than a 3-fold change following infection, were presented as fold change over mock-infected controls (Fig. 3). In agreement with cytokine protein levels shown in Fig. 2, infection with all three viruses resulted in the upregulation of numerous inflammation-related genes. Notably, the fold induction of TNF, IL-5, IL-8, CCL5, CCL16, CCL17, CCL18, CXCL9, CXCL10, and CXCL11 genes was significantly higher in VN/1203-infected cells than in Anhui/1- or Texas/50-infected cells (P < 0.05) (Fig. 3A). Despite this, Anhui/1 virus infection elicited expression levels of several genes comparable to those elicited by VN/1203 virus infection, such as the genes for IL-1A, IL-17C, IL-13, IL-8, CCL2, CCL20, CCL26, and CXCL2 (Fig. 3B), indicating an enhanced ability of this virus to induce innate host responses compared with that of seasonal Texas/50 virus. Numerous inflammation-related genes upregulated >3-fold and with P values of <0.05 following infection with any virus compared to the values in the uninfected group are presented in Table S1 in the supplemental material. In summary, infection with Anhui/1 virus resulted in a higher proinflammatory response than that observed for Texas/50 virus; however, the response was attenuated compared to that in VN/1203 virus infection.
FIG 3.

Fold regulation of selected genes related to human inflammatory cytokines and receptors following influenza virus infection of human lung epithelial cells. Calu-3 cells were infected with influenza viruses at an MOI of 1 in triplicate. Total RNA was isolated from cells at 24 h p.i. and examined by real-time RT-PCR array analysis. Selected genes which exhibited a significant (P < 0.05), >3-fold change over or under that in mock-infected cells are shown. (A) Selected genes (n = 17) for which infection with VN/1203 virus elicited significantly higher transcript levels than did Anhui/1 or Texas/50 virus infection. (B) Selected genes (n = 17) for which infection with Anhui/1 virus elicited transcript levels comparable to those elicited by VN/1203 virus infection.
H7 influenza viruses associated with human ocular disease have been previously shown to downregulate numerous genes related to NF-κB signaling in lung epithelial cells compared with virus subtypes typically associated with respiratory disease (18, 28). As A(H7N9) viruses have been associated with respiratory instead of ocular disease, we examined the induction of NF-κB signaling related genes in Calu-3 cells following Anhui/1 virus infection and compared our findings to previously published data (Fig. 4) (18, 28). Infection of Calu-3 cells with all viruses induced expression of several common genes (red), including those for IFN-β, IL-6, and TNF. However, Anhui/1 virus infection resulted in the significant induction of a broader panel of NF-κB-responsive genes than obtained with the HPAI H7 subtype viruses A/Netherlands/219/03 (H7N7; NL/219) and A/Mexico/7218/12 (H7N3; Mx/7218) (18, 28). While numerous genes associated with NF-κB signaling were downregulated (green) following Anhui/1 virus infection, the breadth of gene downregulation was muted compared with that with the HPAI H7 viruses and more closely resembled that of Thai/16, an HPAI H5N1 virus, and the seasonal Brisbane/59 (H1N1) virus. The data are presented in Table S2 in the supplemental material.
FIG 4.

Fold regulation of selected genes related to NF-κB signaling in human lung epithelial cells following influenza virus infection. Calu-3 cells were infected with virus at an MOI of 1. Total RNA was isolated in triplicate from cells at 24 h p.i. and examined by NF-κB array analysis. Selected genes (n = 61) that were significantly (P < 0.05) upregulated (red) or downregulated (green) (>3-fold) in infected cells compared with uninfected cells are shown using the hierarchical analysis of log2 transformation of fold change. The data can be found in Table S2 in the supplemental material.
A(H7N9) virus elicits elevated levels of proinflammatory cytokine production and gene expression in pulmonary microvascular endothelial cells.
Unlike bronchial epithelial cells, cultured endothelial cells (HULECs) possess an abundance of α2,3-linked sialic acids but do not produce host proteases for extracellular HA cleavage and were found only to support productive replication of the HPAI H5N1 virus (19). Despite the capability to initiate infection in endothelial cells (Fig. 1), Anhui/1 virus did not result in productive replication in HULECs, with titers of <100 PFU/ml detected at 24 h p.i., significantly lower than for VN/1203 virus at this time point (Fig. 5 and data not shown). These findings indicate that A(H7N9) virus is capable of infecting both lung epithelial and endothelial cells but replicates efficiently only in epithelial cells, in contrast with HPAI H5N1 viruses, which can infect and replicate productively in both cell types.
FIG 5.

Cytokine production following influenza virus infection in human lung endothelial cells. HULECs were infected with influenza virus at an MOI of 1. Viral titer was determined by standard plaque assay at 24 h p.i. Supernatants were collected at 24 h p.i., and levels of each analyte were quantified by BioPlex. The means from triplicate independent cultures per virus plus standard deviations are shown. Levels of each analyte in mock-infected cells are indicated with a dotted line. An asterisk indicates a statistically significant difference between groups (P < 0.05).
The role of endothelial cell involvement in lung inflammation was further examined by measuring the production of proinflammatory cytokines and chemokines in culture supernatants of infected HULECs. With the exception of RANTES, infected HULECs produced attenuated levels of cytokine production compared with those of mock-infected Calu-3 cells (Fig. 5). Compared to VN/1203 virus, Anhui/1 virus infection resulted in the production of significantly higher levels of IP-10 and RANTES (P < 0.05), similar levels of IL-6 and IFN-γ, and significantly lower levels of MIP-1β (Fig. 5). Furthermore, production of IL-6, IP-10, and RANTES in Anhui/1 virus-infected cells was significantly higher than those in Texas/50- and shv/Egypt/07-infected cells. Levels of IL-8 (except VN/1203) and TNF-α were not significantly elevated in HULECs following infection with any subtype viruses over uninfected cells (Fig. 5 and data not shown).
