

ABSTRACT
Hepatitis B virus (HBV), a small enveloped DNA virus, chronically infects more than 350 million people worldwide and causes liver diseases from hepatitis to cirrhosis and liver cancer. Here, we report that hepatocyte nuclear factor 6 (HNF6), a liver-enriched transcription factor, can inhibit HBV gene expression and DNA replication. Overexpression of HNF6 inhibited, while knockdown of HNF6 expression enhanced, HBV gene expression and replication in hepatoma cells. Mechanistically, the SP2 promoter was inhibited by HNF6, which partly accounts for the inhibition on S mRNA. Detailed analysis showed that a cis element on the HBV genome (nucleotides [nt] 3009 to 3019) was responsible for the inhibition of the SP2 promoter by HNF6. Moreover, further analysis showed that HNF6 reduced viral pregenomic RNA (pgRNA) posttranscriptionally via accelerating the degradation of HBV pgRNA independent of La protein. Furthermore, by using truncated mutation experiments, we demonstrated that the N-terminal region of HNF6 was responsible for its inhibitory effects. Importantly, introduction of an HNF6 expression construct with the HBV genome into the mouse liver using hydrodynamic injection resulted in a significant reduction in viral gene expression and DNA replication. Overall, our data demonstrated that HNF6 is a novel host factor that can restrict HBV replication via both transcriptional and posttranscriptional mechanisms.
IMPORTANCE HBV is a major human pathogen whose replication is regulated by host factors. Liver-enriched transcription factors are critical for many liver functions, including metabolism, development, and cell proliferation, and some of them have been shown to regulate HBV gene expression or replication in different manners. In this study, we showed that HNF6 could inhibit the gene expression and DNA replication of HBV via both transcriptional and posttranscriptional mechanisms. As HNF6 is differentially expressed in men and women, the current results may suggest a role of HNF6 in the gender dimorphism of HBV infection.
INTRODUCTION
Hepatitis B virus (HBV) is a noncytopathic, hepatotropic virus that targets the liver and replicates in hepatocytes (1). Around 2 billion people have been infected with HBV worldwide, and 350 million people are chronic carriers with a higher risk of chronic liver diseases, including hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) (2). Upon infection of hepatocytes, the viral relaxed circular DNA (rcDNA) genome is delivered into the nucleus and repaired to form covalently closed circular DNA (cccDNA). The cccDNA serves as the transcription template of pregenomic RNA (3.5 kb) and other mRNAs, including precore RNA (3.5 kb), S RNAs (2.4/2.1 kb), and X RNA (0.7 kb). The transcripts are exported to cytoplasm and translated into viral proteins. After being encapsidated by core and polymerase proteins, the pregenomic RNA is reverse transcribed to viral DNA by the viral polymerase. The newly formed relaxed circular DNA is either recycled to the nucleus to amplify the cccDNA pool or enveloped by viral surface proteins and secreted as virions (reviewed in references 3, 4, and 5).
Liver-enriched transcription factors (LETFs) control transcription of liver-specific genes, which are involved in multiple physiological processes, including metabolism, differentiation, and development (6, 7). According to their functional domains, the LETFs are grouped into several families, including hepatocyte nuclear factor 1 (HNF1), forkhead box A2 (FoxA2) (previously called HNF3β), HNF4, HNF6, CCAAT/element binding proteins (C/EBPs), and D site binding protein (6, 7). Apart from their physiological roles in cells, many LETFs, including HNF1, FoxA2, HNF4, and C/EBPs, have been shown to regulate HBV replication transcriptionally or posttranscriptionally (8–10).
HNF6, the prototype of the one-CUT transcription factor family, is characterized by a divergent Homeo domain and a single CUT domain at the C terminus (11, 12). The expression of HNF6 is high in the liver and low in the spleen, brain, and testis (12). HNF6 controls the expression of many liver-specific genes, which are involved in the glucose metabolism, bile homeostasis, and hepatic cell proliferation (13). The expression of HNF6 is regulated by growth hormone (GH), and the GH-HNF6 pathway has been shown to be imperative for liver protection against chronic injury through regulating the expression of genes involved in proliferation (14–16). The GH-HNF6 pathway also contributes to the gender disparity via regulating the expression of some female-predominant genes (17). The molecular mechanisms involved in gene regulation by HNF6 are either by directly binding to the promoter region of its target genes or by cooperating with other proteins (13).
Although many LETFs have been shown to participate in HBV replication, whether HNF6 influences HBV replication is unknown. Here we provide evidence that ectopic expression of HNF6 significantly inhibits HBV gene expression and replication in vitro and in vivo. We show that HNF6-mediated HBV inhibition occurs at both the transcriptional and posttranscriptional levels through inhibiting SP2 promoter-mediated transcription and accelerating the rate of decay of pregenomic RNA (pgRNA).
MATERIALS AND METHODS
Plasmids.
The HBV (genotype D, GenBank accession number V01460.1) replication-competent plasmid pHBV1.3 was a generous gift from Guangxia Gao (Institute of Biophysics, Chinese Academy of Sciences, China). Plasmids pSV-pgRNA, pSV2.4, pSV2.1, pCMV-pgRNA, pCMV-2.4, and pCMV-2.1 containing the indicated fragments of the HBV genome were constructed as previously reported (see Fig. 6A for a schematic illustration) (18, 19). pTRE2-HBV was generated by insertion of an HBV pgRNA fragment (nucleotides [nt] 1812 to 3182/1 to 1997) into the vector pTRE2 (Clontech) between the NheI and HindIII sites. To construct the reporter plasmids, the EnhI/X (nt 950 to 1375), EnhII/C (nt 1415 to 1815), SP1 (nt 2707 to 2849), and SP2 (nt 2937 to 3182) HBV DNA fragments and serial deletion mutants (see Fig. 5A) were PCR amplified from pHBV1.3 and inserted into the SacI and HindIII restriction sites of pGL3-Basic (Promega, Madison, WI). pHBV1.3/S-Luc was constructed based on the pHBV1.3 backbone, in which the S open reading frame (ORF) was replaced with firefly luciferase ORF with the unique restriction sites XhoI and NsiI on pHBV1.3. The N-terminally hemagglutinin (HA)-tagged HNF6 fragment was PCR amplified from HepG2 cDNA and inserted in the pcDNA3.1 vector (Invitrogen, Carlsbad, CA). The deletion mutants of HNF6 were constructed by PCR methods. Two HNF6 short hairpin RNAs (shRNAs) and control shRNA targeting enhanced green fluorescence protein (shGFP) were inserted into vector pLKO.1. The targets of the shRNAs are 5′-AAGACTTAGCTCACCTGCATT-3′ (shHNF6-1), 5′-AACCCTGGAGCAAACTCAAAT-3′ (shHNF6-2), and 5′-GGCGATGCCACCTACGGCAAG-3′ (shGFP).
