

ABSTRACT
In addition to transporting ions, the multisubunit Na+,K+-ATPase also functions by relaying cardiotonic steroid (CTS)-binding-induced signals into cells. In this study, we analyzed the role of Na+,K+-ATPase and, in particular, of its ATP1A1 α subunit during coronavirus (CoV) infection. As controls, the vesicular stomatitis virus (VSV) and influenza A virus (IAV) were included. Using gene silencing, the ATP1A1 protein was shown to be critical for infection of cells with murine hepatitis virus (MHV), feline infectious peritonitis virus (FIPV), and VSV but not with IAV. Lack of ATP1A1 did not affect virus binding to host cells but resulted in inhibited entry of MHV and VSV. Consistently, nanomolar concentrations of the cardiotonic steroids ouabain and bufalin, which are known not to affect the transport function of Na+,K+-ATPase, inhibited infection of cells with MHV, FIPV, Middle East respiratory syndrome (MERS)-CoV, and VSV, but not IAV, when the compounds were present during virus inoculation. Cardiotonic steroids were shown to inhibit entry of MHV at an early stage, resulting in accumulation of virions close to the cell surface and, as a consequence, in reduced fusion. In agreement with an early block in infection, the inhibition of VSV by CTSs could be bypassed by low-pH shock. Viral RNA replication was not affected when these compounds were added after virus entry. The antiviral effect of ouabain could be relieved by the addition of different Src kinase inhibitors, indicating that Src signaling mediated via ATP1A1 plays a crucial role in the inhibition of CoV and VSV infections.
IMPORTANCE Coronaviruses (CoVs) are important pathogens of animals and humans, as demonstrated by the recent emergence of new human CoVs of zoonotic origin. Antiviral drugs targeting CoV infections are lacking. In the present study, we show that the ATP1A1 subunit of Na+,K+-ATPase, an ion transporter and signaling transducer, supports CoV infection. Targeting ATP1A1 either by gene silencing or by low concentrations of the ATP1A1-binding cardiotonic steroids ouabain and bufalin resulted in inhibition of infection with murine, feline, and MERS-CoVs at an early entry stage. Infection with the control virus VSV was also inhibited. Src signaling mediated by ATP1A1 was shown to play a crucial role in the inhibition of virus entry by ouabain and bufalin. These results suggest that targeting the Na+,K+-ATPase using cardiotonic steroids, several of which are FDA-approved compounds, may be an attractive therapeutic approach against CoV and VSV infections.
INTRODUCTION
Despite the wide variety of vaccines already available to prevent viral infections, unexpected epidemics caused by zoonotic viruses, such as severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 to 2003 and the new pandemic H1N1 influenza A virus (IAV) in 2009, underscore the need for additional antiviral measures. Compound and small interfering RNA (siRNA) screening may aid the development of antiviral therapies by the discovery of lead compounds and target proteins (1–3). Elucidating the mechanisms by which such proteins act during infection and how drugs can interfere with the pathogen life cycle is of crucial importance herein.
Coronaviruses (CoVs) are enveloped, plus-strand RNA viruses of the Coronaviridae family in the order Nidovirales. These viruses generally cause respiratory and/or intestinal tract disease. CoVs are important pathogens of domestic livestock, poultry, and companion animals, as exemplified by porcine epidemic diarrhea virus, infectious bronchitis virus, and feline infectious peritonitis virus (FIPV), respectively. In addition, the emergence of new human CoVs of zoonotic origin has shown the potential of CoVs to cause life-threatening disease in humans, as was demonstrated by the 2002-2003 SARS-CoV epidemic and by the recent emergence of Middle East respiratory virus (MERS)-CoV (4, 5). The murine hepatitis coronavirus (MHV) is often employed as a safe model to study CoV infections.
Like all other viruses, CoVs depend on the cellular machinery for efficient infection and replication in their host cells. The CoV infection cycle starts with attachment of the virus to a specific cellular receptor, mediated by the viral spike (S) protein. Upon endocytic uptake, which has been demonstrated to occur via clathrin-mediated endocytosis (CME) for MHV (6), conformational changes in the S protein induce virus-cell fusion. The genomic RNA is thereby released into the cytoplasm and becomes translated, resulting in the formation of RNA replication-transcription complexes associated with rearranged cellular membranes (7). Structural proteins together with newly generated genomic RNAs assemble into progeny virions via budding through the membranes of the endoplasmic reticulum (ER) to the Golgi intermediate compartment. Virions are subsequently released via exocytosis (8).
The Na+,K+-ATPase is perhaps one of the best studied membrane ion transporters. Discovered in 1957 and identified as an ion-activated ATPase in 1965, it is mainly known for its transport function of K+ and Na+ at a ratio of 2:3, creating an electrochemical gradient across the plasma membrane (9). The Na+,K+-ATPase consists of two functional subunits (α and β) and one regulatory subunit (γ subunit, or FXYD protein). The α subunit is a large, catalytic membrane protein, containing 10 transmembrane domains that create five extracellular and four intracellular loops. Four different isoforms of the α subunit exist, which are encoded by ATP1A1 to ATP1A4. The α1 isoform is ubiquitously expressed in almost all tissues. The β subunit is a type II membrane protein that is responsible for the proper translocation of the α subunit into the endoplasmic reticulum and its delivery to the cell surface and that is crucial to the functioning of the pump. Little is known about the function of the regulatory subunit γ (reviewed in reference 10). Specific inhibitors of the Na+,K+-ATPase, so-called cardiotonic steroids (CTSs), can block the transport function of the pump and are used to treat congestive heart failure. Well-known CTSs are the foxglove plant-derived digoxin and ouabain and the vertebrate-derived analogues bufalin and marinobufagenin (11, 12).
In addition to the classical ion-pumping function of the Na+,K+-ATPase, more recent work has demonstrated additional roles of Na+,K+-ATPase in signal transduction. Especially the α subunit appears to be associated with a number of additional proteins and to carry out various signaling functions (reviewed in references 13 and 14), which may differ between the different α-subunit isoforms (15). CTSs (including endogenous CTSs) can trigger the signaling functions of the Na+,K+-ATPase at concentrations that do not affect the pump function or intracellular ion concentration (16–21). There are four main signaling targets of the α subunit known so far: phosphatidylinositol 3-kinase (PI3K), Src, inositol 1,4,5-triphosphate receptor (IP3R), and phospholipase C (PLC). Binding of nanomolar concentrations of ouabain to Na+,K+-ATPase triggers a conformational change in the α subunit, which activates the bound Src protein and results in the recruitment of other signaling factors. Binding of ouabain to Na+,K+-ATPase activates tyrosine phosphorylation of Src and of other proteins. Activation of these targets may lead to a number of downstream signaling effects controlling apoptosis, cell-cell interaction, and gene expression, as well as other processes (16–18, 22–28).
In a high-throughput RNA interference (RNAi) screen, we previously identified ATP1A1 as a protein that supports MHV infection (unpublished results). ATP1A1 is an appealing antiviral target in view of the large number of (FDA-approved) compounds available that target this protein. Therefore, the main goal of the present study was to obtain mechanistic insight into the role of the Na+,K+-ATPase in CoV infection. Targeting ATP1A1 either by gene silencing or by low concentrations of the CTSs ouabain and bufalin resulted in inhibition of CoV infection at an early entry stage. As controls, the well-studied vesicular stomatitis virus (VSV) and influenza A virus (IAV) were included. Src signaling mediated by ATP1A1 was shown to play a crucial role in the inhibition of CoV and VSV entry by CTSs. These results suggest that targeting the Na+,K+-ATPase using CTSs may be an attractive therapeutic approach against CoV and VSV infections.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
Murine LR7 (29) (murine L-2 fibroblast cells; ATCC), stably expressing murine CEACAM1a (mCC1a), and feline FCWF cells (ATCC) were used to propagate the (recombinant) MHV and FIPV viruses, respectively. HEK293T, MDCK-HA, and Huh7 cells were used to propagate pseudotyped VSVΔG/Luc-G*, Renilla luciferase-expressing IAV-WSN pseudovirus (IAV-RLuc), or MERS-CoV, respectively, as described previously (30–32). Cells were maintained as monolayers cultured in Dulbecco's modified Eagle's medium (DMEM; Lonza), supplemented with 10% fetal bovine serum (FBS). HeLa-ATCC cells stably expressing mCC1a (HeLa-mCC1a) (6) HeLa-fAPN cells (33), and HeLa-ATCC cells were used for infection experiments with MHV, FIPV, and VSV, respectively. HeLa-ATCC and HeLa-mCC1a cells stably expressing the defective β-galactosidase ΔM15 [HeLa-(mCC1a-)ΔM15] were used in entry assays (34). Generation of recombinant viruses MHV-EFLM (35), FIPV-Δ3abcRL (36), IAV-RLuc pseudovirus (30), MHV-αN (34), VSVΔG/Luc-Gα* (34), MHV-2aFLSRec (37), and MHV-S2′FCS (6) has been described previously. MHV-2aGFPSRec, which contains a green fluorescent protein (GFP) expression cassette between the 2a and the S genes at the position of the HE pseudogene, was generated similarly as described for MHV-2aFLSRec (37). cDNAs encoding human or mouse ATP1A1 were obtained from Thermo Scientific/Open Biosystems. ATP1A1 cDNAs were subcloned into a pCAGGS expression vector using conventional cloning methods, thereby generating pCAGGS-hATP1A1 and pCAGGS-mATP1A1.
Chemicals.
The MHV fusion inhibitor HR2 peptide has been described before (38) and was synthesized by GenScript. The peptide was diluted in 50 mM Tris-HCl, pH 7.8, and 4 μM EGTA as a 1 mM stock solution and used at a 10 μM final concentration. Stocks of 125 μM bafilomycin A1 (BafA1; Enzo Life Sciences), 15 mM Dyngo-4a (Abcam), 500 μM wortmannin (Enzo Life Sciences), 10 mM PP2 (Sigma), and 10 μM bufalin (Enzo Life Sciences) were prepared in dimethyl sulfoxide (DMSO) and diluted 1:1,000 in the experiments, except when indicated otherwise. Stocks of 10 mM chlorpromazine (Sigma), 20 mM U18666A (Enzo Life Sciences), and 50 μM ouabain (Sigma) were prepared in H2O and diluted 1:1,000 in the experiments, except when indicated otherwise. pNaKtide, an Na+,K+-ATPase-mimetic Src inhibitor peptide (39) that was kindly provided by Z. Xie (Marshall University, Institute for Interdisciplinary Research), was dissolved in phosphate-buffered saline (PBS) at 2 mM and used at a 2 μM final concentration. Solvent DMSO was obtained from Sigma-Aldrich.
siRNA transfections.