Next, the expression profiles of 84 genes related to inflammatory responses were further investigated during infection of HULECs. HULECs were infected with Texas/50, Anhui/1, or VN/1203 virus at an MOI of 1, and total RNA was extracted at 24 h p.i. and analyzed with a cytokine real-time (RT)-PCR array. Unlike that observed in airway epithelial cells, influenza virus infection of pulmonary endothelial cells did not result in a robust induction of inflammatory cytokine and receptor genes (Fig. 6). Only Anhui/1 and VN/1203 virus infection resulted in a significant (>3-fold) upregulation of select cytokine genes compared with mock-infected cells; infection with H3N2 virus did not elicit significant induction of any gene examined (Fig. 6). In summary, influenza virus infection of human pulmonary endothelial cells did not elicit significant cytokine gene expression compared to that observed in epithelial cells; however, A(H7N9) virus was able to induce production of several inflammatory cytokines to a level similar to that for HPAI H5N1 virus.
FIG 6.
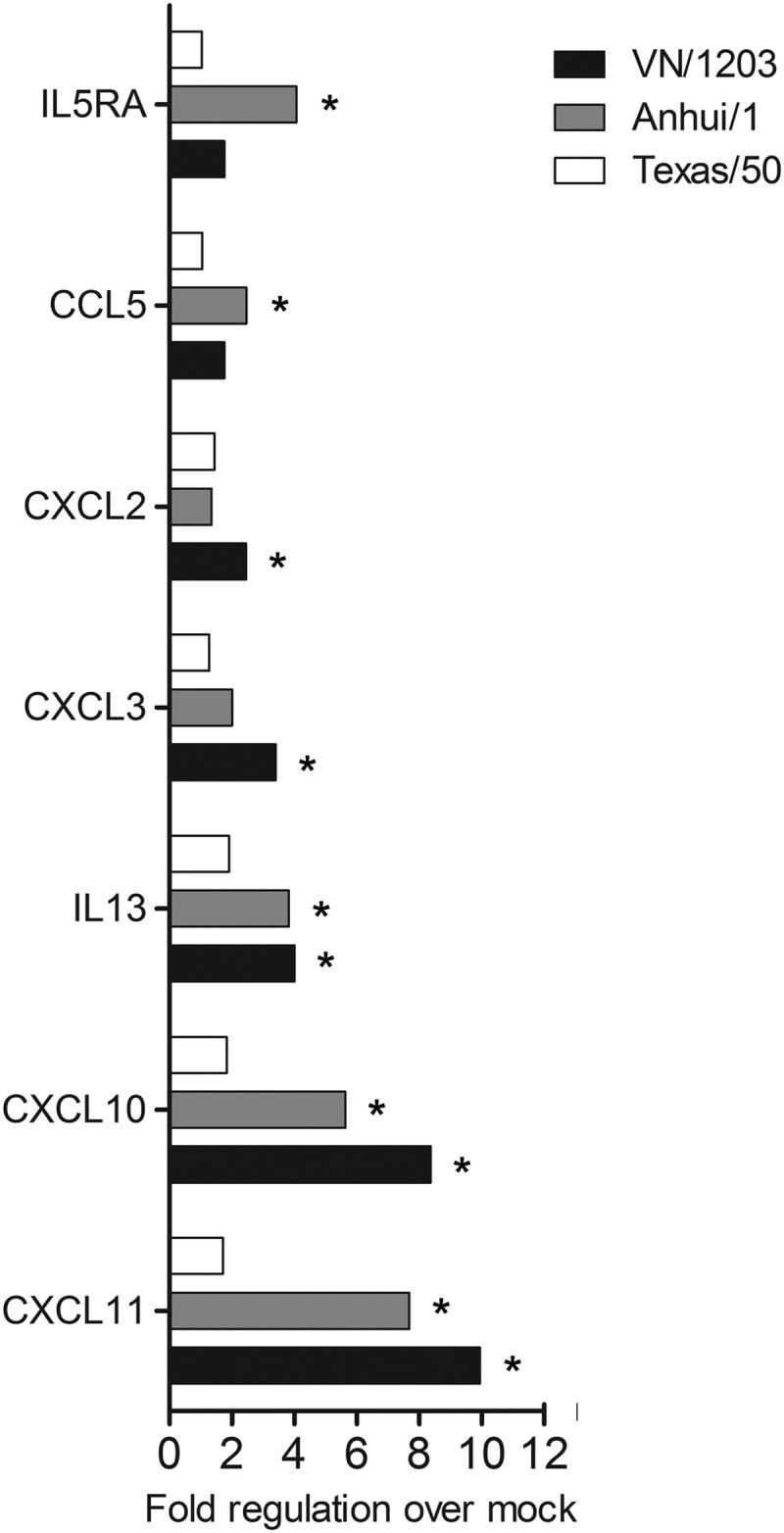
Fold regulation of selected genes related to human inflammatory cytokines and receptors following influenza virus infection of human lung endothelial cells. HULECs were infected with influenza viruses at an MOI of 1. Total RNA was isolated in triplicate in cells at 24 h p.i. and examined by cytokine array analysis. Selected genes which exhibited a significant (P < 0.05), >3-fold change in any conditions compared with uninfected cells are shown. An asterisk indicates that gene expression was significantly induced by virus infection compared to that in uninfected cells (P < 0.05).
A(H7N9) virus induced significantly elevated necrotic cell death in human bronchial epithelial cells.