FIG 6.

HNF6 reduces the HBV RNAs via posttranscriptional mechanisms. (A) Schematic representation of the transcripts under the control of the SV40 or CMV promoter. (B to E) HepG2 cells were cotransfected with the plasmids expressing HBV RNAs under the control of the SV40 (B and D) or CMV (C and E) promoter and HNF6 expression construct. (B and C) Cells were harvested at 48 h after transfection, and the levels of transcripts were determined by Northern blotting. (D and E) HepG2 cells were cotransfected with the β-gal expressing plasmid under the control of SV40 or CMV promoter and HNF6 expression plasmid or its control vector. Cells were harvested 48 h after transfection, and the activities of β-gal were measured.
FIG 5.

HNF6 suppresses the activity of the SP2 promoter, involving the region from nt 3009 to 3019. (A) Schematic diagram of serial deletion mutants of the SP2 reporter. (B) Effects of HNF6 on the serial deletions of SP2. The results represent the mean ± standard error of the mean (SEM) from three independent experiments. (C) Expression and purification of GST-HNF6 from Escherichia coli cells. The arrow indicates the predicted bands of GST-HNF6. The protein from elution 2 was used for the subsequent gel shift assay. (D) EMSA showed that HNF6 did not bind the SP2 promoter. Lanes 1 to 4, a 5′-HEX-labeled HBV (nt 2987 to 3029) probe was incubated with GST-HNF6 protein or not. Lanes 3 and 4, competition with a 100-fold excess of unlabeled cold probe of HBV (nt 2987 to 3029) and HNF6 binding sites in the FoxA2 promoter region (HNF6 cold probe). Lane 5 to 8, a HEX-labeled HNF6 probe derived from the FoxA2 promoter region was incubated with GST-HNF6 protein or not. Lane 7, competition with a 100-fold excess of HNF6 cold probe. Lane 8, competition with a 100-fold excess of HBV cold probe.
Cell cultures and transfection.
Human hepatoma cell-derived HepG2 and Huh7 and human embryonic kidney 293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. HepG2 and Huh7 cells were transfected with Neofect (Neofect Biotech, Beijing, China) according to the manufacturer's instructions. For producing HNF6 shRNAs, HEK 293T cells were cotransfected with shRNA-expressing plasmids separately with packaging plasmids pMD2.G and psPAX using the calcium phosphate precipitation method, and the lentiviruses were harvested at 48 h and 72 h posttransfection.
HBV DNA analysis.
HepG2 cells were cotransfected with pHBV1.3, pSV-β-gal, and HNF6 or its control vector pcDNA3.1 in the amounts as indicated in the related figures or legends. At 96 h posttransfection, the cells were washed with cold phosphate-buffered saline (PBS) and lysed in NP-40 lysis buffer (50 mM Tris-HCl [pH 7.0], and 0.5% NP-40) at 4°C. The same volume of each lysate was subjected to β-galactosidase (β-gal) activity assay and served as a transfection efficiency control. Following centrifugation, the supernatants were digested with DNase I (Promega, Madison, WI) at 37°C for 1 h, and then the enzymes were inactivated at 70°C for 15 min in the presence of 10 mM EDTA. The core-associated DNA was then isolated by proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation and resolved in 20 μl Tris-EDTA (TE) buffer. All of extracted DNA was loaded and separated in a 0.8% agarose gel. After standard denaturation and neutralization procedures, the DNA was transferred onto a positively charged nylon membrane (GE Healthcare, Waukesha, WI) and probed with a digoxigenin (DIG)-labeled HBV RNA probe. The probe preparation and subsequent DIG detection were conducted with the DIG Northern starter kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions.
Extracellular encapsidated HBV DNA was extracted according to the protocol described previously (20) with modifications. Briefly, 10 μl of culture supernatants or mouse sera was treated with DNase I for 1 h at 37°C to remove free DNA and plasmids. After the enzyme was inactivated at 75°C for 15 min in the presence of EDTA, the samples were added into 100 μl lysis buffer (20 mM Tris-HCl, 20 mM EDTA, 50 mM NaCl, and 0.5% sodium dodecyl sulfate [SDS]) containing 27 μg proteinase K. After incubation at 65°C overnight, viral DNA was isolated by phenol-chloroform extraction and ethanol precipitation. The DNA pellet was rinsed with 70% ethanol and resuspended in 10 μl TE. The viral DNA was then analyzed by quantitative PCR (qPCR).
Western blotting.
The transfected cells were lysed in radioimmunoprecipitation assay (RIPA) buffer with SDS. The protein samples were then separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto a nitrocellulose membrane (GE Healthcare, Waukesha, WI). The membrane was then blocked with skim milk and probed with indicated antibodies. The immunoblot signals were detected with enhanced chemiluminescence (ECL) substrate (Millipore, Billerica, MA). Antibodies used in this study included anti-HA (Sigma, St. Louis, US), anti-HNF6 (Proteintech, Wuhan, China), anti-La (Proteintech, Wuhan, China), and anti-β-actin (Proteintech, Wuhan, China).
Real-time PCR.
For mRNA detection, total RNA was isolated from cells or liver tissues using TRIzol reagent. The RNA was reverse transcribed to first-strand cDNA using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega, Madison, WI). The SYBR green master mix (Roche Diagnostics, Indianapolis, IN) was used for real-time PCR. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA was analyzed to serve as an internal control. The primers used in this study were 2RC/CCS (5′-CTCGTGGTGGACTTCTCTC-3′) and 2RC/CCAS (5′-CTGCAGGATGAAGAGGAA-3′) for HBV-DNA amplification (21), 5′-ACCGCCAGTCAGGCTTTCTTTC-3′ and 5′-TCACTTAC GCCAATGTCGTTATCC-3′ for β-gal mRNA detection, and 5′-CCACTCCTCCACCTTTGAC-3′ and 5′-ACCCTGTTGCTGTAGCCA-3′ for GAPDH detection.
Cytoplasmic and nuclear RNA isolation.
Cell fractionation was conducted according to the protocol in reference 22. Briefly, at 48 h posttransfection, cells were harvested and lysed in lysis buffer (50 mM Tris-HCl [pH 8.0], 0.5% NP-40, 100 mM NaCl, 5 mM MgCl2, 1 U/μl RNasin, 1 mM dithiothreitol [DTT]) at 4°C for 5 min. Then the nuclei were precipitated by centrifugation at 12,000 rpm for 2 min. Cytoplasmic RNA in the supernatants was extracted using an RNA clean kit (Tiangen Biotech, Beijing, China). The nuclear RNA was extracted from nuclear pellets using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction. The nuclear and cytoplasmic RNAs were then subjected to Northern blot analysis.