In assays using luciferase-based readouts, 96-well plates were used. For other assays a 24-well plate format was used. A total of 7,500 or 30,000 HeLa-mCC1a or HeLa-fAPN cells were seeded 1 day prior to transfection in each well of the 96-well or 24-well plate, respectively. Using Oligofectamine (Life Technologies) reagent, three independent, nonoverlapping siRNAs (Ambion) targeting ATP1A1 were individually transfected into target cells according to the manufacturer's instructions. The transfection mix for four wells (96-well format) or one well (24-well format) contained 2.5 μl of 1 μM siRNA and 0.5 μl of Oligofectamine in 50 μl of Opti-MEM (Gibco). Transfection was done in a 62.5-μl or 250-μl final volume of Opti-MEM, and at 4 h posttransfection 32 μl or 125 μl of DMEM–30% FBS was added, depending on the plate format used. Cells were infected at 72 h posttransfection.
qRT-PCR of siRNA-mediated gene knockdowns.
HeLa-mCC1a cells were subjected to siRNA-mediated gene knockdown as described above. At 72 h posttransfection, cells were harvested by trypsinization, and a single-cell suspension was counted and collected by centrifugation. Cellular RNA was extracted using an RNeasy minikit (Qiagen). mRNA levels of genes were analyzed by quantitative reverse transcription-PCR (qRT-PCR) using a custom-designed pair of specific primers to the gene, resulting in an approximately 150-bp product. RNA levels were measured using a GoTaq 1-Step RT-qPCR system (Promega) according to the manufacturer's instructions on a LightCycler 480 (Roche). Expression levels were corrected for cell number and viability as determined by a Wst-1 assay (Roche); however, these levels were hardly affected, if at all, by transfection of the siRNAs.
Virus infections.
Cells were inoculated with MHV-EFLM, FIPV-RLuc, IAV-RLuc, VSVΔG/Luc-G*, MHV-S2′FCS, or MHV-2aFLSRec at a multiplicity of infection (MOI) of 0.1 in DMEM–2% FBS for 2 h at 37°C. Cells were lysed at 7 h postinfection (hpi) (MHV, FIPV, and VSV) or 16 hpi (IAV) in passive lysis buffer (Promega). Firefly luciferase expression was assessed using a firefly luciferase assay system from Promega or using a homemade system (50 mM tricine, 100 μM EDTA, 2.5 mM MgSO4, 10 mM dithiothreitol [DTT], 1.25 mM ATP, 12.5 μM d-luciferin). Renilla luciferase expression was assessed using a Renilla luciferase assay system (Promega). Light emission was measured on a Centro LB 960 luminometer. When indicated in the text or figure legends, cells were transfected with siRNAs prior to inoculation as described above. Luciferase expression levels (in relative light units [RLU]) were corrected for cell number and viability as determined by a Wst-1 assay (Roche). When indicated, cells were treated with pharmacological inhibitors starting at 30 min prior to inoculation or 2 h postinoculation.
At 72 h after transfection, siRNA-transfected cells were inoculated with MHV-2aGFPSRec at an MOI of 0.5 (15 to 20% infected cells) in DMEM–2% FBS for 2 h at 37°C. The inoculum was replaced by warm DMEM–10% FBS. At 8 hpi, cells were trypsinized and fixed in 4% formaldehyde solution in PBS. Cells were washed and taken up in fluorescence-activated cell sorting (FACS) buffer (2% FBS, 0.05 M EDTA, 0.2% NaN3 in PBS), and GFP expression was quantified by FACS analysis on a FACSCalibur (Benson Dickson) using FlowJo software. Of each sample at least 10,000 cells were analyzed.
Vero cells were inoculated with MERS-CoV at an MOI of 0.1 in FBS-containing DMEM. At 8 h postinfection, cells were fixed in 4% formaldehyde in PBS. Cells were stained using rabbit anti-SARS-CoV nsp4 antibodies that are cross-reactive for MERS-CoV, according to a standard protocol using a fluorescein isothiocyanate (FITC)-conjugated swine anti-rabbit antibody. The number of infected cells was determined by cell counts on a wide-field fluorescence microscope. Cells were treated with ouabain or bufalin starting at 30 min prior to inoculation or at 2 h postinoculation.
Binding, internalization, and fusion assays using β-galactosidase complementation.
The replication-independent binding, internalization, and fusion assays were performed as described previously (34). The assay is based on complementation of an otherwise defective β-galactosidase ΔM15 protein by a small intravirion peptide that is genetically fused to the N protein. Briefly, in the binding and internalization assay MHV-αN or VSVΔG/Luc-Gα* virus was bound to HeLa-(mCC1a-)ΔM15 target cells at an MOI of 10 for 90 min on ice. In the binding assay unbound virus was removed, and cells and viruses were lysed with NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% NP-40). Complementation was analyzed using a Centro LB 960 luminometer (Berthold Technologies). Beta-Glo reagent (30 μl/well; Promega) was added to each well, the sample was mixed and incubated for 60 min, and light units were measured over 0.1 s. In the internalization assay unbound virus was removed after binding, and cells were shifted to 37°C for 30 or 60 min for VSV or MHV, respectively. Cells were trypsinized to remove surface-bound but not internalized virus. Cells were lysed, and complementation was measured as described above. Depending on the experiment type, cells were transfected with siRNA for 72 h as described above or pretreated with drugs for 30 min prior to binding or internalization experiments.
To assay fusion, cells were preloaded with fluorescein di-β-d-galactopyranoside (FDG) substrate by incubation of adherent target cells with 2.5% FBS, 100 mM FDG, and 50% PBS at room temperature. After a 3-min incubation an excess of 5% FBS in PBS was added, and the supernatant was removed and replaced by growth medium. When pharmacological inhibitors were used, cells were either mock treated or treated with the different inhibitors for 30 min after a recovery period of 30 min at 37°C. MHV-αN or VSVΔG/Luc-Gα* virus was bound to cells in DMEM with 2% FCS (in the absence or presence of inhibitors) at an MOI of 20 for 90 min at 4°C to synchronize infection, after which cells were shifted to 37°C for 2 h. Cells were trypsinized and transferred to Eppendorf tubes, washed, and immediately analyzed by FACS. siRNA transfections were performed 72 h prior to fusion assays.
Ouabain time-of-addition experiment.
MHV-EFLM virus was bound to HeLa-mCC1a cells on ice at an MOI of 0.5 for 90 min. Warm medium containing 10% FBS was added, and cells were incubated for 7 h at 37°C in 5% CO2. At time points indicated in the text or figure legends, the medium was replaced by warm medium containing 50 nM ouabain. At 7 hpi cells were lysed, and luciferase expression was analyzed as described above.
Effect of ouabain on virus entry using fluorescently labeled MHV.
DyLight 488 covalently labeled MHV (MHV-DL488) was made as described before (6). Briefly, MHV strain A59 virus was grown in LR7 cells and purified using a sucrose cushion and gradient purification. After purification virus was labeled using N-hydroxysuccinimide (NHS) ester-activated DyLight 488 (Thermo Scientific) according to the manufacturer's instructions. Infectivity of the labeled virus was confirmed by 50% tissue culture infective dose (TCID50) analysis and qRT-PCR. Fluorescently labeled virus was bound to cells, either mock treated or pretreated with ouabain for 30 min, on ice at an MOI of 10 for 90 min. Unbound virus was removed, and virus was allowed to infect cells for 90 min in the presence or absence of ouabain. Cells were subsequently fixed and stained with 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen) and Alexa Fluor 568-phalloidin (Life Technologies). The samples were analyzed using a confocal laser scanning microscope (Leica SPE-II).
Low-pH bypass of endocytic uptake by VSV via direct fusion at the plasma membrane.
HeLa-ATCC cells were pretreated with medium containing 50 nM ouabain for 30 min at 37°C. Following pretreatment, VSVΔG/Luc-G* was bound to cells at an MOI of 0.3 in the presence of 50 nM ouabain at 4°C. Inoculum was removed, and unbound virus was washed away with ice-cold PBS. Cells were incubated for 2 h at 37°C in the presence of ouabain. At 2 hpi supernatant was removed, and warm buffers at different pHs (7.2, 6.5, 5.5, and 5.0) containing 50 nM ouabain were added for 2 min. Buffers were removed, and cells were incubated at 37°C in medium containing 50 nM ouabain. Infection levels were determined by measuring the luciferase expression levels in the cell lysate at 9 hpi.
RESULTS
RNAi-mediated gene silencing of ATP1A1 inhibits infection with MHV and FIPV but not IAV.
In a high-throughput RNAi screen, ATP1A1 was found to be required for efficient infection of HeLa cells with MHV. To validate this finding and to see whether ATP1A1 is also required for infection with other CoVs, we performed a follow-up analysis using siRNA-mediated gene silencing with oligonucleotides from a different supplier. HeLa cells or HeLa cells carrying the receptor for MHV (HeLa-mCC1a cells) or for FIPV (HeLa-fAPN) were transfected with siRNAs for 72 h. Subsequently, cells were infected with luciferase-expressing MHV (MHV-EFLM) (35), FIPV (FIPV-Δ3abcRL) (36), IAV (IAV-RLuc) (30), or VSV (VSVΔG/Luc-G*) (32) at a multiplicity of infection (MOI) of 0.1. At 7 hpi (MHV, FIPV, and VSV) or 16 hpi (IAV), cells were lysed, and luciferase expression levels were determined. As negative controls, scrambled siRNAs were used. Individual transfection of each of the three siRNAs targeting ATP1A1 resulted in reduced infection of cells with MHV, FIPV, and VSV. IAV infection was not affected by siRNA-mediated gene silencing of ATP1A1 (Fig. 1A). To confirm the efficacies of the siRNAs at the mRNA level, quantitative RT-PCR analysis was performed. All three siRNAs reduced the ATP1A1 mRNA levels by approximately 95% (Fig. 1B). From these results we conclude that ATP1A1 is required for efficient infection of cells with MHV, FIPV, and VSV but not IAV.
FIG 1.