Following virus infection of Calu-3 cells, the extent of transmembrane resistance was clearly dependent on virus strain. We observed that Calu-3 cells maintained better transmembrane resistance, monolayer integrity, and cell barrier function during the course of infection with the seasonal Texas/50 virus than observed in avian (Anhui/1 and VN/1203) virus-infected cultures. Programmed cell death, or apoptosis, resulting from virus infection is induced as a host innate immune response. To examine cell death (both apoptosis and necrosis) during virus infection, polarized Calu-3 monolayers and HULECs were infected with virus at an MOI of 1. At 40 h p.i., the supernatant and cell lysate were analyzed using a Cell Death Detection ELISAPLUS kit for necrosis and apoptosis. In Calu-3 cells, infection with all viruses resulted in comparable levels of apoptosis which were significantly higher than in uninfected cells (P < 0.05), whereas the proportion of cell death due to necrosis varied. Infection with Anhui/1 virus resulted in a level of necrosis similar to that obtained with VN/1203 virus and significantly higher than that obtained with Texas/50 and Shv/Egypt/07 viruses (Fig. 7). The monolayers of HULECs retained their integrity, and no significant cell death (apoptosis or necrosis) was detected following infection with any virus (data not shown). In summary, infection with A(H7N9) virus resulted in apoptosis and a higher level of necrosis than with H3N2 virus infection in human bronchial epithelial cells, which may contribute to the severe clinical manifestations associated with A(H7N9) virus infection.
FIG 7.

Infection of A(H7N9) virus results in both apoptosis and necrosis in human bronchial epithelial cells. Calu-3 cells grown on transwells were infected apically with viruses at an MOI of 1. At 40 h p.i., cell lysates and supernatants were analyzed using a Cell Death Detection ELISAPLUS kit for apoptosis and necrosis, respectively. Values represent the mean OD values of three independent experiments plus standard deviations. An asterisk indicates that Anhui/1 virus induced a significantly higher level of necrosis than did the other tested viruses (P < 0.05).
Cellular tropism of A(H7N9) virus and impact of temperature on the replication of A(H7N9) virus in differentiated primary human airway epithelial cells.
The potential for further adaptation of A(H7N9) virus in humans is a public health concern. In this study, we utilized differentiated human primary bronchial and tracheal epithelial (HTE) cells to further investigate A(H7N9) virus infection. This in vitro cell model, containing stratified epithelium structure with the presence of tight junctions and ciliated and mucus-secretory cells, serves as a rigorous representative of the environment of the human airway (29). The model has been used previously to study viral replication and host responses to seasonal and pandemic influenza virus (29). We first examined the cellular tropism of A(H7N9) virus during infection. HTE cells were washed and infected with Anhui/1 virus apically at an MOI of 1, and cells were fixed at 24 h p.i. and analyzed by transmission electron microscopy. As shown in Fig. 8, HTE cells preserved the pseudostratified epithelium structure present in the human trachea (Fig. 8A). During infection, Anhui/1 virions with a uniform spheroidal shape distribution were released from both goblet cells (Fig. 8B) and ciliated cells (Fig. 8C), indicating that A(H7N9) virus is capable of infecting both goblet and ciliated cells.
FIG 8.

Transmission electron microscopy demonstrates release and cellular tropism of A(H7N9) virus in differentiated primary human bronchial and tracheal epithelial cells. HTE cells were infected apically with Anhui/1 virus at an MOI of 1 and fixed at 24 h p.i. for examination by transmission electron microscopy. (A) Low-magnification view showing multiple layers of cells growing on a membranous insert (I). The arrow points to an area seen at higher magnification in panel B. G, goblet cell; C, ciliated cell. Bar, 2 μm. (B) Goblet cell with numerous extracellular spherical virions and one budding particle (arrow). M, mucin-containing granule. Bar, 100 nm. (C) Ciliated cell with released virions, and a particle (arrow) budding from microvillus. C, cilium. Bar, 500 nm.
Previous studies have suggested an association between the transmissibility of influenza viruses in ferrets and the ability of the virus to replicate efficiently at the lower temperature (33°C) found in the mammalian upper airway (30). Thus, we next assessed the ability of the A(H7N9) virus to replicate at two different temperatures compared with that of seasonal and HPAI H5N1 virus. HTE cells were infected apically with Brisbane/59, VN/1203, or Anhui/1 virus at an MOI of 0.01, and replication kinetics were evaluated at either 37°C or 33°C. All viruses examined replicated to similar titers through 72 h p.i. when cells were maintained at 37°C (Fig. 9). However, differential kinetics of replication were observed among tested viruses at 33°C. Only Brisbane/59 virus replicated equally well at both temperatures and reached similar titers at all time points examined. In contrast, it was determined that both Anhui/1 and VN/1203 viruses replicated to significantly reduced titers at 33°C (light gray) than at 37°C (dark gray) from 2 to 48 h p.i. (P < 0.05) (Fig. 9) when the area under the curve (AUC) was analyzed. Brisbane/59 possessed 98% replication capacity (AUC at 33°C/AUC at 37°C), followed by Anhui/1 virus at 72% and VN/1203 virus at 58%. These results demonstrate that in human airway cells, A(H7N9) virus does not replicate as well as human influenza viruses at the lower temperatures found in the upper airway of mammals. However, A(H7N9) virus was able to replicate with greater efficacy at 33°C than was the HPAI H5N1 virus.
FIG 9.

Replication kinetics study of both human and avian influenza viruses in differentiated primary human bronchial and tracheal epithelial cells. HTE cells were infected apically with virus at an MOI of 0.01 and cultured at either 37°C or 33°C for the duration of the time course. At each time point, 200 μl of medium was added to the apical surface for 20 min and collected for viral titer determination. Values represent the means of three independent experiments plus standard deviations. (A) Replication of viruses at 37°C; (B) replication of Brisbane/59 virus at 37°C and 33°C; (C) replication of Anhui/01 virus at 37°C and 33°C; (D) replication of VN/1203 virus at 37°C and 33°C. *, virus replicated to a significantly higher titer at 37°C than at 33°C (P < 0.05) at 24 h and 48 h p.i. according to the area under the curve (AUC).