Analysis of secreted HBV antigens.
At the indicated time points, cell culture supernatants or mouse sera were collected to detect the levels of hepatitis B surface antigen (HBsAg) and hepatitis B E antigen (HBeAg) with a commercial enzyme-linked immunosorbent assay (ELISA) kit (Kehua Bio-Engineering, Shanghai, China). All the values were normalized against the β-galactosidase activities in the cell lysates as measured with the Beta-Glo kit (Promega, Madison, WI).
Immunohistochemistry.
Paraffin-embedded liver sections were treated with 3% hydrogen peroxide and blocked with 5% bovine serum albumin (BSA). The sections were then incubated sequentially with anti-HBc antibody (Sigma, St. Louis, MO, USA), biotin-labeled secondary antibody, and avidin-biotin complex. Peroxidase stain was developed with 3,3′-diaminobenzidine solution and counterstained with hematoxylin.
Dual-luciferase assays.
HepG2 cells were cotransfected with HBV reporters (200 ng) together with indicated amounts of HNF6 expression plasmids or control vector, and pRL-TK (50 ng) was included as a transfection efficiency control. At 48 h posttransfection, the cells were lysed and subjected to luciferase activity assays using the Dual-Glo system (Promega, Madison, WI).
Northern blot analysis.
Total cellular RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Five micrograms of total RNA was resolved in a 1% agarose gel containing 2.2 M formaldehyde and transferred onto a positively charged nylon membrane (GE Healthcare, Waukesha, WI). To detect HBV transcripts, the membrane was probed with a DIG-labeled plus-strand-specific RNA probe corresponding to nucleotides 156 to 1061 of the HBV genome. The probe preparation and subsequent DIG detection were conducted with the DIG Northern starter kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instruction. The amounts of 28S and 18S rRNAs were used as loading controls.
Hydrodynamics-based transfection in mice.
For the in vivo experiments, 5-week-old male BALB/c mice were separated to 2 groups (8 each). pHBV1.3 (10 μg) and pSV-β-gal (5 μg) were injected into the tail veins of mice together with HNF6 expression construct or empty vector pcDNA3 (20 μg) within 8 s in a volume of saline equivalent to 10% of the mouse body weight. Animals were sacrificed after 4 days. Sera were taken for analysis of HBsAg, HBeAg, HBV DNA, and alanine aminotransferase (ALT). For Northern blot analysis, a piece of liver tissue was homogenized in 1 ml of TRIzol reagent and total RNA was isolated following the manufacturer's protocol. Ten micrograms of total RNA of each mouse was subjected to Northern blot analysis as described above. The level of β-gal mRNA in the liver was determined by real-time PCR and served as control for the injection efficiencies. All mice were housed in a pathogen-free mouse colony, and the animal experiments were performed according to the Guide for the Care and Use of Medical Laboratory Animals (Ministry of Health, People's Republic of China, 1998).
RESULTS
Overexpression of HNF6 inhibits HBV gene expression and replication in hepatoma cell lines.
To investigate the role of HNF6 in HBV gene expression, the hepatoma cell line HepG2 was cotransfected with an HBV replication-competent plasmid (pHBV1.3) and different doses of HNF6 expression vector (pcDNA-HA-HNF6). The expression levels of HNF6 were detected by Western blotting (Fig. 1A). The HBV transcripts in the transfected cells were analyzed by Northern blotting. As shown in Fig. 1B, the steady-state levels of HBV RNA were remarkably reduced upon HNF6 overexpression. The 3.5-kb transcripts consisting of the pgRNA and the longer precore RNA were inhibited in a dose-dependent manner (Fig. 1B). Moreover, the 2.4- and 2.1-kb RNAs were inhibited more potently (Fig. 1B). Consistent with the reduction of HBV RNAs, the secreted viral protein levels in the extracellular culture medium were reduced as well (Fig. 1D and E). In accordance with the more potent effects on S mRNA, the inhibitory effect of HNF6 on the production of HBsAg was more potent than that of HBeAg. The modest effects on HBeAg might be attributable to the differential regulation of precore RNA and pgRNA, as shown previously for another LETF, HNF3β (9, 23).
FIG 1.

HNF6 inhibits HBV gene expression and replication in hepatoma cells. HepG2 cells were transfected as indicated and harvested at 48 h after transfection (A, B, D, and E) or 96 h posttransfection (C and F). (A) The expression of HNF6 was determined by Western blotting using an HA antibody. (B) Northern blot analysis of HBV transcripts. The rRNAs (18S and 28S) were used as loading controls. (C) Southern blot analysis of HBV DNA. RC, relaxed circular DNA; RI, replication intermediate DNA. (D to F) The levels of HBsAg (D), HBeAg (E), and HBV DNA (F) in the culture medium were measured by ELISA and qPCR and normalized against β-gal activities. (G and H) Huh7 (G) and HEK 293T (H) cells were transfected with the indicated amounts of plasmids. At 48 h posttransfection, total cellular RNA was extracted and analyzed by Northern blotting. The data are averages from three independent experiments and were statistically analyzed with a t test. *, P < 0.05; **, P < 0.01.
To further investigate the effect of HNF6 on HBV replication, the replication intermediate DNA in the transfected cells and secreted HBV DNA in the culture supernatants were isolated and detected by qPCR. As shown in Fig. 1C and E, both the cellular and extracellular core-associated HBV DNAs were reduced upon HNF6 overexpression. Taken together, these results demonstrate that overexpression of HNF6 inhibits the gene expression and replication of HBV.
To determine whether the suppressive effect of HNF6 was limited to hepatocyte-derived cell lines, pHBV1.3 and HNF6 plasmids were cotransfected into other hepatic or nonhepatic cell lines. The results showed that the levels of HBV RNA were reduced by HNF6 overexpression in another hepatoma cell line, Huh7 (Fig. 1G), but not in the nonhepatic cell line HEK 293T (Fig. 1H). These results suggested that HNF6 might inhibit HBV replication through hepatocyte-specific genes or pathways.
As the activity of HNF6 may be affected by the expression level of HNF6, we measured a more detailed dose effect of HNF6 on HBV biosynthesis. As shown in Fig. 2, cotransfection of 10 ng or 40 ng HNF6 expression plasmid (the HBV DNA/HNF6 ratios are 40:1 and 10:1, respectively) was able to reduce HBV RNA (about 3-fold) and replication intermediates (about 5-fold), and larger amounts of HNF6 expression plasmid exerted more profound inhibition effects. The expression levels of HNF6 in the transfected cells were determined by Western blotting with an HNF6 antibody (Fig. 2C). These results indicate that HNF6 can inhibit HBV replication in a dose-dependent manner.
FIG 2.