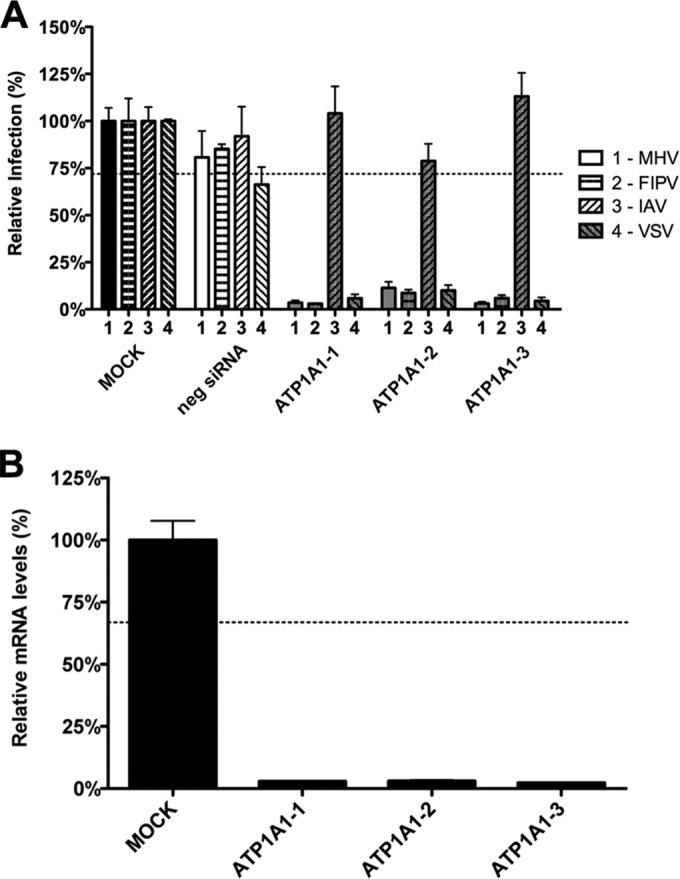
RNAi-mediated downregulation of ATP1A1 affects MHV, FIPV, and VSV but not IAV. (A) Effect of RNAi-mediated downregulation of ATP1A1 on MHV-ERLM, FIPV-RLuc, IAV-RLuc, and VSV-FLuc. Gene silencing was performed using individual transfection of three different siRNAs targeting ATP1A1 (ATP1A1-1 to ATP1A1-3) in HeLa cells expressing the appropriate virus receptors. Negative siRNA (neg siRNA) was included as a control. Cells were infected with luciferase-expressing viruses at an MOI of 0.1 for 7 h or overnight for IAV. Infection levels were determined by assaying the luciferase activity in cell lysates relative to lysates of infected cells that had been mock treated. Infection levels were corrected for cell number and viability as determined by the Wst-1 assay. Error bars represent standard errors of the means (n = 3 replicates of 3). (B) Confirmation of siRNA-mediated reduction in mRNA levels. mRNA levels at 72 h posttransfection were measured by qRT-PCR relative to mock-transfected cells. Expression levels were corrected for cell number and viability as determined by the Wst-1 assay. Error bars represent standard errors of the means (n = 3 replicates of 3). Dotted lines indicate the lower 95% confidence interval of negative siRNA controls (A) or mock treatment (B).
RNAi-mediated gene silencing of ATP1A1 inhibits the fusion signal of MHV and VSV.
To investigate whether the siRNA-mediated silencing of ATP1A1 affected entry of MHV, we made use of a recently developed, replication-independent binding, internalization, and fusion assay (34). The assay is based on minimal complementation of defective β-galactosidase (β-galactosidase ΔM15) with the short α-peptide (40) that is genetically fused to the intravirion N protein in MHV-αN. Prior to virus binding, ΔM15-expressing cells were transfected with siRNAs for 72 h. After binding of virus particles to cells on ice, unbound viruses were removed, and cells and viruses were lysed (binding assay). In the internalization assay MHV-αN was bound to cells on ice, unbound virus was removed, and virus was subsequently allowed to internalize at 37°C for 60 min, after which cell surface-bound virus particles were removed by protease treatment prior to lysis of cells and viruses. Virus particle binding and internalization into cells were quantified by measuring the amount of luminescence generated after addition of Beta-Glo substrate to the cell lysate. As shown in Fig. 2, both virus binding and internalization did not appear to be affected by siRNA-mediated silencing of ATP1A1. To measure fusion, MHV-αN was bound to cells preloaded with fluorescein-di-β-d-galactopyranoside (FDG). After binding, virus was allowed to internalize and fuse. Conversion of the nonfluorescent substrate FDG by reconstituted β-galactosidase into the green fluorophore fluorescein (FIC) in intact cells was measured by FACS. Please note that viral fusion signals can also be inhibited by interference with essential processes that precede viral fusion. In contrast to virus binding and internalization, fusion of MHV was inhibited by the lack of ATP1A1 relative to the negative-control siRNAs (Fig. 2A). siRNA-mediated gene silencing of ATP1A1 also inhibited the fusion signal of VSV as determined with a VSV fusion assay (34) that, just as for MHV, is based on minimal complementation of defective β-galactosidase (Fig. 2B).
FIG 2.
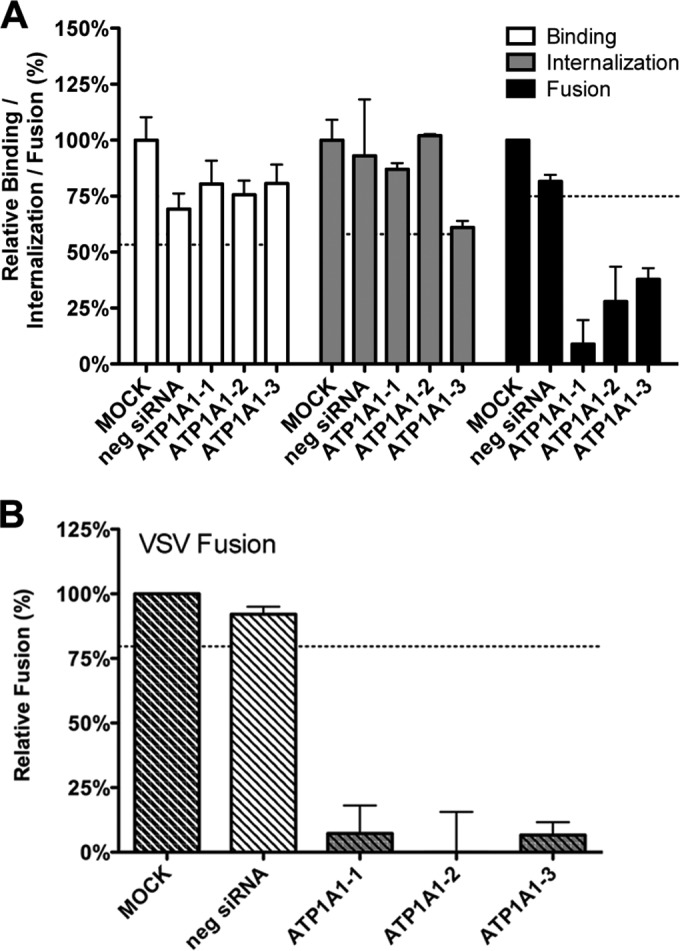
Knockdown of ATP1A1 affects MHV and VSV fusion. (A) Effects of siRNA-mediated gene silencing on viral binding, internalization, and fusion using replication-independent assays. Three different siRNAs against ATP1A1 (ATP1A1-1 to ATP1A1-3) were transfected individually into HeLa-(mCC1a-)ΔM15 cells. Negative siRNA (neg siRNA) was included as a control. At 72 h posttransfection MHV-αN was allowed to bind to the cells on ice at an MOI of 20 for 90 min. Unbound virus was washed off. For the binding assay, cells and viruses were subsequently lysed, and complementation of ΔM15 by αN was determined relative to mock-treated samples using Beta-Glo substrate and a luminometer. For internalization and fusion assays, the cells were warmed to 37°C, and virus was allowed to enter cells for 60 and 100 min, respectively. To assay internalization, cell surface-bound virus was removed using trypsin, and cells and viruses were subsequently lysed. Complementation of ΔM15 by αN was determined relative to mock-treated samples using Beta-Glo substrate and a luminometer. For the fusion assay, cells were preloaded with FDG by hypotonic shock before inoculation. Upon infection for 100 min, cells were collected and analyzed by FACS. Fusion was determined relative to the number of FIC-positive cells observed upon mock treatment of infected cells. Error bars represent standard errors of the means (n = 3 replicates of 3) for binding and internalization; n = 3 for fusion). (B) VSV fusion was determined as described in panel A using VSV-ΔG/Luc-Gα. Dotted lines indicate the lower 95% confidence intervals of the mock treatment.
RNAi-mediated gene silencing of ATP1A1 inhibits infection with MHV independent of the intracellular site of fusion or the identity of the receptor.
Trafficking of MHV and FIPV to lysosomes is a prerequisite for proteolytic activation of the S protein and for efficient virus-cell fusion to occur (6). To study whether downregulation of ATP1A1 inhibits MHV infection by negatively affecting the trafficking of MHV to lysosomes, we made use of a mutant MHV (MHV-S2′FCS), which is cleavage activated by furin rather than by lysosomal proteases and which, hence, fuses in early endosomes (6). Thus, HeLa-mCC1a cells were transfected with siRNAs for 72 h, followed by inoculation with luciferase-expressing MHV (MHV-EFLM) or MHV-S2′FCS at an MOI of 0.1. At 7 hpi cells were lysed, and firefly luciferase expression levels were determined. As shown in Fig. 3A, transfection of siRNAs targeting ATP1A1 reduced luciferase expression levels to the same extent for both viruses. From these results we conclude that infection with MHV is negatively affected by downregulation of ATP1A1, regardless of the intracellular site of fusion.
FIG 3.