DISCUSSION
Human infection with A(H7N9) virus can cause severe disease of the lower respiratory tract, and many patients progress to moderate to severe ARDS (3, 5). Although multiple studies have examined A(H7N9) virus infection in human bronchial epithelial cells, and host responses have been investigated in human alveolar epithelial cells and macrophages (9, 10), the role of pulmonary endothelial cells in A(H7N9) pathogenesis is largely unknown. In this study, we found that A(H7N9) virus infected bronchial epithelial and pulmonary endothelial cells with comparable efficiencies. A(H7N9) virus infection of bronchial epithelial cells led to productive virus replication but elicited an attenuated production of proinflammatory cytokines compared with that of H5N1 virus-infected cells. In contrast, A(H7N9) virus infection of pulmonary endothelial cells resulted in levels of host inflammatory mediators comparable to those induced by H5N1 virus despite the lack of productive virus replication.
The ongoing detection of human cases suggests that A(H7N9) virus possesses a degree of adaptation to mammalian hosts and only a few mutations are likely to be required for further mammal or human adaptation. Receptor specificity is a known determinant of host range restriction and can modulate cellular tropism, pathogenesis, and transmission in mammals (31). A(H7N9) viruses exhibit an increased affinity for human-like α2,6-linked SA receptors compared with other avian H5 and H7 viruses, while maintaining an avian-like α2,3-linked SA receptor binding preference (32–36). The abundant attachment of A(H7N9) viruses to epithelial cells distributed throughout the human respiratory tract suggests that A(H7N9) viruses not only are capable of causing severe pulmonary disease (similar to H5N1 viruses) but also concurrently possess an enhanced ability for upper airway infection, similar to seasonal influenza viruses (37). Furthermore, A(H7N9) viruses have been shown to readily infect and replicate in ex vivo and in vitro cultures derived from numerous sites throughout the human respiratory tract, including the trachea, bronchus, and lung (9, 10, 38). In agreement with these observations, we showed that A(H7N9) virus can infect and replicate efficiently in differentiated primary bronchial and tracheal epithelial cells, targeting both ciliated and mucin-secretory cells, providing greater insight into the cellular tropism of this novel virus. In contrast to H5 and H7 subtype viruses isolated from avian species that possess PB2 627E and 701D, A(H7N9) viruses isolated from humans bear either 627K or 701N, which are commonly detected among human viruses (34). The presence of amino acids 627K and 701N in PB2 has been associated with host range, increased virulence, increased transmission in mammals, and efficient viral replication at 33°C (30, 39–42). In the current study, we found that A(H7N9) virus possessed increased infectivity and replication efficiency and induced higher host responses in human bronchial epithelial cells compared with a precursor avian H7N9 virus at 37°C. However, despite the presence of molecular determinants (in HA and PB2) associated with mammalian host adaptation (43), this virus was unable to replicate as efficiently as seasonal influenza virus at the temperature (33°C) representing the upper respiratory tract. This suggests that A(H7N9) viruses have not yet acquired all the features required for mammalian host adaptation, including efficient respiratory droplet transmission (10, 11, 34, 44). However, heightened replication of A(H7N9) virus at 33°C compared with that of H5N1 virus indicates increased adaptation to the human upper airway environment and highlights the need for continued surveillance of this emerging virus.
A(H7N9) virus elicits a cytokine transcription profile different from that of other H7 subtype viruses in human bronchial epithelial cells (17, 18, 20, 28). Moreover, A(H7N9) viruses do not display an ocular tropism typically associated with subtype H7 influenza viruses (6). It was previously shown that the transcriptomic response in human bronchial epithelial cells to A(H7N9) virus was more similar to the response to human H3N2 virus than to the response to either avian H5 or H7 subtype virus (45). We and others have shown previously that H7 subtype viruses associated with ocular disease in humans are generally highly infectious in human bronchial epithelial cells but elicit delayed and weakened induction of host inflammatory responses and NF-κB signaling compared with H5N1 viruses (17, 18, 20, 28). In contrast, while A(H7N9) viruses maintained the high infectivity and replicative ability in human bronchial epithelial cells observed with other H7 viruses (Fig. 1 and 2), this virus induced a differential expression profile of NF-κB signaling genes compared with prior H7 viruses examined. A(H7N9) viruses more closely resemble H5N1 virus and human H1N1 virus in their ability to elicit heightened expression of genes associated with NF-κB signal transduction compared to that elicited by other H7 viruses associated with conjunctivitis in human bronchial epithelial cells (Fig. 4) (18). Collectively, these findings suggest that A(H7N9) viruses have the ability to induce a proinflammatory response (NF-κB) similar to that elicited by seasonal or H5N1 viruses, but less so than other H7 subtype viruses (9).
Engagement of pulmonary endothelial cells in severe pulmonary disease, including HPAI H5N1 virus infection, plays an important role in lung pathogenesis and potential systemic infection (19, 46, 47). The normal human lung typically consists of 30% pulmonary endothelial cells, which play a central role in mounting aggressive innate responses with early recruitment of inflammatory leukocytes to the lung during influenza virus infection (47). Pulmonary endothelial cells have been shown to support HPAI H5N1 virus replication, with heightened expression of proinflammatory mediators and decreased cell viability and proliferation following virus infection (19). However, the infectivity and replication of A(H7N9) viruses had not been previously examined in this cell type. In the current study, we determined that both H5N1 and seasonal influenza viruses exhibit reduced infectivity of pulmonary endothelial cells compared to bronchial epithelial cells. However, our unexpected finding of comparable infectivities in the two cell types following A(H7N9) virus infection (Fig. 1) demonstrates an enhanced ability of this virus subtype to infect pulmonary endothelial cells and warrants further investigation to determine if this property is shared by other viruses within the H7 subtype. Unlike HPAI H5N1 viruses, A(H7N9) viruses lack a polybasic HA cleavage site for intracellular HA cleavage and exhibit a low-pathogenicity (LPAI) phenotype in poultry (1). Productive replication of A(H7N9) viruses in pulmonary endothelial cells was not observed, likely attributable to the lack of appropriate extracellular enzymes for HA cleavage in this cell type. The absence of a polybasic HA cleavage site among A(H7N9) viruses may contribute to the reduced viral replication in endothelial cells and the lack of systemic spread of the virus in mammals.