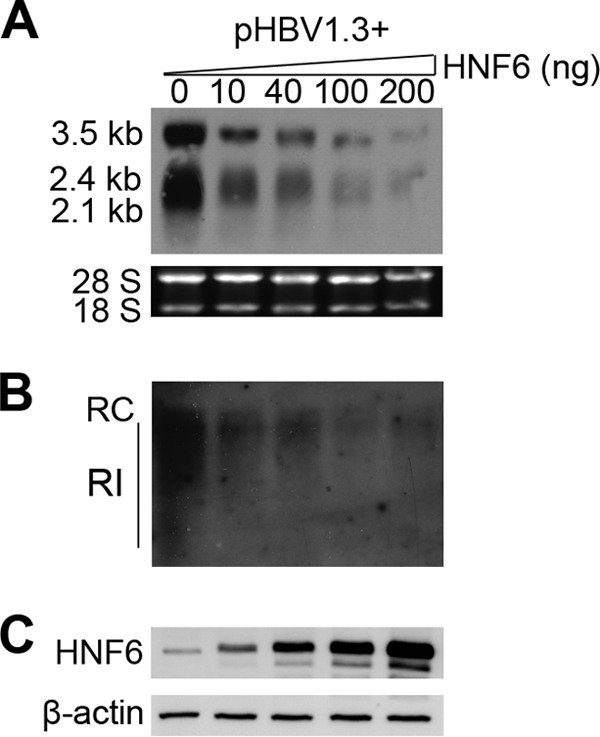
Dosage effects of HNF6 on HBV replication. At 24 h before transfection, HepG2 cells were seeded into 12-well plates. The cells were cotransfected with 400 ng of pHBV1.3 and the indicated amounts of HNF6 expression plasmids. In all transfection experiments, pcDNA vector was included to ensure that the same total amounts of plasmids were added in each well. (A) At 48 h after transfection, the cells were harvested and analyzed for HBV RNA levels by Northern blotting. (B) At 96 h after transfection, the cells were harvested and analyzed for HBV DNA levels by Southern blotting. RC, relaxed circular DNA; RI, replication intermediate DNA. (C) At 48 h after transfection, the cells were harvested and analyzed for HNF6 expression with an HNF6 antibody.
Knockdown of endogenous HNF6 leads to increased HBV replication.
To further illustrate the role of endogenous HNF6 in HBV, the HNF6 expression in HepG2 cells was depleted by transducing with lentiviral vector-based short hairpin RNAs (shRNAs). As shown in Fig. 3A, the protein levels of HNF6 were markedly reduced in cells transduced with shHNF6-1 or shHNF6-2 compared with the control, and the HNF6 depletion was associated with increased expression of HBV RNAs and replication intermediate DNA (Fig. 3B and C). Accordingly, the secreted HBsAg and HBeAg levels in the culture medium of shHNF6-1 and shHNF6-2 were also elevated significantly (Fig. 3D and E). Overall, these results indicated that endogenous HNF6 possesses the ability to restrict HBV gene expression.
FIG 3.
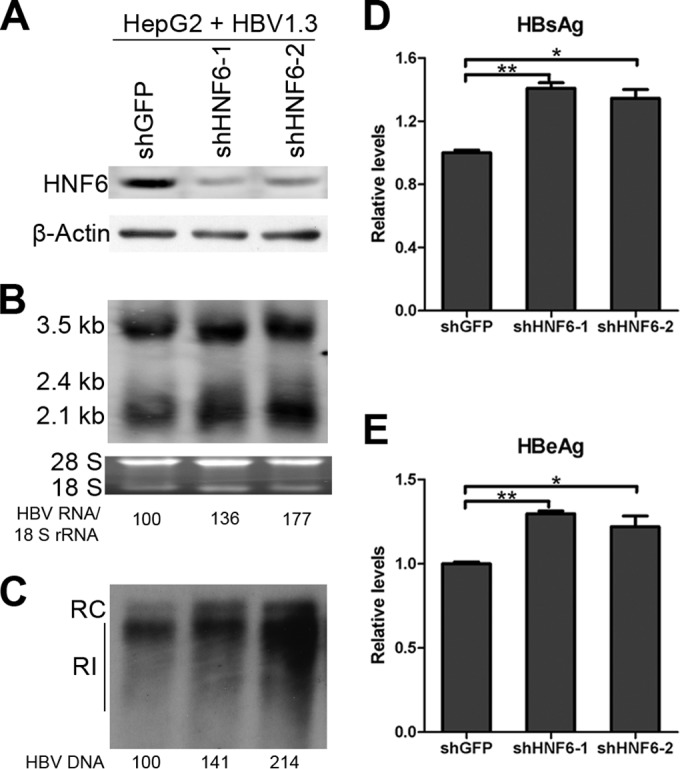
Knockdown of endogenous HNF6 expression enhances HBV gene expression. HepG2 cells were transfected with pHBV1.3, and 24 h later the cells were transduced with lentiviruses expressing shRNAs against HNF6; an shRNA against eGFP was used as irrelevant negative control. (A to C) The cells were harvested at 3 days posttransduction and analyzed for HNF6 expression by Western blotting (A), HBV transcripts by Northern blotting (B), and HBV replication intermediates DNA by Southern blotting (C). RC, relaxed circular DNA; RI, replication intermediate DNA. (D and E) The levels of HBsAg (D) and HBeAg (E) in the culture supernatants were measured by ELISA. The data are averages from three independent experiments and were statistically analyzed with a t test. *, P < 0.05; **, P < 0.01.
HNF6 suppresses the activity of the pre-S2/S promoter.
To investigate the mechanisms involved in HNF6-mediated HBV suppression, the effects of HNF6 on four HBV promoters were determined by a luciferase-based reporter assay. HepG2 cells were cotransfected with HNF6 expression plasmid and the reporters driven by EnhI/Xp, EnhII/Cp, SP1, and SP2 promoters. As shown in Fig. 4A, HNF6 overexpression did not affect the activity of the HBV enhancer I/X, enhancer II/C, or SP1 promoter (Fig. 4A). However, the activity of the SP2 promoter was inhibited by HNF6 in a dose-dependent manner (Fig. 4A and B). To further validate its inhibitory effect on the S promoter, another reporter, pHBV1.3/S-Luc, was constructed based on pHBV1.3, in which the major S ORF was replaced by the firefly luciferase ORF. Consistent with the inhibitory effect described above, HNF6 suppressed the activity of pHBV1.3/S-Luc in a dose-dependent manner (Fig. 4C). These results indicated that HNF6 could suppress HBsAg expression transcriptionally.
FIG 4.