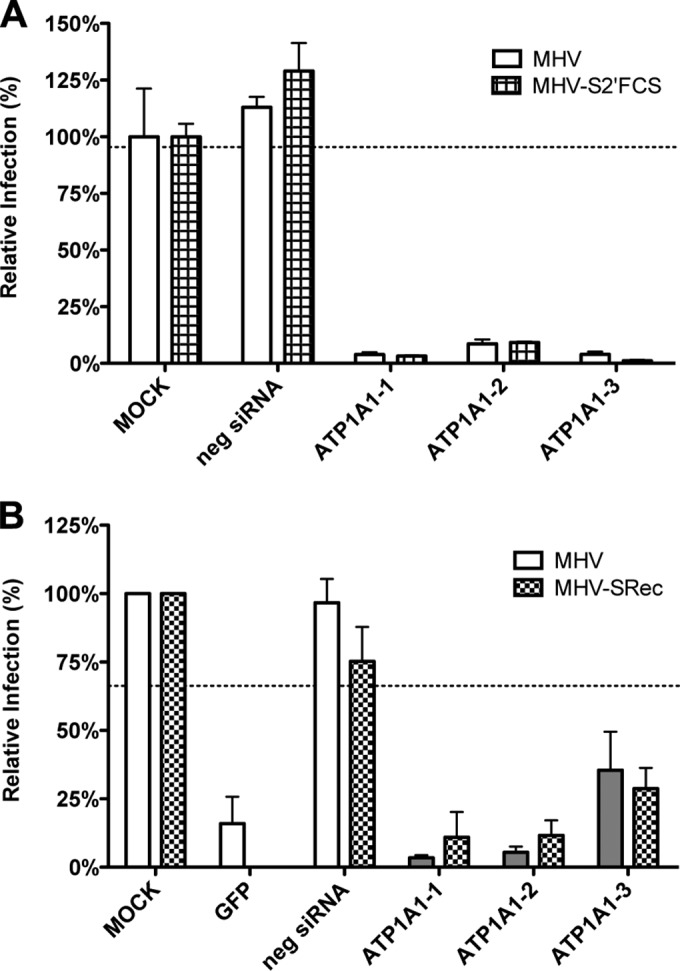
Knockdown of ATP1A1 inhibits infection with MHV independent of the intracellular site of fusion or the receptor used. Gene silencing was performed as described in the legend to Fig. 1. (A) Cells were infected with luciferase-expressing MHV or MHV-S2′FCS at an MOI of 0.1 for 7 h. Infection levels were determined by assaying the luciferase activity in cell lysates relative to lysates of infected cells that had been mock treated. Infection levels were corrected for cell number and viability as determined by the Wst-1 assay. Error bars represent standard errors of the means (n = 3 replicates of 3). (B) Cells were infected with GFP-expressing MHV or MHV-SRec at an MOI of 0.5 for 8 h. Cells were collected, and virus replication and cell viability were analyzed by FACS relative to mock-treated samples. Negative siRNA and an siRNA targeting GFP were included as controls. Error bars represent standard errors of the means (n = 3).
CEACAM1 has been reported to interact with ATP1A1 in porcine cells (41). Since murine CEACAM1a, the natural receptor of MHV, is a homologue thereof, we investigated whether the positive effect of ATP1A1 on MHV infection is somehow linked to MHV binding to CEACAM1a. We made use of a mutant of MHV (MHV-SRec) (37), which enters cells in a CEACAM1a-independent but heparan sulfate-dependent manner. Transfection of HeLa or HeLa-mCC1a (expressing murine CEACAM1a) cells with three different siRNAs against ATP1A1 was followed by low-MOI inoculation with GFP-expressing MHV-SRec (MHV-2aGFP-SRec) or MHV (MHV-EGFPM), respectively. After 8 h of infection, cells were collected, and GFP expression was analyzed by fluorescence-activated cell sorting (FACS). As controls, an siRNA silencing GFP and a negative-control siRNA were used. Infection with MHV-SRec of cells lacking the MHV receptor was reduced to the same extent as MHV infection of receptor-expressing cells by all three siRNAs targeting ATP1A1 (Fig. 3B). These results indicate that, irrespective of the entry receptor, infection with MHV depends on ATP1A1.
Nanomolar concentrations of CTSs inhibit infection with CoVs and VSV but not with IAV.
High concentrations of CTSs are known to inhibit the ion-pumping function of Na+,K+-ATPase (42–44). However, recent research has revealed that CTSs, in particular, ouabain and bufalin, can trigger various signal transduction pathways mediated by Na+,K+-ATPase (14, 16–21, 45, 46) at much lower concentrations. In view of the critical role of ATP1A1 on infection of MHV and FIPV, we investigated to what extent CTSs affect infection of CoVs. Therefore, HeLa cells (MHV, FIPV [indicated as FIPV-H in Fig. 4], VSV, and IAV), Huh-7 cells (MERS-CoV), and feline FCWF cells (FIPV [FIPV-F in Fig. 4]) were treated with ouabain or bufalin at high or low concentrations for 30 min and then inoculated with the indicated viruses in the presence of the drugs, after which the CTSs were present until cells were lysed or fixed. CTSs were also added to cells at 2 h postinfection (hpi) to assess the effects of these drugs on postentry steps. At the indicated time points, cells were lysed or fixed, and luciferase expression levels or numbers of virus-infected cells were determined. Addition of relatively high concentrations of ouabain (250 nM) or bufalin (50 nM) had severe negative effects on infection with all viruses tested, both when the drug was added prior to and after inoculation (data not shown). Also translation of transfected synthetic, capped reporter mRNA was inhibited at these high concentrations (data not shown). Addition of small amounts of ouabain (50 nM) or bufalin (10 to 15 nM) inhibited infection with MHV, FIPV, MERS-CoV, and VSV, but only when the drug was added prior to inoculation. Infection was not affected when the drugs were added at 2 hpi. Infection with IAV was not affected by the addition of low concentrations of ouabain or bufalin (Fig. 4A and B). These results show that low concentrations of CTSs inhibit infection with different CoVs but not with IAV. CTSs most likely affect CoV infection during the entry stage as no effect was observed when they were added after inoculation.
FIG 4.
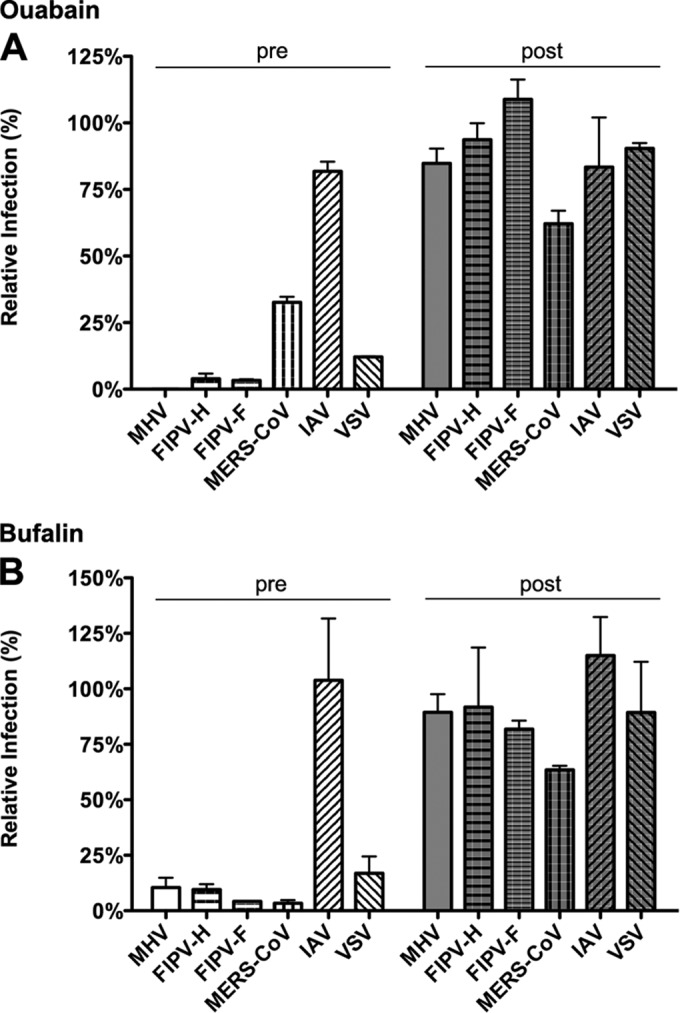
Low levels of ouabain and bufalin affect entry of CoVs and VSV but not of IAV. (A) HeLa (MHV, FIPV [FIPV-H], VSV, and IAV), Huh7 (MERS-CoV), or FCWF (FIPV [FIPV-F]) cells were inoculated with the indicated viruses at an MOI of 0.1 for 2 h. Cells were treated with 50 nM ouabain from 30 min prior to (pre) or 2 h after (post) inoculation until 7 h (MHV and FIPV), 8 h (MERS-CoV), or 16 h (IAV) postinfection. Infection levels were determined by measuring the luciferase activity in cell lysates or by determining the number of infected cells (MERS-CoV) by immunocytochemistry relative to levels in mock-treated cells. Error bars represent standard errors of the means (n = 3 replicates of 3). (B) Effect of low doses of bufalin on MHV, FIPV, MERS-CoV, IAV, and VSV infection. Cells were infected and treated as described in panel A with 10 nM bufalin instead of ouabain. Error bars represent standard errors of the means (n = 3 replicates of 3).
Effect of ouabain on MHV and VSV infection is linked to ATP1A1.
To confirm that the effect of ouabain on CoV infection is indeed linked to ATP1A1, we made use of the fact that rodent ATP1A1-encoded Na+,K+-ATPase is much more resistant to ouabain due to severely decreased binding of the drug to the protein caused by two amino acid mutations in the ectodomain (47). HeLa cells were transfected with plasmids encoding either human or murine ATP1A1. Transfected cells were pretreated with nanomolar concentrations of ouabain and subsequently inoculated with luciferase-expressing MHV, IAV, or VSV in the presence of the drug. Ouabain was kept present until cells were lysed, and luciferase expression levels were determined. As a control, ouabain was also added to cells only from 2 hpi onwards. IAV infection was not affected by ouabain treatment when either human or murine ATP1A1 was overexpressed (Fig. 5). MHV and VSV infection of cells transfected with a plasmid expressing human ATP1A1 was inhibited by ouabain. However, when the ouabain-insensitive murine ATP1A1 was overexpressed, the inhibitory effect of ouabain on infection was abolished (Fig. 5). These results demonstrate that the inhibitory effect of ouabain on CoV infection is directly linked to ATP1A1.
FIG 5.
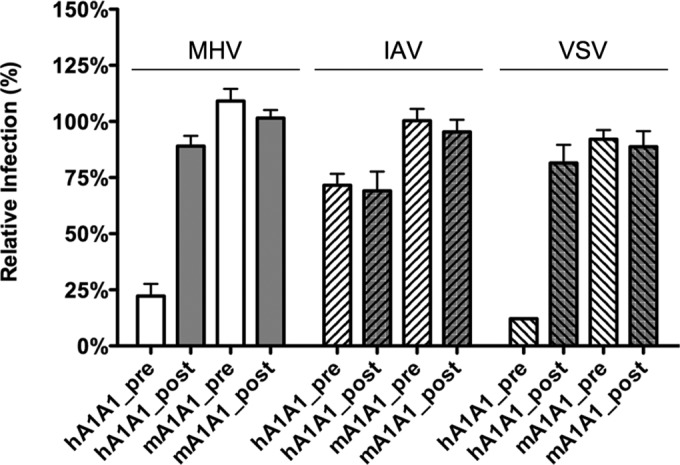
Effect of ouabain on virus entry is linked to ATP1A1-encoded α1 subunit. HeLa cells were transfected with plasmids expressing either human- or murine-derived ATP1A1 (hATP1A1 and mATP1A1, respectively). Cells were treated with 50 nM ouabain from 30 min prior to (pre) or 2 h after (post) inoculation with luciferase-expressing MHV, IAV, or VSV at an MOI of 0.1 until 7 h (MHV and VSV) or 16 h (IAV) postinfection. Infection levels were determined by measuring the luciferase activity in cell lysates relative to that in lysates of mock-treated cells. Infection levels were corrected for cell number and viability as determined by the Wst-1 assay prior to infection. Error bars represent standard errors of the means (n = 3 replicates of 3).