Substantial viral shedding and elevated cytokine and chemokine production have been observed in patients infected with A(H7N9) virus (15, 48, 49). Persistent high viral loads have been detected in throat swabs and endotracheal aspirates from patients with severe disease outcomes compared with those with mild symptoms (49, 50). Hypercytokinemia has been detected in fatal cases of H5N1 human infection, identifying an important role for elevated levels of proinflammatory mediators in detrimental immunopathology in the host (13, 26). Similar to the case with H5N1 virus infection, elevated levels of numerous cytokines and chemokines, such as IP-10, MIP-1β, IL-6, and IL-8, were detected in sera from A(H7N9) virus-infected patients during the acute phase of infection, correlating with their serious outcomes (38). Significantly elevated levels of IP-10 and IL-6 were also observed in sera from A(H7N9) virus-infected patients compared to those in H3N2 virus-infected patients (38, 48). In our study, despite the potent viral replication detected in infected human bronchial epithelial cells, the absence of high cytokine production, with the exception of IL-8 and RANTES, is in accord with previous studies with macrophages and alveolar epithelial cells (9). These data suggest that A(H7N9) viruses employ alternate, unknown mechanisms to cause severe disease. However, detection of significant levels of proinflammatory mediators in human lung endothelial cells during virus infection provides evidence that A(H7N9) viruses are capable of eliciting elevated levels of proinflammatory mediators (IL-6, IP-10, and RANTES), similar to the HPAI H5N1 virus (Fig. 3 and 5), even though these cells do not support productive viral replication of this virus. In the human lung, epithelial and endothelial cells are the main structural cell types, and alveolar epithelial cells are in close proximity to the underlying endothelium (46, 47). We found that infection of A(H7N9) in epithelial cells resulted in the release of a substantial amount of virus with cleaved HA and damaged epithelial monolayer from cell death, especially via necrosis. Thus, pulmonary endothelial cells become accessible to infection with A(H7N9) virus following release from neighboring epithelial cells, which, in turn, can produce raised levels of cytokines and chemokines, similar to the case with the HPAI H5N1 virus. Elevated inflammatory cytokines and chemokines can recruit lymphocytes to the lung and facilitate viral clearance. However, an overwhelming hyper-cytokine response can result in enhanced lymphocyte infiltration and lung inflammation and damage, leading to pneumonia and ARDS. Our data suggest that in contrast to seasonal influenza virus infection, A(H7N9) virus infection is capable of inducing elevated levels of inflammatory cytokines in pulmonary endothelial cells, which may contribute to the severe lung pathology observed among A(H7N9) virus-infected patients.
A(H7N9) virus continues to cause human infections with an aggressive clinical course and respiratory failure, resulting in a high percentage of hospitalizations and thus posing a public concern and burden to the region, primarily in China. This virus possesses characteristics of both human and avian influenza viruses during its infection. Our study suggests that A(H7N9) virus is capable of causing lower respiratory tract infection and targeting both epithelial cells and endothelial cells in the human lung, resulting in a higher viral load and elevated proinflammatory cytokine responses compared with those seen with human influenza viruses. Compared to HPAI H5N1, A(H7N9) virus exhibits better replication efficiency at lower temperatures, a feature associated with respiratory droplet transmission in mammals. With additional molecular changes, this newly emerging virus has the potential of further adaptation to humans and other mammalian species. Increased understanding of the mechanisms of viral pathogenesis and human adaptation of the virus is urgently required.
Supplementary Material
ACKNOWLEDGMENTS
We thank the China CDC for facilitating access to viruses.
The findings and conclusions in this report are those of the authors and do not necessarily reflect the views of the funding agency.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03095-14.