Effects of HNF6 on HBV promoter activity. (A) The activities of SP2 but not other promoters of HBV are inhibited by HNF6 overexpression. HepG2 cells were cotransfected with four reporter plasmids individually and the HNF6 expression plasmid or its control vector (250 ng). The cells were lysed at 48 h posttransfection and subjected to luciferase activity assay. For each transfection, pRL-TK was included as control for transfection efficiencies. (B and C) The activities of SP2 (B) and pHBV1.3/S-Luc (C) were suppressed by HNF6 in a dose-dependent manner.
In order to further investigate the mechanisms of the inhibitory effects of HNF6 on the HBV SP2 promoter, a series of SP2 deletion mutants was constructed (Fig. 5A). As shown in Fig. 5B, deletion of region from nt 3008 to 3019 rendered the SP2 promoter resistance to the inhibition by HNF6. Thus, the region from nt 3008 to 3019 of the HBV genome might be responsible for the inhibitory effects of HNF6. However, computer-based bioinformatic analysis did not reveal any HNF6 binding site in the SP2 promoter and the flanking region of 200 bp. Gel shift experiments also confirmed that HNF6 did not bind the SP2 promoter directly (Fig. 5C and D). These results suggested that HNF6 does not affect the activity of SP2 directly and that it may function through some undefined factor(s) involving a cis element of the HBV genome (nt 3008 to 3019).
HNF6 reduces the steady-state levels of HBV RNA via posttranscriptional mechanisms.
The pgRNA of HBV serves as the template for reverse transcription and is critical for viral replication. The transcription of pgRNA is controlled by the EnhI/X and EnhII/C promoters. Since HNF6 does not influence the activity of these two elements (Fig. 4A), we sought to clarify whether the intrinsic promoters of HBV are required for the inhibition by HNF6. To this end, we generated three constructs (pSV-HBV, pSV-2.4, and pSV-2.1) in which the pgRNA, 2.4-kb RNA, and 2.1-kb RNA of HBV are transcribed under the control of the simian virus 40 (SV40) promoter, as depicted in Fig. 6A. These three constructs were transfected into HepG2 cells along with HNF6 or its control vector. As shown in Fig. 6B, the levels of HBV RNA transcribed from SV40 promoters were also dramatically inhibited by HNF6. To exclude the possibility that HNF6 might affect the activity of SV40 promoter directly, a construct in which the β-gal ORF is under the control of the SV40 promoter was transfected into cells along with HNF6 or its control vector. Interestingly, HNF6 did not inhibit the activities of β-gal, indicating that HNF6 does not influence the activity of the SV40 promoter (Fig. 6D). Similar results were obtained when the SV40 promoter was replaced with the cytomegalovirus (CMV) immediate early (IE) promoter (Fig. 6C and E). These results suggest that HNF6 suppresses the levels of 3.5- and 2.4-kb RNAs via posttranscriptional mechanisms. Since the SP2 promoter was also inhibited (Fig. 4D), it is suggested that the 2.1-kb RNA might be suppressed through both transcriptional and posttranscriptional mechanisms.
HNF6 accelerates the rates of decay of HBV RNA independent of La.
As the HNF6-mediated HBV RNA reduction occurs posttranscriptionally, we suggest that HNF6 may promote the turnover rate of HBV pgRNA. To this end, we directly measured the decay rates of HBV pgRNA in the absence or presence of HNF6 overexpression. HepG2 cells were cotransfected with HNF6 and pTRE2-HBV, in which the HBV pgRNA is transcribed from a tetracycline-controlled promoter. Doxycycline was added to culture supernatants 36 h after transfection to turn off the HBV pgRNA transcription, and then the cells were harvested at 0, 3, 6, and 9 h after the doxycycline addition and subjected to RNA extraction and Northern blot analysis. As shown in Fig. 7A and B, the half-life of HBV pgRNA was shortened by HNF6 by about 3 h in comparison with that of cells transfected with the empty control plasmid (pcDNA3). This result demonstrated that HNF6 could increase the decay rates of HBV pgRNA.
FIG 7.

HNF6 promotes HBV pgRNA decay in the nucleus. (A and B) HepG2 cells were cotransfected with pTet-Off, pTRE2-HBV, and HNF6 expression vector or its control vector for 36 h. Doxycycline hyclate (dox) was then added to the culture medium to shut down the pgRNA transcription. Cells were harvested 0, 3, 6, and 9 h after dox addition. (A) Cellular RNA was extracted and analyzed for HBV transcripts. The result is representative of two separate trials. (B) Kinetic analysis of HBV pgRNA decay in the absence or presence of HNF6 expression. The relative levels of HBV RNA from each sample were normalized against 18S rRNA densities and represented as the percentage of the RNA signals of the corresponding samples at time zero. (C) HNF6 did not affect the expression of La. HepG2 cells were transfected with pcDNA or HNF6 for 48 h. The transfected cells were harvested and subjected to Western blotting with antibody against La, HA tag, or β-actin. (D) The La binding sites in the HBV genome do not account for the inhibitory effects of HNF6. HepG2 cells were cotransfected with the La binding-deficient mutant pSV-HBV M2, pSV-2.4 M2, or pSV-2.1 M2 and HNF6 expression vector for 48 h. Total RNA was extracted and analyzed by Northern blotting. (E) Subcellular analysis of antiviral activity of HNF6. HepG2 cells were cotransfected with pHBV1.3 and HNF6 expression plasmid or its control vector. At 48 h posttransfection, the cells were fractioned for Northern blot analysis of HBV RNA. (F) The experiments shown in panel E were repeated three times independently. Viral pgRNA levels were quantified and plotted as inhibition rates (HNF6/pcDNA). The data shown are averages and SEMs and were statistically analyzed with a t test.
It is well characterized that upon treatment with cytotoxic T lymphocytes and some cytokines, the La protein became fragmented and left the HBV RNA vulnerable to endonuclease degradation (24, 25). We therefore sought to determine whether the HNF6-mediated pgRNA decay is mediated by La. Our results showed that overexpression of HNF6 did not affect the expression or fragmentation status of La (Fig. 7C). To further verify the effects of La during HNF6-mediated pgRNA degradation, the La binding-deficient HBV pgRNA and 2.4- and 2.1-kb RNAs constructs were constructed as depicted previously (25, 26). However, HNF6 was also able to inhibit the levels of La binding-deficient HBV pgRNA and 2.4- and 2.1-kb RNAs (Fig. 7D). These results suggested that the HNF6-mediated HBV RNA degradation is independent of the La protein.
HNF6-mediated HBV RNA inhibition occurs primarily in the nucleus.
To further determine the cellular localization where the inhibitory effect occurs, nuclear and cytoplasmic RNAs were fractioned and extracted from cells cotransfected with pHBV1.3 and HNF6. As shown in Fig. 7E and F, the levels of HBV transcripts were reduced in both the nuclear and cytoplasmic portions with similar inhibition rates. Since HBV RNA is transcribed in the nucleus and then exported to the cytoplasm, it is suggested that HNF6-mediated HBV RNA inhibition is primarily a nuclear event.