CTSs decrease the fusion signal of MHV and VSV.
Next, we investigated the inhibition of MHV infection by low levels of ouabain by performing an ouabain time-of-addition experiment (Fig. 6A). Luciferase-expressing MHV was bound to HeLa-mCC1a cells for 90 min on ice. Unbound virus was removed, and cells were shifted to 37°C to allow infection. At the indicated time points, cell culture medium was replaced by warm, ouabain-containing medium. At 7 hpi cells were lysed, and luciferase expression levels were determined. Addition of ouabain affected MHV infection only when the drug was added during the first 2 h of infection (Fig. 6A), indicating that ouabain specifically inhibits MHV infection during entry.
FIG 6.

Low levels of ouabain and bufalin prevent fusion of MHV and VSV. (A) Time-of-addition experiment using 50 nM ouabain. Luciferase-expressing MHV was bound to HeLa-mCC1a cells at an MOI of 0.5 for 90 min on ice. Unbound virus was washed off, and incubation continued at 37°C. At the indicated time points medium was replaced by warm medium containing 50 nM ouabain. Luciferase expression levels were determined relative to those of mock-treated cells. Error bars represent standard errors of the means (n = 3). (B) Binding, internalization, and fusion assays of MHV upon ouabain or bufalin treatment were performed as described in the legend to Fig. 2. HeLa-(mCC1a-)ΔM15 cells were pretreated with 50 nM ouabain or 10 nM bufalin. (C) Binding, internalization, and fusion assays of VSV were performed using VSV-ΔG/Luc-Gα as described for MHV in the legend to Fig. 2. In panels B and C, error bars represent standard errors of the means (n = 3 replicates of 3 for binding and internalization; n = 3 for fusion). (D) Effect of ouabain treatment on infection with MHV-SRec or MHV-S2′FCS. Cells were treated with ouabain as described in the legend to Fig. 5 and inoculated with luciferase-expressing MHV, MHV-S2′FCS, or MHV-SRec at an MOI of 0.1. As a control, cells were treated with U18666A. Infection levels were determined by measuring the luciferase expression levels in cells at 7 h postinfection relative to those in mock-treated cells. Error bars represent standard errors of the means (n = 3 replicates of 3).
To dissect which CoV entry step is affected by the addition of low concentrations of bufalin or ouabain, we again made use of the replication-independent binding, internalization, and fusion assays. MHV-αN was bound to ΔM15-expressing cells that were pretreated for 30 min with either ouabain or bufalin. Binding, internalization, and fusion of MHV-αN were determined as described above. Binding and internalization of MHV did not appear to be affected by ouabain or bufalin treatment. However, the fusion signal of MHV was clearly reduced (Fig. 6B). Very similar results were obtained for VSV (Fig. 6C).
In addition, we analyzed whether the inhibition of MHV infection by ouabain is dependent on the nature of the entry receptor used or on the depth of MHV trafficking into the endo-lysosomal pathway. Therefore, HeLa-mCC1a and HeLa cells were inoculated with luciferase-expressing MHV (virus dependent on CEACAM1a and lysosomal trafficking), MHV-S2′FCS (virus fuses in early endosomes), or MHV-SRec (virus dependent on heparan sulfate rather than CEACAM1a) in the presence of ouabain, which was retained until cell lysis. To control for any postentry effects of ouabain, the drug was added only at 2 h postinoculation and retained. As an additional control, cells were treated with U18666A, which inhibits late endosome-to-lysosome trafficking (6, 48). Ouabain negatively affected infection with both MHV and MHV-S2′FCS (Fig. 6D), indicating that it inhibits infection regardless of the intracellular site of fusion. In contrast, infection with MHV, but not with MHV-S2′FCS, was affected by U18666A. Also CEACAM1a-independent infection of MHV-SRec was inhibited to the same extent as MHV.
Ouabain inhibits virus entry at an early stage.
The replication-independent binding, internalization, and fusion assays indicate that nanomolar levels of ouabain decrease the MHV fusion but not the internalization signal. To get more insight into the inhibition of virus entry by ouabain, we analyzed whether MHV infection could recover during an overnight incubation upon removal of ouabain at 2 hpi. Our results (Fig. 7A) show that this is indeed the case. No inhibition of virus infection was observed when ouabain was removed after virus inoculation. Virus infection was only inhibited when ouabain was present during both inoculation and the overnight incubation. This allowed us to study whether the block induced by ouabain inhibited entry of MHV upstream or downstream of the inhibitory effects of known inhibitors of virus entry (6, 34, 49). Cells were either mock treated or treated with ouabain prior to and during inoculation with luciferase-expressing MHV. After removal of the inoculum, cells were incubated for another 16 h in the absence or presence of inhibitory agents known to affect MHV entry (6, 34). Subsequently, cells were lysed, and luciferase expression levels were determined. Luciferase expression levels obtained after ouabain treatment prior to and during inoculation but not thereafter were set to 100% (Fig. 7A, black bar). Overnight incubation in the presence of the cell-impermeable MHV fusion inhibitor peptide HR2 (38) did not inhibit MHV infection, regardless of the absence or presence of ouabain during virus inoculation. However, virus infection was severely reduced when HR2 peptide was present during virus inoculation, confirming the ability of the HR2 peptide to inhibit entry (Fig. 7B). Addition of inhibitors of dynamin-2 (Dyngo-4A), clathrin-mediated endocytosis (chlorpromazine), or endosomal maturation (bafilomycin A1; BafA1) reduced infection with MHV when the inhibitor was added after removal of ouabain (Fig. 7C). The smaller inhibition observed after addition of BafA1 compared to that of Dyngo-4A and chlorpromazine is in agreement with the reported inhibition of MHV entry by these compounds (6, 34). The inhibitors did not affect luciferase expression levels without prior incubation with ouabain, indicating that they do not affect MHV infection at postentry stages. These results indicate that ouabain inhibits infection with MHV upstream of the inhibitory effects of Dyngo-4A, chlorpromazine, and BafA1.
FIG 7.

Ouabain inhibits virus entry upstream of inhibitors of CME. (A) The inhibitory effect of ouabain is not observed when the compound is removed after virus inoculation. Cells were mock treated or treated with 50 nM ouabain (Ou) starting at 30 min prior to and during inoculation (the period 0 to 2 h) and/or after removal of the inoculum (the period 2 to 18 h). Cells were inoculated with luciferase-expressing MHV at an MOI of 0.1. (B) Virus infection is not inhibited when the fusion-inhibitory peptide HR2 is added after removal of ouabain. Cells were mock treated or treated with the indicated compound (ouabain [Ou] or HR2 peptide) starting at 30 min prior to and during inoculation (0 to 2 h) and/or after removal of the inoculum (2 to 18 h). (C) After the removal of ouabain, virus infection was inhibited by the addition of CME inhibitors. Cells were mock treated or treated with 50 nM ouabain (Ou) starting at 30 min prior to and during inoculation (0 to 2 h). After removal of the inoculum the medium was replaced by drug-containing medium (Dyngo, Dyngo-4A; CPZ, chlorpromazine; and BafA1, bafilomycin A1). In panels A to C, after overnight infection cells were lysed, and infection levels were determined by measuring the luciferase activity in cell lysates relative to that of control cells that were treated only with ouabain prior to and during inoculation (Ou, 0 to 2 h only; black bar). Error bars represent standard errors of the means (n = 3 replicates of 3).
MHV particles remain associated close to the cell surface.
To confirm and visualize the early block in infection by ouabain, MHV covalently labeled with DyLight 488 (MHV-DL488) (6) was bound to ouabain- or mock-treated cells for 90 min at an MOI of 20 on ice. After removal of unbound virus particles, cells were incubated for 90 min at 37°C in the presence or absence of ouabain. Cells were then fixed and analyzed by confocal microscopy. The contours of the cells were visualized using phalloidin, which stains the actin cytoskeleton. In mock-treated cells, relatively few fluorescent virions were visible inside the cells (Fig. 8A, upper panel). On the other hand, in ouabain-treated cells a larger number of virions were observed which appeared, however, to remain associated close to the cell surface (Fig. 8A, lower panel), in agreement with ouabain inhibiting virus entry at an early stage. The larger number of virions observed in the presence of ouabain is probably explained by the inhibition of virus uptake and subsequent trafficking of virus particles to lysosomes, where they fuse and/or are broken down (6).
FIG 8.

Ouabain inhibits virus entry at an early stage. (A) MHV particles accumulate close to the cell surface in the presence of ouabain. Images show ouabain-treated cells inoculated with DyLight 488-labeled MHV by confocal microscopy. Cells were mock treated (upper two rows) or treated with 50 nM ouabain (lower two rows) throughout the experiment starting at 30 min prior to inoculation. MHV covalently labeled with DyLight 488 (MHV particles) was bound to cells at an MOI of 20 for 70 min on ice. Unbound virus was removed, and cell-bound virus was allowed to infect at 37°C for 90 min. Cells were fixed, stained with DAPI (nuclei) and phalloidin (actin), and analyzed by confocal microscopy. Single z-slices are shown. (B) The inhibitory effect of ouabain on VSV entry can be bypassed by low-pH shock-induced fusion. Cells were pretreated with 50 nM ouabain. VSV-FLuc virus was bound to the pretreated cells at an MOI of 0.3 in the presence of 50 nM ouabain on ice for 90 min. Unbound virus was removed, and cells were incubated for 2 h at 37°C in the presence of ouabain. At 2 hpi the inoculum was removed, and cells were incubated for 2 min with warm buffers at different pHs (7.2, 6.5, 5.5, and 5.0) that contained 50 nM ouabain. Incubation at 37°C in ouabain-containing medium was continued until 11 hpi. Infection levels were determined by measuring the luciferase expression levels in cell lysates relative to those in mock-treated cells. Error bars represent standard errors of the means (n = 3 replicates of 3).