REFERENCES
- 1.WHO. 1 February 2015. Human infection with avian influenza A (H7N0) virus—Canada. WHO, Geneva, Switzerland: http://www.who.int/csr/don/01-february-2015-avian-influenza/en/. [Google Scholar]
- 2.WHO. 2 October 2014. Human infections with avian influenza A(H7N9) virus. WHO, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/influenza_h7n9/riskassessment_h7n9_2Oct14.pdf?ua=1. [Google Scholar]
- 3.Gao HN, Lu HZ, Cao B, Du B, Shang H, Gan JH, Lu SH, Yang YD, Fang Q, Shen YZ, Xi XM, Gu Q, Zhou XM, Qu HP, Yan Z, Li FM, Zhao W, Gao ZC, Wang GF, Ruan LX, Wang WH, Ye J, Cao HF, Li XW, Zhang WH, Fang XC, He J, Liang WF, Xie J, Zeng M, Wu XZ, Li J, Xia Q, Jin ZC, Chen Q, Tang C, Zhang ZY, Hou BM, Feng ZX, Sheng JF, Zhong NS, Li LJ. 2013. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med 368:2277–2285. doi: 10.1056/NEJMoa1305584. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Cowling BJ, Feng L, Lau EH, Liao Q, Tsang TK, Peng Z, Wu P, Liu F, Fang VJ, Zhang H, Li M, Zeng L, Xu Z, Li Z, Luo H, Li Q, Feng Z, Cao B, Yang W, Wu JT, Wang Y, Leung GM. 2013. Human infection with avian influenza A H7N9 virus: an assessment of clinical severity. Lancet 382:138–145. doi: 10.1016/S0140-6736(13)61207-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, Xiang N, Chen E, Tang F, Wang D, Meng L, Hong Z, Tu W, Cao Y, Li L, Ding F, Liu B, Wang M, Xie R, Gao R, Li X, Bai T, Zou S, He J, Hu J, Xu Y, Chai C, Wang S, Gao Y, Jin L, Zhang Y, Luo H, Yu H, He J, Li Q, Wang X, Gao L, Pang X, Liu G, Yan Y, Yuan H, Shu Y, Yang W, Wang Y, Wu F, Uyeki TM, Feng Z. 2014. Epidemiology of human infections with avian influenza A(H7N9) virus in China. N Engl J Med 370:520–532. doi: 10.1056/NEJMoa1304617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belser JA, Bridges CB, Katz JM, Tumpey TM. 2009. Past, present, and possible future human infection with influenza virus A subtype H7. Emerg Infect Dis 15:859–865. doi: 10.3201/eid1506.090072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowling BJ, Jin L, Lau EH, Liao Q, Wu P, Jiang H, Tsang TK, Zheng J, Fang VJ, Chang Z, Ni MY, Zhang Q, Ip DK, Yu J, Li Y, Wang L, Tu W, Meng L, Wu JT, Luo H, Li Q, Shu Y, Li Z, Feng Z, Yang W, Wang Y, Leung GM, Yu H. 2013. Comparative epidemiology of human infections with avian influenza A H7N9 and H5N1 viruses in China: a population-based study of laboratory-confirmed cases. Lancet 382:129–137. doi: 10.1016/S0140-6736(13)61171-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. 2013. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan MC, Chan RW, Chan LL, Mok CK, Hui KP, Fong JH, Tao KP, Poon LL, Nicholls JM, Guan Y, Peiris JM. 2013. Tropism and innate host responses of a novel avian influenza A H7N9 virus: an analysis of ex-vivo and in-vitro cultures of the human respiratory tract. Lancet Respir Med 1:534–542. doi: 10.1016/S2213-2600(13)70138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belser JA, Gustin KM, Pearce MB, Maines TR, Zeng H, Pappas C, Sun X, Carney PJ, Villanueva JM, Stevens J, Katz JM, Tumpey TM. 2013. Pathogenesis and transmission of avian influenza A(H7N9) virus in ferrets and mice. Nature 501:556–559. doi: 10.1038/nature12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu H, Wang D, Kelvin DJ, Li L, Zheng Z, Yoon SW, Wong SS, Farooqui A, Wang J, Banner D, Chen R, Zheng R, Zhou J, Zhang Y, Hong W, Dong W, Cai Q, Roehrl MH, Huang SS, Kelvin AA, Yao T, Zhou B, Chen X, Leung GM, Poon LL, Webster RG, Webby RJ, Peiris JS, Guan Y, Shu Y. 2013. Infectivity, transmission, and pathology of human-isolated H7N9 influenza virus in ferrets and pigs. Science 341:183–186. doi: 10.1126/science.1239844. [DOI] [PubMed] [Google Scholar]
- 12.Knepper J, Schierhorn KL, Becher A, Budt M, Tonnies M, Bauer TT, Schneider P, Neudecker J, Ruckert JC, Gruber AD, Suttorp N, Schweiger B, Hippenstiel S, Hocke AC, Wolff T. 2013. The novel human influenza A(H7N9) virus is naturally adapted to efficient growth in human lung tissue. mBio 4(5):e00601-13. doi: 10.1128/mBio.00601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, Hoang DM, Chau NV, Khanh TH, Dong VC, Qui PT, Cam BV, Ha do Q, Guan Y, Peiris JS, Chinh NT, Hien TT, Farrar J. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med 12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Zhang A, Wan Y, Liu X, Qiu C, Xi X, Ren Y, Wang J, Dong Y, Bao M, Li L, Zhou M, Yuan S, Sun J, Zhu Z, Chen L, Li Q, Zhang Z, Zhang X, Lu S, Doherty PC, Kedzierska K, Xu J. 2014. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc Natl Acad Sci U S A 111:769–774. doi: 10.1073/pnas.1321748111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Z, Chen Z, Li X, Xu L, Guan W, Cao Y, Hu Y, Zhang J. 2014. Host immunological response and factors associated with clinical outcome in patients with the novel influenza A H7N9 infection. Clin Microbiol Infect 20(8):O493–O500. doi: 10.1111/1469-0691.12505. [DOI] [PubMed] [Google Scholar]