The N-terminal domain of HNF6 is required for the anti-HBV activity of HNF6.
HNF6 contains the CUT and Homeo domains at the C-terminal half, which are both involved in DNA binding and protein interactions. The N-terminal part of HNF6 has been shown to interact with cellular factors (27). To further delineate the mechanism involved in the inhibitory effects of HNF6, the contributions of different domains of HNF6 were determined. As shown in Fig. 8A and B, the CUT-Homeodomain of HNF6 alone was not able to reduce HBV transcripts, while the deletion of the Homeo domain, the CUT domain, or both could suppress the levels of HBV transcripts. The immunofluorescence results showed that the N-terminal part of HNF6 is localized in both the nucleus and cytoplasm and that the wild-type HNF6 is localized exclusively in nucleus (Fig. 8C). In accordance with the Northern blot results, the N-terminal domain of HNF6 was also able to inhibit the activity of the SP2 promoter (Fig. 8D) and reduce the amount of HBV nuclear mRNA (Fig. 8E). These results suggested that the N-terminal domain accounts for the anti-HBV activity of HNF6.
FIG 8.

Mapping of the functional antiviral domains of HNF6. (A) Schematic diagram of HNF6 truncation mutants. (B) Effects of different mutants of HNF6 on the HBV RNA levels were analyzed by Northern blotting at 2 days posttransfection. (C) Subcellular localization of HNF6 and its N-terminal mutant was determined by staining with HA antibody fluorescein isothiocyanate and (FITC)-conjugated secondary antibody. The cell nucleus was stained with Hoechst stain. The subcellular localization was examined by confocal microscopy. (D and E) The effects of HNF6 N-terminal mutants on the SP2 promoter (D) and nuclear HBV RNA (E) were determined.
HNF6 inhibits HBV gene expression and replication in mice.
Because HNF6 was shown to inhibit HBV replication in hepatoma cells, we further investigate the effects of HNF6 expression on HBV replication in a mouse model. pHBV1.3 and HNF6 expression plasmids or control vector were codelivered into mice through hydrodynamic injection, and the mice were sacrificed at 4 days postinjection. The expression of HNF6 in mouse livers was determined by Western blotting (Fig. 9E). Consistent with the results in the hepatoma cell lines, the titers of HBsAg, HBeAg, and HBV DNA in mouse sera were reduced significantly upon HNF6 injection (Fig. 9A and B). The Northern blot result showed that the HBV transcripts were reduced significantly upon HNF6 overexpression (Fig. 9C). Immunohistochemical staining showed that HNF6 could inhibit the HBcAg expression in mouse livers (Fig. 9D). Taken together, these results demonstrated that HNF6 is able to inhibit HBV gene expression and replication in vivo.
FIG 9.
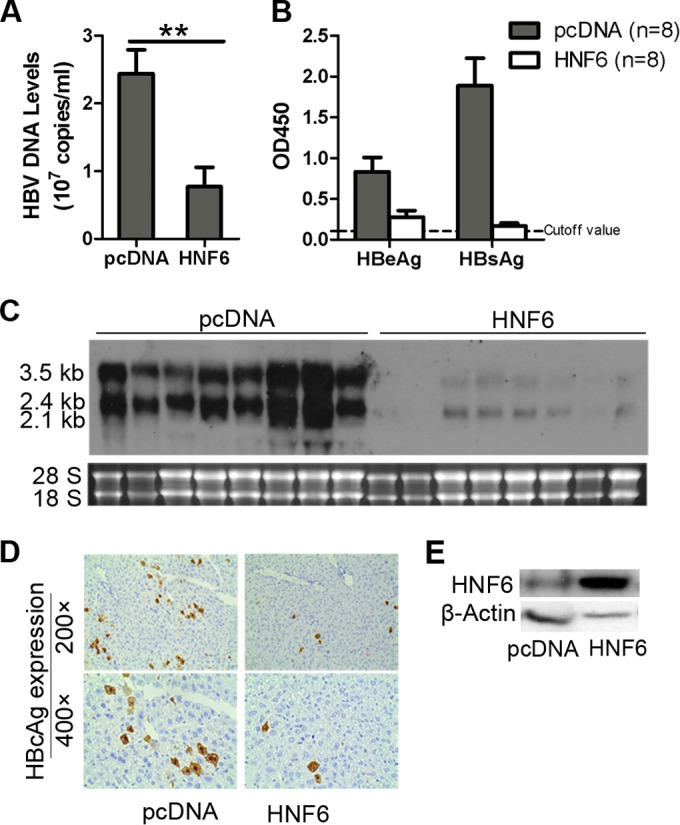
HNF6 inhibits HBV gene expression and replication in vivo. Two groups of mice (8 in each group) were coinjected with pHBV1.3 and HNF6 expression plasmid or its control vector (pcDNA). At 4 days after injection, the mice were sacrificed. (A and B) The titers of HBV DNA (A) and HBeAg and HBsAg (B) in the mouse sera were determined by qPCR and ELISA. (C) HBV RNA transcripts in the liver tissues were analyzed by Northern blotting. (D) Immunohistochemical staining for HBcAg in the liver tissues. (E) Western blot analysis of HNF6 expression in mouse livers. The graphic results represent the mean of each group with SEM and were statistically analyzed with a t test. *, P < 0.05; **, P < 0.01.
DISCUSSION
LETFs are important transcription factors involved in liver development, differentiation, and metabolism (6, 7). Many of them have been shown to regulate HBV gene expression or replication. HNF1α activates the transcription from the EnhII/core promoter and thus promotes HBV replication (8). HNF4α activates transcription from the EnhII/C, SP1, and SP2 promoters and supports HBV replication in nonhepatic cells (9, 28). FoxA2 was shown to inhibit HBV replication posttranscriptionally in hepatoma cells and transgenic mice (9, 10). In this study, we demonstrated that another LETF, HNF6, could inhibit expression of HBV secreted antigens and DNA replication. The overexpression of HNF6 reduced, whereas the suppression of HNF6 expression increased, the levels of HBV transcripts and proteins (Fig. 1 and 3). Importantly, the HNF6-mediated HBV suppression was a posttranscriptional event via accelerating the decay of pgRNA in the nucleus (Fig. 6 and 7). The role of HNF6 in HBV gene expression was also confirmed in vivo using a mouse model, in which the overexpression of HNF6 led to a marked reduction of HBV proteins and DNA in the mouse sera and livers (Fig. 9).