To assess whether also for VSV the block in infection is caused by an early entry block, we made use of the ability of VSV to bypass the endocytic uptake route when it was exposed to a low extracellular pH upon binding. Cells were pretreated with 50 nM ouabain. Cells were inoculated with luciferase-expressing VSV at an MOI of 0.1 in the presence of 50 nM ouabain. At 2 hpi the inoculum was removed, and unbound viruses were washed away with ice-cold PBS. Cells were incubated for 2 min in warm buffers at different pHs (7.2, 6.5, 5.5, and 5.0) containing 50 nM ouabain. Cells were subsequently incubated for another 7 h in ouabain-containing medium. Infection levels were determined by measuring the luciferase expression levels of cell lysates relative to those in mock-treated cells. In agreement with VSV entry being inhibited by ouabain at an early stage, infection could be rescued by a low-pH shock (Fig. 8B).
Inhibitory effects of ouabain on CoV infection can be rescued by inhibitors of Src but not PI3K.
CoV infection is inhibited by low concentrations of CTSs known to trigger different Na+,K+-ATPase-mediated signaling pathways but not to affect the ion pump function (16–21). Two of the main signaling pathways induced by CTSs and mediated through Na+,K+-ATPase involve the activation of Src or PI3K (50). In order to elucidate whether these signaling pathways are involved in the antiviral action of ouabain, we used mock-treated cells or cells treated with ouabain alone or in combination with either the PI3K inhibitor wortmannin (51), Src inhibitor PP2 (52), or an Na+,K+-ATPase-mimetic Src-inhibitor peptide (pNaKtide). pNaKtide binds and inhibits the Na+,K+-ATPase-interacting pool of Src (39). As a control, cells were treated with the kinase inhibitors in the absence of ouabain. The cells were inoculated with luciferase-expressing MHV, FIPV, or VSV in the presence of the inhibitors, after which the drugs were present until cell lysis. To check for inhibitory effects after virus entry, cells were also treated with inhibitors starting at 2 hpi. At 7 hpi cells were lysed, and luciferase expression levels were determined. For reasons unknown, treatment of cells with wortmannin or PP2 during virus inoculation reduced MHV and VSV infection by about 75% (Fig. 9A and C), while the pNaKtide had a smaller negative effect. Infection with FIPV was not affected by these inhibitors (Fig. 9B). pNaKtide has a much smaller inhibitory effect than PP2 because pNaKtide, in contrast to PP2, specifically targets the Na+,K+-ATPase-interacting pool of Src and has fewer (off-target) effects than PP2 (39, 53). MHV-, FIPV-, and VSV-driven luciferase expression levels were severely reduced by ouabain when the drug was present during virus inoculation yet not when it was added at 2 hpi only, as observed earlier. The combined treatment with ouabain and wortmannin did not positively affect MHV, FIPV, or VSV infection compared to treatment with ouabain alone. However, combined treatment of ouabain with PP2 or pNaKtide almost completely restored MHV, FIPV, and VSV infection to the levels observed after treatment with PP2 or pNaKtide alone (Fig. 9A, B, and C). These results show that the negative effect of ouabain on the entry of MHV, FIPV, and VSV can be relieved by inhibition of Src.
FIG 9.

Inhibition of infection by ouabain is rescued by inhibition of Src. HeLa cells were inoculated with luciferase-expressing MHV, FIPV, or VSV at an MOI of 0.1 for 2 h. Cells were treated with 50 nM ouabain (Ou), wortmannin (Wort), PP2, pNaKtide or a combination thereof as indicated from 30 min prior to (pre) or 2 h after (post) inoculation. The drugs were present until cell lysis at 7 h postinoculation. Infection levels were determined by measuring the luciferase activity in lysates of drug-treated cells relative to that in mock-treated cells. Error bars represent standard errors of the means (n = 3 replicates of 3).
DISCUSSION
This study provides an extensive analysis of the role of the ATP1A1-encoded α1 subunit of Na+,K+-ATPase in CoV infection. Using gene silencing, we showed that ATP1A1 is important for infection of cells with MHV and FIPV but not IAV. Also entry of VSV was shown to depend on ATP1A1. Lack of ATP1A1 was found not to affect MHV binding to cells and did not appear to inhibit endosomal uptake although fusion with cellular membranes was reduced. Consistently, nanomolar concentrations of CTSs inhibited infection of cells with MHV, FIPV, and MERS-CoV when the compounds were present during virus entry. Similar results were obtained with VSV but not with IAV. CTSs were shown to inhibit entry of MHV at an early stage, resulting in the accumulation of virions close to the cell surface and, as a consequence, in reduced fusion. Viral RNA replication per se was not affected by these compounds at the concentrations used. In agreement with low concentrations of CTSs not affecting the ion transport function of Na+,K+-ATPase (16–21), the anti-coronaviral effect could be relieved by the addition of inhibitors of Src kinases, indicating that Src signaling mediated via ATP1A1 plays a crucial role in the inhibition of infection with CoVs.
Knockdown of ATP1A1 or additions of low concentrations of CTSs inhibit CoV and VSV infection during the virus entry stage as we could demonstrate using our recently developed replication-independent entry assays (34). CoV and VSV replication was not affected, as revealed by the addition of the CTSs after inoculation. Inhibition of MHV was found to be independent of the particular virus receptor being used by the virus, despite the reported interaction between CEACAM1 and ATP1A1 (41). This is in line with FIPV and MERS-CoV being similarly inhibited although they use entirely different entry receptors (54, 55). FIPV, on the one hand, and MHV and MERS-CoV, on the other hand, belong to the Alphacoronovirus (α-CoV) and Betacoronovirus (β-CoV) genera, respectively, suggesting that CTSs may function as pan-CoV inhibitors.
Interpreting our combined results, we developed the model shown in Fig. 10. In this model, two elements are addressed. First, it recapitulates the early stage of CoV entry as we and others have described (6, 56–59): uptake of CoV in a preendosome that pinches off to form an early endosome. Based on our data, we propose a model in which the uptake of MHV particles is arrested in preendosomal structures by transfection of siRNAs targeting ATP1A1 or by the addition of CTSs rather than having a direct effect on virus-cell fusion. This model explains the apparent paradoxical observations that internalization of MHV particles was not affected by ATP1A1 interference, while, on the other hand, ouabain was shown to inhibit a very early step in MHV entry, upstream of the inhibitory effect of compounds affecting dynamin-2 and/or clathrin-mediated endocytosis (6). The internalization assay depends on the removal of cell surface-bound virions by proteases. The lack of internalization inhibition observed with this assay can be explained by MHV particles accumulating in preendosomal invaginations, which are not accessible by the membrane-impermeable protease in the presence of ouabain. In agreement with this model, we previously showed that also the dynamin-2 inhibitor Dynasore has little effect on the internalization of MHV and VSV virions, while fusion was severely hampered (34). Also the inability of the membrane-impermeable inhibitory HR2 peptide to prevent MHV infection after ouabain removal can be explained by the inability of the HR2 compound to access the preendosomal structures. The inhibition of MHV entry by inhibitors of clathrin-mediated endocytosis after ouabain removal indicates that further internalization of the preendosomal invaginations is sensitive to these inhibitors. In agreement with our model, interference with the Na+,K+-ATPase (either by ATP1A1 knockdown or addition of CTSs) inhibited CoV entry, regardless whether viruses fusing in early endosomes (MHV-2′FCS and MERS-CoV) or lysosomes (MHV and FIPV) were used (6). These results indicate that interference with the ATP1A1 subunit acts prior to the formation of early endosomes and does not result from a defect in endosome maturation. Our model is supported by confocal microscopy analysis, which showed MHV particles to accumulate close to the cell surface in the presence of ouabain. As for the CoVs, entry of VSV, but not of IAV, was also inhibited by ouabain. Apparently, treatment of cells with oubain does not affect the low pH in the endosomes, which is essential for entry of both IAV and VSV (60–62). The ability to bypass the VSV entry block induced by ouabain by low-pH shock indicates that fusion per se of VSV is not affected but, rather, results from ouabain preventing uptake of VSV in early endosomes where fusion can take place (49, 60, 61).
FIG 10.

Model of the effect of ATP1A1 knockdown and CTSs treatment on entry of CoVs and VSV. siRNA-mediated gene silencing of ATP1A1 encoding the α1 subunit of the Na+,K+-ATPase or treatment of cells with CTSs inhibits infection with CoVs and VSV at an early entry stage, resulting in reduced virus-cell fusion. In the presence of CTSs or after siRNA-mediated gene silencing of ATP1A1, virus particles accumulate in preendosomal invaginations that are not accessible to the membrane-impermeable HR2 peptide or trypsin. For VSV, this block in entry can be bypassed by low-pH shock. Knockdown of ATP1A1 leads to release of an Na+,K+-ATPase-bound subset of Src, Src activation, and increased Src signaling (20, 39, 63). Ouabain binding to the α1 subunit of Na+,K+-ATPase triggers a conformational change in this subunit, which also results in release of Src from Na+,K+-ATPase and its concomitant activation (20, 86). Activated Src induces yet unknown downstream signaling, which inhibits virus entry at an early stage upstream of the inhibitory effects of inhibitors of clathrin-mediated endocytosis.
The second element represented in our model (Fig. 10) addresses the mechanism by which interference with the ATP1A1 subunit activity blocks CoV and VSV entry. Our results indicate that CoV infection is inhibited by low concentrations of CTSs via Na+,K+-ATPase-mediated Src signaling. CoV infection of HeLa cells expressing the ouabain-insensitive murine ATP1A1-encoded α1 subunit (47) was unaffected by ouabain treatment. These results show that ouabain mediates its antiviral effect via the α1 subunit and not via an off-target effect, in agreement with the literature (reviewed in references 13 and 14). Ample evidence exists in the literature demonstrating that nanomolar concentrations of CTSs induce α1-subunit-mediated signaling pathways, including the activation of Src. At these concentrations, ouabain binding to the α1 subunit triggers a conformational change in this subunit, which results in release of Src from Na+,K+-ATPase and its concomitant activation (16–18, 22–28). The alleviation of inhibition of CoV infection by ouabain with two chemically different Src inhibitors, PP2 and pNaKtide, but not by an inhibitor of PI3K shows that the activation of Src via the α1 subunit is the inhibitory mode of action of this compound on CoV infection. In agreement with the inhibitory effect of Na+,K+-ATPase-mediated Src signaling on CoV infection, gene silencing of ATP1A1 has also been shown to result in activation of Src (20, 39, 63).
