

ABSTRACT
It is hypothesized that targeting stable cellular factors involved in viral replication instead of virus-specific proteins may raise the barrier for development of resistant mutants, which is especially important for highly adaptable small (+)RNA viruses. However, contrary to this assumption, the accumulated evidence shows that these viruses easily generate mutants resistant to the inhibitors of cellular proteins at least in some systems. We investigated here the development of poliovirus resistance to brefeldin A (BFA), an inhibitor of the cellular protein GBF1, a guanine nucleotide exchange factor for the small cellular GTPase Arf1. We found that while resistant viruses can be easily selected in HeLa cells, they do not emerge in Vero cells, in spite that in the absence of the drug both cultures support robust virus replication. Our data show that the viral replication is much more resilient to BFA than functioning of the cellular secretory pathway, suggesting that the role of GBF1 in the viral replication is independent of its Arf activating function. We demonstrate that the level of recruitment of GBF1 to the replication complexes limits the establishment and expression of a BFA resistance phenotype in both HeLa and Vero cells. Moreover, the BFA resistance phenotype of poliovirus mutants is also cell type dependent in different cells of human origin and results in a fitness loss in the form of reduced efficiency of RNA replication in the absence of the drug. Thus, a rational approach to the development of host-targeting antivirals may overcome the superior adaptability of (+)RNA viruses.
IMPORTANCE Compared to the number of viral diseases, the number of available vaccines is miniscule. For some viruses vaccine development has not been successful after multiple attempts, and for many others vaccination is not a viable option. Antiviral drugs are needed for clinical practice and public health emergencies. However, viruses are highly adaptable and can easily generate mutants resistant to practically any compounds targeting viral proteins. An alternative approach is to target stable cellular factors recruited for the virus-specific functions. In the present study, we analyzed the factors permitting and restricting the establishment of the resistance of poliovirus, a small (+)RNA virus, to brefeldin A (BFA), a drug targeting a cellular component of the viral replication complex. We found that the emergence and replication potential of resistant mutants is cell type dependent and that BFA resistance reduces virus fitness. Our data provide a rational approach to the development of antiviral therapeutics targeting host factors.
INTRODUCTION
Morbidity and mortality associated with positive-strand RNA [(+)RNA] viruses represent a significant public health burden worldwide. Vaccines are available for some of these viruses, such as poliovirus, hepatitis A virus, yellow fever virus, and a few others, and yet in most cases for the diseases induced by (+)RNA viruses modern medicine can offer nothing more than supportive therapies. For many viruses with high antigenic diversity, such as for rhinoviruses with more than a hundred known serotypes, the vaccination approach is not a viable option (1, 2). Moreover, vaccination always requires a lengthy period before the protective response is mounted, and it is practically inapplicable to immunocompromised patients.
Antiviral drugs may provide a much needed alternative to vaccination. They are the only option for the diseases associated with viruses that cannot be currently controlled with vaccines, such as hepatitis C virus. Even in the case of poliovirus, for which arguably the best known vaccines are available, the development of antipoliovirus drugs is considered an important component of the end-game strategy of the Global Polio Eradication initiative (3).
Traditionally, the development of antiviral therapeutics is focused on virus-specific targets (direct-acting antivirals) such as capsid proteins, polymerases, and proteases. However, one of the main obstacles for the development of clinically effective drugs is the genetic plasticity of (+)RNA viruses and thus their ability to readily generate resistant mutants and escape therapeutic pressure. Due to the low fidelity of the viral RNA-dependent RNA polymerase, every cycle of replication generates a multitude of similar but not identical genomes (quasispecies), providing a substrate for the rapid selection of resistant variants (4, 5). Indeed, the selection of polioviruses resistant to virtually any compound targeting viral proteins has been reported (6, 7). An alternative approach is to target host proteins hijacked for the viral replication. Although drugs interacting with the host proteins are much more likely to exert adverse effects on host metabolism, they potentially offer a significant advantage over the direct-acting antivirals. Related viruses are likely to rely on the same host machinery, thus providing an opportunity for the development of broad-spectrum antiviral therapeutics. Moreover, targeting the genetically stable host factors instead of highly adaptable viral proteins is believed to raise the resistance barrier. However, the accumulated data show that the establishment of resistance to compounds targeting cellular proteins involved in the viral replication cycle varies greatly. Inhibition of a cellular chaperone Hsp90 required for proper assembly of the viral capsid could not be overcome by poliovirus (8). At the same time, mutants of poliovirus and of another related enterovirus, coxsackievirus B3 (CVB3), resistant to the inhibition of another cellular factor involved in the viral replication, PI4KIIIβ, can be selected relatively easily (9, 10).