Regulation of HBsAg expression is crucial for the pathogenesis of HBV, as the envelope proteins is essential for the viral particle formation, infectivity, and immune response (2, 4). In the present study, an approximately 10-fold inhibition of HBsAg expression was observed upon HNF6 overexpression in mice (Fig. 9B). By performing reporter assays, we demonstrated that the activity of the SP2 promoter is inhibited by HNF6 (Fig. 4A). Further analysis showed that the region from nt 3009 to 3019 of the HBV genome is responsible for the HNF6-mediated SP2 promoter inhibition in an indirect manner (Fig. 5). However, HNF6 does not bind directly to the SP promoter region (nt 3009 to 3019) (Fig. 5D), and this suggests that HNF6 may function through some undefined factors that could bind directly to the SP2 promoter. As no regulatory protein was previously reported to bind with this region, we analyzed the transcription factors that can potentially bind to this region by a bioinformatic method (MatInspector). Potential binding sites could be predicted for four cellular factors: glial cells missing homolog 1, heat shock factor 1, Kruppel-like factor (KLF15), and sterol regulatory element binding protein. Among these factors, KLF15 has been shown to activate the SP2 promoter of HBV in a previous study, but the precise binding site of KLF15 has not been defined (29). Therefore, it will be interesting to investigate whether KLF15 binds to this region and whether HNF6 can suppress the KLF15-mediated SP2 activation in future work. Moreover, the S mRNA (2.1-kb RNA) driven by the SV40 or CMV promoter was also inhibited by HNF6 overexpression (Fig. 6), which suggests that both transcriptional and posttranscriptional mechanisms may account for the profound suppression activity of HNF6 on HBsAg expression synergistically.
By using the pSV40-HBV and pCMV-HBV constructs, the HNF6-mediated HBV pgRNA inhibition was shown to be independent of the nature of the HBV promoters (Fig. 6). Therefore, it is likely that HNF6 might influence the transcriptional elongation or the stability of HBV RNAs. By using the Tet-controlled system, we demonstrated that HNF6 could promote the degradation of HBV pgRNA, which is the template for viral reverse transcription. Since HNF6 does not possess RNA binding or nuclease activity, it is reasonable to hypothesize that a downstream effector accounts for the HNF6-mediated HBV pgRNA decay. It has been shown that La protein and zinc finger antiviral protein (ZAP) can regulate HBV RNA stability through direct binding to the their RNA target via recognition sites (25, 30). La is capable of binding HBV RNAs and protecting them from endonuclease cleavage. However, our results showed that the HNF6-mediated HBV RNA decay is independent of La. ZAP binds the ZAP-responsive element in HBV RNA and recruits exosomes or other factors, resulting in HBV RNA degradation. ZAP is not likely the downstream effector of HNF6, for ZAP reduces HBV transcript levels in both hepatic and nonhepatic cell lines, which is different from the manner of hepatocyte-specific inhibition by HNF6 (30). Therefore, there might be other host factors which serve as the downstream effectors of HNF6, directly regulating the stability of HBV RNAs. Actually, a previous report has showed that Myd88 promotes the decay of HBV pgRNA in hepatocyte-specific manner through a cryptic effector other than the La antigen (31). Although these data have demonstrated that HNF6 could accelerate the decay of HBV pregenomic RNA, the possibility that HNF6 affects HBV RNA elongation cannot be excluded. Therefore, more studies are required to explore this possibility.
As a transcriptional factor, HNF6 could directly modulate the promoter activity of its target genes. HNF6 has also been shown to regulate some gene expression via interaction with other cellular factors (32). The C-terminal CUT and Homeo domain of HNF6 is responsible for the promoter binding and protein-interacting activity, while the N-terminal part was shown to be involved in transcriptional activation of some promoters (27). Using serial deletion mutants of HNF6, we identified that the N-terminal region is critical for the inhibitory effects on HBV RNA. The N-terminal region alone was able to reduce the level of HBV RNA (Fig. 8). Hence, the direct transcriptional regulatory activity of HNF6 was not required for the HNF6-mediated pgRNA inhibition, while the protein interaction activity of the N terminus might be required.
HNF6 regulates various important cellular functions, including liver and pancreas development, differentiation, cell proliferation, and metabolisms (13). The expression of HNF6 shows a female-predominant mode and is controlled by GH, whose secretion is pulsive in male mice while constant in female mice (17). HNF6 was believed to be the dominant effector for GH-stimulated pathways (16, 33). A previous report showed that female-specific expression of CYP2C12 is attributable to the regulation of the GH-HNF6 pathway (17). Interestingly, the gender disparity of HBV has also been well documented. Male HBV carriers usually have higher viral load and a higher possibility of progressing to HCC than female carriers (34, 35). Hence, it is reasonable to speculate that the GH/HNF6 axis might also partly account for the gender dimorphism of HBV. However, more studies are warranted to demonstrate the role of HNF6 in the sex disparity of HBV gene expression and replication.
ACKNOWLEDGMENTS
This study was supported by the National Basic Research Program of China (2010CB911800) and the National Science Foundation of China (31221061). D.G. is also supported by Hubei Province's Outstanding Medical Academic Leader Program.
We thank Zhang Hui, Li Shun, and Liu Ye for technical assistance and Ma Chunqiang, Su Ceyang, Li Shun, Chen Shiyou, and Chen Shuliang for meaningful discussions.