We speculate that ATP1A1-mediated Src signaling somehow interferes with clathrin-mediated uptake of CoVs and VSV. Similar to MHV (6, 56–59) and VSV (49, 60), MERS-CoV and FIPV also appear to be taken up via clathrin-mediated endocytosis as infection with these viruses is inhibited by chlorpromazine in a dose-dependent manner (64; unpublished results) although FIPV has also been reported to enter monocytes via a clathrin- and caveola-independent endocytic pathway (65). An explanation for the lack of inhibition of IAV could be that this virus is able to enter cells via multiple fully redundant endocytic routes (62, 66–69). ATP1A1-mediated Src signaling has been shown to induce phosphorylation of dynamin-2 and caveolar endocytosis (63, 70). However, it is not yet clear how ATP1A1-mediated Src signaling could interfere with clathrin-mediated entry of CoVs and VSV. Other studies have shown the importance of Src-mediated phosphorylation of dynamin-2 in clathrin-mediated as well as caveola-mediated endocytic uptake of cargo (71–73). For reoviruses, which are probably taken up via clathrin-mediated endocytosis, Src phosphorylation has also been shown to mediate endocytic sorting of these viruses; internalization of reovirus virions, however, was not affected by inhibition of Src signaling (74). Spatiotemporal compartmentalization and regulation of Src signaling may be an important determinant of the specificity of Src signaling and of the different biological outcomes observed (75, 76).
Inhibition of infection by CTSs has been reported earlier for several other viruses, including Sindbis virus (77), Sendai virus (78), Semliki Forest virus (79), several herpesviruses (80), and porcine reproductive and respiratory syndrome virus (PRRSV) (81). Most of these studies, however, employed relatively high concentrations of the CTSs (micromolar range) which inhibit the Na+,K+-ATPase pump function and affect intracellular ion concentrations (82, 83). In the present study, the low levels of CTSs did not affect infection with IAV. However, at high concentrations, infections by CoVs, VSV, and IAVs were inhibited also when the compounds were only present after virus entry. This more general inhibitory effect at these concentrations may result from side effects of the drugs, such as inhibition of mRNA translation (data not shown). Indeed, intracellular levels of Na+ and K+ have been implicated previously in the regulation of cellular protein synthesis (84, 85). Interestingly, for human cytomegalovirus, low-nanomolar concentrations of ouabain were shown to inhibit an early step in the infection cycle of this virus prior to DNA replication but following binding to cellular receptors, suggesting that also for this virus entry may be impaired (80).
Ouabain and several other CTSs are FDA-approved compounds. Targeting host factors using FDA-approved compounds to combat viral infections is certainly attractive. Drugs targeting host rather than viral factors may lower the probability of generating drug-resistant viral variants since mutation of the drug target is not possible. In addition, the repurposing of FDA-approved compounds may enable relatively fast clinical application. Elucidation of the action mechanism of the antiviral compounds as exemplified here by the anti-CoV and -VSV effects of low concentrations of CTSs may aid the development and design of new compounds with improved therapeutic efficacy and fewer side effects.
ACKNOWLEDGMENTS
We thank Zijian Xie, Marshall University, Institute of Interdisciplinary Research, Huntington, VA, for providing us with pNaKtide.
This work was supported by the EU 7th Framework Programme (Virus Entry, project 235649, to P.J.M.R) and by a Utrecht University High potential grant to C.A.M.D.H.
REFERENCES
- 1.Alvarez-Salas LM. 2008. Nucleic acids as therapeutic agents. Curr Top Med Chem 8:1379–1404. doi: 10.2174/156802608786141133. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch AJ. 2010. The use of RNAi-based screens to identify host proteins involved in viral replication. Future Microbiol 5:303–311. doi: 10.2217/fmb.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pauli I, Timmers LF, Caceres RA, Soares MB, de Azevedo WF Jr. 2008. In silico and in vitro: identifying new drugs. Curr Drug Targets 9:1054–1061. doi: 10.2174/138945008786949397. [DOI] [PubMed] [Google Scholar]
- 4.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 6.Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, Pelkmans L, Rottier PJ, Bosch BJ, de Haan CA. 2014. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog 10:e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagemeijer MC, Rottier PJ, de Haan CA. 2012. Biogenesis and dynamics of the coronavirus replicative structures. Viruses 4:3245–3269. doi: 10.3390/v4113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Haan CA, Rottier PJ. 2005. Molecular interactions in the assembly of coronaviruses. Adv Virus Res 64:165–230. doi: 10.1016/S0065-3527(05)64006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skou JC. 2004. The identification of the sodium pump. Biosci Rep 24:436–451. doi: 10.1007/s10540-005-2740-9. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan JH. 2002. Biochemistry of Na,K-ATPase. Annu Rev Biochem 71:511–535. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 11.Withering W. 1785. An account of the foxglove and some of its medical uses with practical remarks on dropsy and other diseases. G. G. J. and J. Robinson, London, United Kingdom. [Google Scholar]
- 12.Schoner W, Scheiner-Bobis G. 2007. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs 7:173–189. doi: 10.2165/00129784-200707030-00004. [DOI] [PubMed] [Google Scholar]
- 13.Reinhard L, Tidow H, Clausen MJ, Nissen P. 2013. Na+,K+-ATPase as a docking station: protein-protein complexes of the Na+,K+-ATPase. Cell Mol Life Sci 70:205–222. doi: 10.1007/s00018-012-1039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aperia A. 2007. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J Intern Med 261:44–52. doi: 10.1111/j.1365-2796.2006.01745.x. [DOI] [PubMed] [Google Scholar]
- 15.Karpova LV, Bulygina ER, Boldyrev AA. 2010. Different neuronal Na+/K+-ATPase isoforms are involved in diverse signaling pathways. Cell Biochem Funct 28:135–141. doi: 10.1002/cbf.1632. [DOI] [PubMed] [Google Scholar]
- 16.Aizman O, Uhlen P, Lal M, Brismar H, Aperia A. 2001. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci U S A 98:13420–13424. doi: 10.1073/pnas.221315298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haas M, Askari A, Xie Z. 2000. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 275:27832–27837. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 18.Mohammadi K, Kometiani P, Xie Z, Askari A. 2001. Role of protein kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J Biol Chem 276:42050–42056. doi: 10.1074/jbc.M107892200. [DOI] [PubMed] [Google Scholar]
- 19.Su CT, Hsu JT, Hsieh HP, Lin PH, Chen TC, Kao CL, Lee CN, Chang SY. 2008. Anti-HSV activity of digitoxin and its possible mechanisms. Antiviral Res 79:62–70. doi: 10.1016/j.antiviral.2008.01.156. [DOI] [PubMed] [Google Scholar]
- 20.Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang XY, Xie ZJ. 2006. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell 17:317–326. doi: 10.1091/mbc.E05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian J, Gong X, Xie Z. 2001. Signal-transducing function of Na+-K+-ATPase is essential for ouabain's effect on [Ca2+]i in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 281:H1899–H1907. [DOI] [PubMed] [Google Scholar]
- 22.Barwe SP, Anilkumar G, Moon SY, Zheng Y, Whitelegge JP, Rajasekaran SA, Rajasekaran AK. 2005. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol Biol Cell 16:1082–1094. doi: 10.1091/mbc.E04-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belusa R, Wang ZM, Matsubara T, Sahlgren B, Dulubova I, Nairn AC, Ruoslahti E, Greengard P, Aperia A. 1997. Mutation of the protein kinase C phosphorylation site on rat α1 Na+,K+-ATPase alters regulation of intracellular Na+ and pH and influences cell shape and adhesiveness. J Biol Chem 272:20179–20184. doi: 10.1074/jbc.272.32.20179. [DOI] [PubMed] [Google Scholar]
- 24.Contreras RG, Shoshani L, Flores-Maldonado C, Lazaro A, Cereijido M. 1999. Relationship between Na+,K+-ATPase and cell attachment. J Cell Sci 112:4223–4232. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Zelenin S, Aperia A, Aizman O. 2006. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-κB. J Am Soc Nephrol 17:1848–1857. doi: 10.1681/ASN.2005080894. [DOI] [PubMed] [Google Scholar]
- 26.Kometiani P, Li J, Gnudi L, Kahn BB, Askari A, Xie Z. 1998. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J Biol Chem 273:15249–15256. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. 2000. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem 275:27838–27844. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- 28.Miyakawa-Naito A, Uhlen P, Lal M, Aizman O, Mikoshiba K, Brismar H, Zelenin S, Aperia A. 2003. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J Biol Chem 278:50355–50361. doi: 10.1074/jbc.M305378200. [DOI] [PubMed] [Google Scholar]
- 29.Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. 2000. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J Virol 74:1393–1406. doi: 10.1128/JVI.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473-12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tani H, Komoda Y, Matsuo E, Suzuki K, Hamamoto I, Yamashita T, Moriishi K, Fujiyama K, Kanto T, Hayashi N, Owsianka A, Patel AH, Whitt MA, Matsuura Y. 2007. Replication-competent recombinant vesicular stomatitis virus encoding hepatitis C virus envelope proteins. J Virol 81:8601–8612. doi: 10.1128/JVI.00608-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wurdinger T, Verheije MH, Raaben M, Bosch BJ, de Haan CA, van Beusechem VW, Rottier PJ, Gerritsen WR. 2005. Targeting non-human coronaviruses to human cancer cells using a bispecific single-chain antibody. Gene Ther 12:1394–1404. doi: 10.1038/sj.gt.3302535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burkard C, Bloyet LM, Wicht O, van Kuppeveld FJ, Rottier PJ, de Haan CA, Bosch BJ. 2014. Dissecting virus entry: replication-independent analysis of virus binding, internalization, and penetration using minimal complementation of β-galactosidase. PLoS One 9:e101762. doi: 10.1371/journal.pone.0101762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Haan CA, van Genne L, Stoop JN, Volders H, Rottier PJ. 2003. Coronaviruses as vectors: position dependence of foreign gene expression. J Virol 77:11312–11323. doi: 10.1128/JVI.77.21.11312-11323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Haan CA, Haijema BJ, Boss D, Heuts FW, Rottier PJ. 2005. Coronaviruses as vectors: stability of foreign gene expression. J Virol 79:12742–12751. doi: 10.1128/JVI.79.20.12742-12751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Haan CA, Li Z, te Lintelo E, Bosch BJ, Haijema BJ, Rottier PJ. 2005. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J Virol 79:14451–14456. doi: 10.1128/JVI.79.22.14451-14456.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Z, Cai T, Tian J, Xie JX, Zhao X, Liu L, Shapiro JI, Xie Z. 2009. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biol Chem 284:21066–21076. doi: 10.1074/jbc.M109.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langley KE, Villarejo MR, Fowler AV, Zamenhof PJ, Zabin I. 1975. Molecular basis of beta-galactosidase alpha-complementation. Proc Natl Acad Sci U S A 72:1254–1257. doi: 10.1073/pnas.72.4.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta S, Yan Y, Malhotra D, Liu J, Xie Z, Najjar SM, Shapiro JI. 2012. Ouabain and insulin induce sodium pump endocytosis in renal epithelium. Hypertension 59:665–672. doi: 10.1161/HYPERTENSIONAHA.111.176727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blaustein MP, Juhaszova M, Golovina VA. 1998. The cellular mechanism of action of cardiotonic steroids: a new hypothesis. Clin Exp Hypertens 20:691–703. doi: 10.3109/10641969809053247. [DOI] [PubMed] [Google Scholar]