- 16.Matthay MA, Ware LB, Zimmerman GA. 2012. The acute respiratory distress syndrome. J Clin Invest 122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belser JA, Zeng H, Katz JM, Tumpey TM. 2011. Infection with highly pathogenic H7 influenza viruses results in an attenuated proinflammatory cytokine and chemokine response early after infection. J Infect Dis 203:40–48. doi: 10.1093/infdis/jiq018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belser JA, Zeng H, Katz JM, Tumpey TM. 2011. Ocular tropism of influenza A viruses: identification of H7 subtype-specific host responses in human respiratory and ocular cells. J Virol 85:10117–10125. doi: 10.1128/JVI.05101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng H, Pappas C, Belser JA, Houser KV, Zhong W, Wadford DA, Stevens T, Balczon R, Katz JM, Tumpey TM. 2012. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J Virol 86:667–678. doi: 10.1128/JVI.06348-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesenhagen J, Boergeling Y, Hrincius E, Ludwig S, Roth J, Viemann D. 2012. Highly pathogenic avian influenza viruses inhibit effective immune responses of human blood-derived macrophages. J Leukoc Biol 92:11–20. doi: 10.1189/jlb.0911479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan MC, Chan RW, Yu WC, Ho CC, Yuen KM, Fong JH, Tang LL, Lai WW, Lo AC, Chui WH, Sihoe AD, Kwong DL, Wong DS, Tsao GS, Poon LL, Guan Y, Nicholls JM, Peiris JS. 2010. Tropism and innate host responses of the 2009 pandemic H1N1 influenza virus in ex vivo and in vitro cultures of human conjunctiva and respiratory tract. Am J Pathol 176:1828–1840. doi: 10.2353/ajpath.2010.091087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maines TR, Jayaraman A, Belser JA, Wadford DA, Pappas C, Zeng H, Gustin KM, Pearce MB, Viswanathan K, Shriver ZH, Raman R, Cox NJ, Sasisekharan R, Katz JM, Tumpey TM. 2009. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325:484–487. doi: 10.1126/science.1177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng H, Goldsmith C, Thawatsupha P, Chittaganpitch M, Waicharoen S, Zaki S, Tumpey TM, Katz JM. 2007. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type I interferon response in polarized human bronchial epithelial cells. J Virol 81:12439–12449. doi: 10.1128/JVI.01134-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chosewood LC, Wilson DE, Centers for Disease Control and Prevention, National Institutes of Health. 2009. Biosafety in microbiological and biomedical laboratories, 5th ed US Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institutes of Health, Washington, DC. [Google Scholar]
- 25.Walls HH, Harmon MW, Slagle JJ, Stocksdale C, Kendal AP. 1986. Characterization and evaluation of monoclonal antibodies developed for typing influenza A and influenza B viruses. J Clin Microbiol 23:240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan MC, Cheung CY, Chui WH, Tsao SW, Nicholls JM, Chan YO, Chan RW, Long HT, Poon LL, Guan Y, Peiris JS. 2005. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res 6:135. doi: 10.1186/1465-9921-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam TT, Wang J, Shen Y, Zhou B, Duan L, Cheung CL, Ma C, Lycett SJ, Leung CY, Chen X, Li L, Hong W, Chai Y, Zhou L, Liang H, Ou Z, Liu Y, Farooqui A, Kelvin DJ, Poon LL, Smith DK, Pybus OG, Leung GM, Shu Y, Webster RG, Webby RJ, Peiris JS, Rambaut A, Zhu H, Guan Y. 2013. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 502:241–244. doi: 10.1038/nature12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belser JA, Davis CT, Balish A, Edwards LE, Zeng H, Maines TR, Gustin KM, Martinez IL, Fasce R, Cox NJ, Katz JM, Tumpey TM. 2013. Pathogenesis, transmissibility, and ocular tropism of a highly pathogenic avian influenza A (H7N3) virus associated with human conjunctivitis. J Virol 87:5746–5754. doi: 10.1128/JVI.00154-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerlach RL, Camp JV, Chu YK, Jonsson CB. 2013. Early host responses of seasonal and pandemic influenza A viruses in primary well-differentiated human lung epithelial cells. PLoS One 8:e78912. doi: 10.1371/journal.pone.0078912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Hoeven N, Pappas C, Belser JA, Maines TR, Zeng H, Garcia-Sastre A, Sasisekharan R, Katz JM, Tumpey TM. 2009. Human HA and polymerase subunit PB2 proteins confer transmission of an avian influenza virus through the air. Proc Natl Acad Sci U S A 106:3366–3371. doi: 10.1073/pnas.0813172106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai M, Kawaoka Y. 2012. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr Opin Virol 2:160–167. doi: 10.1016/j.coviro.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong X, Martin SR, Haire LF, Wharton SA, Daniels RS, Bennett MS, McCauley JW, Collins PJ, Walker PA, Skehel JJ, Gamblin SJ. 2013. Receptor binding by an H7N9 influenza virus from humans. Nature 499:496–499. doi: 10.1038/nature12372. [DOI] [PubMed] [Google Scholar]
- 33.Tharakaraman K, Jayaraman A, Raman R, Viswanathan K, Stebbins NW, Johnson D, Shriver Z, Sasisekharan V, Sasisekharan R. 2013. Glycan receptor binding of the influenza A virus H7N9 hemagglutinin. Cell 153:1486–1493. doi: 10.1016/j.cell.2013.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, Kong H, Gu C, Li X, Liu J, Wang G, Chen Y, Liu L, Liang L, Li Y, Fan J, Wang J, Li W, Guan L, Li Q, Yang H, Chen P, Jiang L, Guan Y, Xin X, Jiang Y, Tian G, Wang X, Qiao C, Li C, Bu Z, Chen H. 2013. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 341:410–414. doi: 10.1126/science.1240532. [DOI] [PubMed] [Google Scholar]