REFERENCES
- 1.Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol Mol Biol Rev 64:51–68. doi: 10.1128/MMBR.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arzumanyan A, Reis HM, Feitelson MA. 2013. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nature reviews. Cancer 13:123–135. doi: 10.1038/nrc3449. [DOI] [PubMed] [Google Scholar]
- 3.Beck J, Nassal M. 2007. Hepatitis B virus replication. World J Gastroenterol 13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schädler S, Hildt E. 2009. HBV life cycle: entry and morphogenesis. Viruses 1:185–209. doi: 10.3390/v1020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grimm D, Thimme R, Blum HE. 2011. HBV life cycle and novel drug targets. Hepatol Int 5:644–653. doi: 10.1007/s12072-011-9261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrem H, Klempnauer J, Borlak J. 2002. Liver-enriched transcription factors in liver function and development. I. The hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol Rev 54:129–158. [DOI] [PubMed] [Google Scholar]
- 7.Schrem H, Klempnauer J, Borlak J. 2004. Liver-enriched transcription factors in liver function and development. II. The C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol Rev 56:291–330. doi: 10.1124/pr.56.2.5. [DOI] [PubMed] [Google Scholar]
- 8.Wang WX, Li M, Wu X, Wang Y, Li ZP. 1998. HNF1 is critical for the liver-specific function of HBV enhancer II. Res Virol 149:99–108. doi: 10.1016/S0923-2516(98)80085-X. [DOI] [PubMed] [Google Scholar]
- 9.Tang H, McLachlan A. 2001. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci U S A 98:1841–1846. doi: 10.1073/pnas.98.4.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang H, McLachlan A. 2002. Mechanisms of inhibition of nuclear hormone receptor-dependent hepatitis B virus replication by hepatocyte nuclear factor 3. J Virol 76:8572–8581. doi: 10.1128/JVI.76.17.8572-8581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buckwold VE, Xu Z, Chen M, Yen TS, Ou JH. 1996. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J Virol 70:5845–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemaigre FP, Durviaux SM, Truong O, Lannoy VJ, Hsuan JJ, Rousseau GG. 1996. Hepatocyte nuclear factor 6, a transcription factor that contains a novel type of homeodomain and a single cut domain. Proc Natl Acad Sci U S A 93:9460–9464. doi: 10.1073/pnas.93.18.9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K, Holterman AX. 2012. Pathophysiologic role of hepatocyte nuclear factor 6. Cell Signal 24:9–16. doi: 10.1016/j.cellsig.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Holterman AX, Tan Y, Kim W, Yoo KW, Costa RH. 2002. Diminished hepatic expression of the HNF-6 transcription factor during bile duct obstruction. Hepatology 35:1392–1399. doi: 10.1053/jhep.2002.33680. [DOI] [PubMed] [Google Scholar]
- 15.Wang M, Tan Y, Costa RH, Holterman AX. 2004. In vivo regulation of murine CYP7A1 by HNF-6: a novel mechanism for diminished CYP7A1 expression in biliary obstruction. Hepatology 40:600–608. doi: 10.1002/hep.20349. [DOI] [PubMed] [Google Scholar]
- 16.Wang M, Chen M, Zheng G, Dillard B, Tallarico M, Ortiz Z, Holterman AX. 2008. Transcriptional activation by growth hormone of HNF-6-regulated hepatic genes, a potential mechanism for improved liver repair during biliary injury in mice. Am J Physiol Gastrointest Liver Physiol 295:G357–G366. doi: 10.1152/ajpgi.00581.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lahuna O, Fernandez L, Karlsson H, Maiter D, Lemaigre FP, Rousseau GG, Gustafsson J, Mode A. 1997. Expression of hepatocyte nuclear factor 6 in rat liver is sex-dependent and regulated by growth hormone. Proc Natl Acad Sci U S A 94:12309–12313. doi: 10.1073/pnas.94.23.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schormann W, Kraft A, Ponsel D, Bruss V. 2006. Hepatitis B virus particle formation in the absence of pregenomic RNA and reverse transcriptase. J Virol 80:4187–4190. doi: 10.1128/JVI.80.8.4187-4190.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo H, Jiang D, Ma D, Chang J, Dougherty AM, Cuconati A, Block TM, Guo JT. 2009. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J Virol 83:847–858. doi: 10.1128/JVI.02008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian Y, Chen WL, Ou JH. 2011. Effects of interferon-alpha/beta on HBV replication determined by viral load. PLoS Pathog 7:e1002159. doi: 10.1371/journal.ppat.1002159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, Trepo C, Marcellin P, Goodman Z, Delaney WE 4th, Xiong S, Brosgart CL, Chen SS, Gibbs CS, Zoulim F. 2004. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 22.Gilman M. 2002. Preparation of cytoplasmic RNA from tissue culture cells. Curr Protoc Mol Biol Chapter 4:Unit 4.1. doi: 10.1002/0471142727.mb0401s58. [DOI] [PubMed] [Google Scholar]
- 23.Banks KE, Anderson AL, Tang H, Hughes DE, Costa RH, McLachlan A. 2002. Hepatocyte nuclear factor 3beta inhibits hepatitis B virus replication in vivo. J Virol 76:12974–12980. doi: 10.1128/JVI.76.24.12974-12980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsui LV, Guidotti LG, Ishikawa T, Chisari FV. 1995. Posttranscriptional clearance of hepatitis B virus RNA by cytotoxic T lymphocyte-activated hepatocytes. Proc Natl Acad Sci U S A 92:12398–12402. doi: 10.1073/pnas.92.26.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heise T, Guidotti LG, Chisari FV. 1999. La autoantigen specifically recognizes a predicted stem-loop in hepatitis B virus RNA. J Virol 73:5767–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehlers I, Horke S, Reumann K, Rang A, Grosse F, Will H, Heise T. 2004. Functional characterization of the interaction between human La and hepatitis B virus RNA. J Biol Chem 279:43437–43447. doi: 10.1074/jbc.M402227200. [DOI] [PubMed] [Google Scholar]
- 27.Lannoy VJ, Rodolosse A, Pierreux CE, Rousseau GG, Lemaigre FP. 2000. Transcriptional stimulation by hepatocyte nuclear factor-6. Target-specific recruitment of either CREB-binding protein (CBP) or p300/CBP-associated factor (p/CAF). J Biol Chem 275:22098–22103. doi: 10.1074/jbc.M000855200. [DOI] [PubMed] [Google Scholar]
- 28.Zheng Y, Li J, Ou JH. 2004. Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J Virol 78:6908–6914. doi: 10.1128/JVI.78.13.6908-6914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou J, Tan T, Tian Y, Zheng B, Ou JH, Huang EJ, Yen TS. 2011. Kruppel-like factor 15 activates hepatitis B virus gene expression and replication. Hepatology 54:109–121. doi: 10.1002/hep.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mao R, Nie H, Cai D, Zhang J, Liu H, Yan R, Cuconati A, Block TM, Guo JT, Guo H. 2013. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog 9:e1003494. doi: 10.1371/journal.ppat.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Lin S, Chen Q, Peng L, Zhai J, Liu Y, Yuan Z. 2010. Inhibition of hepatitis B virus replication by MyD88 involves accelerated degradation of pregenomic RNA and nuclear retention of pre-S/S RNAs. J Virol 84:6387–6399. doi: 10.1128/JVI.00236-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rausa FM, Tan Y, Costa RH. 2003. Association between hepatocyte nuclear factor 6 (HNF-6) and FoxA2 DNA binding domains stimulates FoxA2 transcriptional activity but inhibits HNF-6 DNA binding. Mol Cell Biol 23:437–449. doi: 10.1128/MCB.23.2.437-449.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan Y, Yoshida Y, Hughes DE, Costa RH. 2006. Increased expression of hepatocyte nuclear factor 6 stimulates hepatocyte proliferation during mouse liver regeneration. Gastroenterology 130:1283–1300. doi: 10.1053/j.gastro.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka M, Katayama F, Kato H, Tanaka H, Wang J, Qiao YL, Inoue M. 2011. Hepatitis B and C virus infection and hepatocellular carcinoma in China: a review of epidemiology and control measures. J Epidemiol 21:401–416. doi: 10.2188/jea.JE20100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El-Serag HB. 2012. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142:1264–1273.e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]