- 43.Dvela M, Rosen H, Feldmann T, Nesher M, Lichtstein D. 2007. Diverse biological responses to different cardiotonic steroids. Pathophysiology 14:159–166. doi: 10.1016/j.pathophys.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 44.Wasserstrom JA, Aistrup GL. 2005. Digitalis: new actions for an old drug. Am J Physiol Heart Circ Physiol 289:H1781–H1793. doi: 10.1152/ajpheart.00707.2004. [DOI] [PubMed] [Google Scholar]
- 45.Kurosawa M, Numazawa S, Tani Y, Yoshida T. 2000. ERK signaling mediates the induction of inflammatory cytokines by bufalin in human monocytic cells. Am J Physiol Cell Physiol 278:C500–C508. [DOI] [PubMed] [Google Scholar]
- 46.Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. 1996. The cooperative interaction of two different signaling pathways in response to bufalin induces apoptosis in human leukemia U937 cells. J Biol Chem 271:14067–14072. doi: 10.1074/jbc.271.24.14067. [DOI] [PubMed] [Google Scholar]
- 47.Jewell-Motz EA, Lingrel JB. 1993. Site-directed mutagenesis of the Na,K-ATPase: consequences of substitutions of negatively charged amino acids localized in the transmembrane domains. Biochemistry 32:13523–13530. doi: 10.1021/bi00212a018. [DOI] [PubMed] [Google Scholar]
- 48.Huynh KK, Gershenzon E, Grinstein S. 2008. Cholesterol accumulation by macrophages impairs phagosome maturation. J Biol Chem 283:35745–35755. doi: 10.1074/jbc.M806232200. [DOI] [PubMed] [Google Scholar]
- 49.Johannsdottir HK, Mancini R, Kartenbeck J, Amato L, Helenius A. 2009. Host cell factors and functions involved in vesicular stomatitis virus entry. J Virol 83:440–453. doi: 10.1128/JVI.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J, Xie ZJ. 2010. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim Biophys Acta 1802:1237–1245. doi: 10.1016/j.bbadis.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yano H, Nakanishi S, Kimura K, Hanai N, Saitoh Y, Fukui Y, Nonomura Y, Matsuda Y. 1993. Inhibition of histamine secretion by wortmannin through the blockade of phosphatidylinositol 3-kinase in RBL-2H3 cells. J Biol Chem 268:25846–25856. [PubMed] [Google Scholar]
- 52.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem 271:695–701. [DOI] [PubMed] [Google Scholar]
- 53.Brandvold KR, Steffey ME, Fox CC, Soellner MB. 2012. Development of a highly selective c-Src kinase inhibitor. ACS Chem Biol 7:1393–1398. doi: 10.1021/cb300172e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, Thiel V, Drosten C, Rottier PJ, Osterhaus AD, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tresnan DB, Levis R, Holmes KV. 1996. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. J Virol 70:8669–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eifart P, Ludwig K, Bottcher C, de Haan CA, Rottier PJ, Korte T, Herrmann A. 2007. Role of endocytosis and low pH in murine hepatitis virus strain A59 cell entry. J Virol 81:10758–10768. doi: 10.1128/JVI.00725-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qiu Z, Hingley ST, Simmons G, Yu C, Das Sarma J, Bates P, Weiss SR. 2006. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J Virol 80:5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stauber R, Pfleiderera M, Siddell S. 1993. Proteolytic cleavage of the murine coronavirus surface glycoprotein is not required for fusion activity. J Gen Virol 74:183–191. doi: 10.1099/0022-1317-74-2-183. [DOI] [PubMed] [Google Scholar]
- 59.Sturman LS, Ricard CS, Holmes KV. 1985. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: activation of cell-fusing activity of virions by trypsin and separation of two different 90K cleavage fragments. J Virol 56:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White J, Matlin K, Helenius A. 1981. Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J Cell Biol 89:674–679. doi: 10.1083/jcb.89.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blumenthal R, Bali-Puri A, Walter A, Covell D, Eidelman O. 1987. pH-dependent fusion of vesicular stomatitis virus with Vero cells. Measurement by dequenching of octadecyl rhodamine fluorescence. J Biol Chem 262:13614–13619. [PubMed] [Google Scholar]
- 62.Lakadamyali M, Rust MJ, Zhuang X. 2004. Endocytosis of influenza viruses. Microbes Infect 6:929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cai T, Wang H, Chen Y, Liu L, Gunning WT, Quintas LE, Xie ZJ. 2008. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol 182:1153–1169. doi: 10.1083/jcb.200712022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Wilde AH, Jochmans D, Posthuma CC, Zevenhoven-Dobbe JC, van Nieuwkoop S, Bestebroer TM, van den Hoogen BG, Neyts J, Snijder EJ. 2014. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother 58:4875–4884. doi: 10.1128/AAC.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Hamme E, Dewerchin HL, Cornelissen E, Verhasselt B, Nauwynck HJ. 2008. Clathrin- and caveolae-independent entry of feline infectious peritonitis virus in monocytes depends on dynamin. J Gen Virol 89:2147–2156. doi: 10.1099/vir.0.2008/001602-0. [DOI] [PubMed] [Google Scholar]
- 66.de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matlin KS, Reggio H, Helenius A, Simons K. 1981. Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 91:601–613. doi: 10.1083/jcb.91.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patterson S, Oxford JS, Dourmashkin RR. 1979. Studies on the mechanism of influenza virus entry into cells. J Gen Virol 43:223–229. doi: 10.1099/0022-1317-43-1-223. [DOI] [PubMed] [Google Scholar]
- 69.Sieczkarski SB, Whittaker GR. 2002. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 76:10455–10464. doi: 10.1128/JVI.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harris TL. 2012. Ouabain regulates caveolin-1 vesicle trafficking by a Src-dependent mechanism. Ph.D. thesis. The University of Toledo, Ann Arbor, MI. [Google Scholar]
- 71.Cao H, Chen J, Krueger EW, McNiven MA. 2010. SRC-mediated phosphorylation of dynamin and cortactin regulates the “constitutive” endocytosis of transferrin. Mol Cell Biol 30:781–792. doi: 10.1128/MCB.00330-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD. 2004. Role of Src-induced dynamin-2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. J Biol Chem 279:20392–20400. doi: 10.1074/jbc.M308710200. [DOI] [PubMed] [Google Scholar]
- 73.Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. 1999. Src-mediated tyrosine phosphorylation of dynamin is required for β2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J Biol Chem 274:1185–1188. doi: 10.1074/jbc.274.3.1185. [DOI] [PubMed] [Google Scholar]
- 74.Mainou BA, Dermody TS. 2011. Src kinase mediates productive endocytic sorting of reovirus during cell entry. J Virol 85:3203–3213. doi: 10.1128/JVI.02056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Veracini L, Franco M, Boureux A, Simon V, Roche S, Benistant C. 2005. Two functionally distinct pools of Src kinases for PDGF receptor signalling. Biochem Soc Trans 33:1313–1315. doi: 10.1042/BST20051313. [DOI] [PubMed] [Google Scholar]
- 76.Veracini L, Simon V, Richard V, Schraven B, Horejsi V, Roche S, Benistant C. 2008. The Csk-binding protein PAG regulates PDGF-induced Src mitogenic signaling via GM1. J Cell Biol 182:603–614. doi: 10.1083/jcb.200705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mento SJ, Stollar V. 1978. Effect of ouabain on Sindbis virus replication in ouabain-sensitive and ouabain-resistant Aedes albopictus cells (Singh). Virology 87:58–65. doi: 10.1016/0042-6822(78)90157-5. [DOI] [PubMed] [Google Scholar]
- 78.Nagai Y, Maeno K, Iinuma M, Yoshida T, Matsumoto T. 1972. Inhibition of virus growth by ouabain: effect of ouabain on the growth of HVJ in chick embryo cells. J Virol 9:234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Helenius A, Kielian M, Wellsteed J, Mellman I, Rudnick G. 1985. Effects of monovalent cations on Semliki Forest virus entry into BHK-21 cells. J Biol Chem 260:5691–5697. [PubMed] [Google Scholar]
- 80.Kapoor A, Cai H, Forman M, He R, Shamay M, Arav-Boger R. 2012. Human cytomegalovirus inhibition by cardiac glycosides: evidence for involvement of the HERG gene. Antimicrob Agents Chemother 56:4891–4899. doi: 10.1128/AAC.00898-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karuppannan AK, Wu KX, Qiang J, Chu JJ, Kwang J. 2012. Natural compounds inhibiting the replication of porcine reproductive and respiratory syndrome virus. Antiviral Res 94:188–194. doi: 10.1016/j.antiviral.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson-Couterie B, Benecke A, Quinkler M, Fallo F, Plouin PF, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro MC, Strom TM, Reincke M. 2013. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 45:440–444. doi: 10.1038/ng.2550. [DOI] [PubMed] [Google Scholar]
- 83.Chang JT, Lowery LA, Sive H. 2012. Multiple roles for the Na,K-ATPase subunits, Atp1a1 and Fxyd1, during brain ventricle development. Dev Biol 368:312–322. doi: 10.1016/j.ydbio.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frugulhetti IC, Rebello MA. 1989. Na+ and K+ concentration and regulation of protein synthesis in L-A9 and Aedes albopictus cells infected with Marituba virus (Bunyaviridae). J Gen Virol 70:3493–3499. doi: 10.1099/0022-1317-70-12-3493. [DOI] [PubMed] [Google Scholar]
- 85.Pauw PG, Kaffer CR, Petersen RJ, Semerad SA, Williams DC. 2000. Inhibition of myogenesis by ouabain: effect on protein synthesis. In vitro Cell Dev Biol Anim 36:133–138. doi:. [DOI] [PubMed] [Google Scholar]
- 86.Ye J, Chen S, Maniatis T. 2011. Cardiac glycosides are potent inhibitors of interferon-beta gene expression. Nat Chem Biol 7:25–33. doi: 10.1038/nchembio.476. [DOI] [PMC free article] [PubMed] [Google Scholar]