- 35.Ramos I, Krammer F, Hai R, Aguilera D, Bernal-Rubio D, Steel J, García-Sastre A, Fernandez-Sesma A. 2013. H7N9 influenza viruses interact preferentially with alpha2,3-linked sialic acids and bind weakly to alpha2,6-linked sialic acids. J Gen Virol 94(Part 11):2417–2423. doi: 10.1099/vir.0.056184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H, Carney PJ, Chang JC, Villanueva JM, Stevens J. 2013. Structural analysis of the hemagglutinin from the recent 2013 H7N9 influenza virus. J Virol 87:12433–12446. doi: 10.1128/JVI.01854-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Riel D, Leijten LM, de Graaf M, Siegers JY, Short KR, Spronken MI, Schrauwen EJ, Fouchier RA, Osterhaus AD, Kuiken T. 2013. Novel avian-origin influenza A (H7N9) virus attaches to epithelium in both upper and lower respiratory tract of humans. Am J Pathol 183:1137–1143. doi: 10.1016/j.ajpath.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou J, Wang D, Gao R, Zhao B, Song J, Qi X, Zhang Y, Shi Y, Yang L, Zhu W, Bai T, Qin K, Lan Y, Zou S, Guo J, Dong J, Dong L, Zhang Y, Wei H, Li X, Lu J, Liu L, Zhao X, Li X, Huang W, Wen L, Bo H, Xin L, Chen Y, Xu C, Pei Y, Yang Y, Zhang X, Wang S, Feng Z, Han J, Yang W, Gao GF, Wu G, Li D, Wang Y, Shu Y. 2013. Biological features of novel avian influenza A (H7N9) virus. Nature 499:500–503. doi: 10.1038/nature12379. [DOI] [PubMed] [Google Scholar]
- 39.Liu Q, Lu L, Sun Z, Chen GW, Wen Y, Jiang S. 2013. Genomic signature and protein sequence analysis of a novel influenza A (H7N9) virus that causes an outbreak in humans in China. Microbes Infect 15:432–439. doi: 10.1016/j.micinf.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Hatta M, Hatta Y, Kim JH, Watanabe S, Shinya K, Nguyen T, Lien PS, Le QM, Kawaoka Y. 2007. Growth of H5N1 influenza A viruses in the upper respiratory tracts of mice. PLoS Pathog 3:1374–1379. doi: 10.1371/journal.ppat.0030133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J Virol 67:1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao Y, Zhang Y, Shinya K, Deng G, Jiang Y, Li Z, Guan Y, Tian G, Li Y, Shi J, Liu L, Zeng X, Bu Z, Xia X, Kawaoka Y, Chen H. 2009. Identification of amino acids in HA and PB2 critical for the transmission of H5N1 avian influenza viruses in a mammalian host. PLoS Pathog 5:e1000709. doi: 10.1371/journal.ppat.1000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neumann G, Macken CA, Kawaoka Y. 2014. Identification of amino acid changes that may have been critical for the genesis of A(H7N9) influenza viruses. J Virol 88:4877–4896. doi: 10.1128/JVI.00107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe T, Kiso M, Fukuyama S, Nakajima N, Imai M, Yamada S, Murakami S, Yamayoshi S, Iwatsuki-Horimoto K, Sakoda Y, Takashita E, McBride R, Noda T, Hatta M, Imai H, Zhao D, Kishida N, Shirakura M, de Vries RP, Shichinohe S, Okamatsu M, Tamura T, Tomita Y, Fujimoto N, Goto K, Katsura H, Kawakami E, Ishikawa I, Watanabe S, Ito M, Sakai-Tagawa Y, Sugita Y, Uraki R, Yamaji R, Eisfeld AJ, Zhong G, Fan S, Ping J, Maher EA, Hanson A, Uchida Y, Saito T, Ozawa M, Neumann G, Kida H, Odagiri T, Paulson JC, Hasegawa H, Tashiro M, Kawaoka Y. 2013. Characterization of H7N9 influenza A viruses isolated from humans. Nature 501:551–555. doi: 10.1038/nature12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Josset L, Zeng H, Kelly SM, Tumpey TM, Katze MG. 2014. Transcriptomic characterization of the novel avian-origin influenza A (H7N9) virus: specific host response and responses intermediate between avian (H5N1 and H7N7) and human (H3N2) viruses and implications for treatment options. mBio 5(1):e01102-13. doi: 10.1128/mBio.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ware LB. 2006. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med 27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 47.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. 2011. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chi Y, Zhu Y, Wen T, Cui L, Ge Y, Jiao Y, Wu T, Ge A, Ji H, Xu K, Bao C, Zhu Z, Qi X, Wu B, Shi Z, Tang F, Xing Z, Zhou M. 2013. Cytokine and chemokine levels in patients infected with the novel avian influenza A (H7N9) virus in China. J Infect Dis 208:1962–1967. doi: 10.1093/infdis/jit440. [DOI] [PubMed] [Google Scholar]
- 49.Hu Y, Lu S, Song Z, Wang W, Hao P, Li J, Zhang X, Yen HL, Shi B, Li T, Guan W, Xu L, Liu Y, Wang S, Zhang X, Tian D, Zhu Z, He J, Huang K, Chen H, Zheng L, Li X, Ping J, Kang B, Xi X, Zha L, Li Y, Zhang Z, Peiris M, Yuan Z. 2013. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet 381:2273–2279. doi: 10.1016/S0140-6736(13)61125-3. [DOI] [PubMed] [Google Scholar]
- 50.Diao H, Cui G, Wei Y, Chen J, Zuo J, Cao H, Chen Y, Yao H, Tian Z, Li L. 2014. Severe H7N9 infection is associated with decreased antigen-presenting capacity of CD14+ cells. PLoS One 9:e92823. doi: 10.1371/journal.pone.0092823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richard M, Schrauwen EJ, de Graaf M, Bestebroer TM, Spronken MI, van Boheemen S, de Meulder D, Lexmond P, Linster M, Herfst S, Smith DJ, van den Brand JM, Burke DF, Kuiken T, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2013. Limited airborne transmission of H7N9 influenza A virus between ferrets. Nature 501:560–563. doi: 10.1038/nature12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maines TR, Lu XH, Erb SM, Edwards L, Guarner J, Greer PW, Nguyen DC, Szretter KJ, Chen LM, Thawatsupha P, Chittaganpitch M, Waicharoen S, Nguyen DT, Nguyen T, Nguyen HH, Kim JH, Hoang LT, Kang C, Phuong LS, Lim W, Zaki S, Donis RO, Cox NJ, Katz JM, Tumpey TM. 2005. Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J Virol 79:11788–11800. doi: 10.1128/JVI.79.18.11788-11800.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.