To understand the limiting factors allowing or restricting the establishment of viral resistance to an inhibitor of a cellular protein, we investigated in detail the establishment of poliovirus resistance to brefeldin A (BFA), a fungal metabolite that strongly inhibits replication of poliovirus and other related picornaviruses by targeting cellular protein GBF1 (11–17). Poliovirus belongs to the Enterovirus genus and is a prototype member of the Picornaviridae family of small (+)RNA viruses, which are broadly distributed among animal hosts, including humans (18). A single genome RNA of poliovirus is directly translated into one polyprotein, which undergoes a multistep maturation cascade driven in cis and in trans by three virus-encoded proteases. Both viral and cellular proteins are necessary for effective viral RNA replication. One of such cellular proteins is GBF1, a guanine nucleotide exchange factor for a subset of small cellular GTPases of the Arf family. GBF1 orchestrates COPI trafficking between the Golgi and the endoplasmic reticulum, as well as participates in the Golgi morphogenesis and lipid droplet metabolism (3, 19–21). BFA inhibits regeneration of Arf-GTP from Arf-GDP by GBF1 by stabilizing the transient complex formed by the Sec7 domain of GBF1 and Arf-GDP (22–24). GBF1 was found to interact with poliovirus and CVB3 nonstructural protein 3A, and its overexpression relieved the BFA block of enterovirus replication, while knockdown of GBF1 expression severely inhibited viral propagation (11, 12, 25).
We found that in HeLa cells, poliovirus easily generates BFA-resistant mutants in accordance with previously reported data (26); however, it was impossible to select a population of resistant viruses in Vero cells, in spite of the fact that both cell types support robust poliovirus replication without the drug. Moreover, the mutants that were BFA resistant in HeLa cells were BFA sensitive in Vero cells. The limited availability of GBF1 in Vero cells for the viral replication complexes was the limiting factor preventing the replication of BFA-resistant mutants in this cell culture. We also identified the level of 3A-mediated GBF1 recruitment as the defining parameter permitting or restricting BFA resistance in HeLa cells. The selection and growth of resistant mutants was precluded by reduced interaction between 3A and GBF1 which minimally affected poliovirus replication in the absence of the drug. Thus, BFA-resistant mutants still require GBF1 protein for replication rather than engage a completely different GBF1-independent mechanism of functioning of the replication complexes. Interestingly, the viral replication was much more resilient to BFA inhibition than was operation of the cellular secretory pathway. These data, in combination with previous findings (27), strongly suggest that the GBF1 protein is an essential component of the replication complexes but question the importance of its Arf activating property for the virus. BFA-resistant poliovirus demonstrated different levels of resistance to the inhibitor in human cell lines of different origin, emphasizing the importance of assessment of the cell-type-specific efficiency of antiviral drugs. We also show that BFA-resistant mutations reduce the efficiency of RNA replication, slowing the development of infection and making it more sensitive to antiviral pressure. Our findings provide a rationale for a drug development strategy targeting host factors involved in viral replication.
MATERIALS AND METHODS
Cells.
HeLa and SH-S5Y5 cells were grown in high-glucose Dulbecco minimum essential medium supplemented with 1 mM sodium pyruvate and 10% heat-inactivated fetal bovine serum. Vero cells were maintained in Eagle minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum.
Plasmids.
Plasmid pVenus-GBF1 was kindly provided by Catherine L. Jackson from Université Paris, Diderot, France. pCI-GBF1A795E expressing the full-length GBF1 with a BFA-resistant mutation in the Sec7 domain has been previously described (27). Vero cell-specific mutations were introduced into this plasmid by replacing XhoI/EcoRI and KasI/KpnI fragments with the corresponding fragments from GBF1 PCR fragments obtained from mRNA isolated from Vero cells. Plasmid pCMV-GLUC coding for naturally secreted Gaussia luciferase (GLUC) was from New England BioLabs. The pXpA-RenR plasmid, encoding a poliovirus replicon with the Renilla luciferase gene substituting the capsid coding sequence was previously described (11). Point mutations were first introduced into the plasmid pXpAP2P3 coding for the P2P3 fragment of poliovirus using QuikChange II XL mutagenesis kit (Stratagene), and then the SpeI/BglII fragment was recloned into pXpA-RenR. Constructs pT7pPV1 3A-HA and pT7PV1 3A-FLAG-Y coding for poliovirus cDNA with hemagglutinin (HA) and FLAG-Y inserts in the 3A sequence were kindly provided by Natalya Teterina, National Institutes of Health (NIH). These inserts were introduced into pXpA-RenR by recloning the SpeI/BglII fragment. All constructs were verified by sequencing.
Antibodies.
Rabbit polyclonal anti-GBF1 antibodies were a gift from Nihal Altan-Bonnet, NIH. Mouse monoclonal anti-GBF1 antibodies were obtained from Becton Dickinson. Anti-polio 3A mouse monoclonal antibody was a gift from K. Bienz, University of Basel, Basel, Switzerland. Rabbit polyclonal antibodies against polio 3D protein were described previously (27). Anti-FLAG affinity gel used for immunoprecipitation was obtained from Sigma-Aldrich. The anti-FLAG rabbit monoclonal antibody used in immunofluorescence studies was obtained from Cell Signaling. Alexa Fluoro-conjugated secondary antibodies were obtained from Molecular Probes.
Sequencing of GBF1 from Vero cells.
Poly(A)-containing RNA from Vero was isolated with Oligotex mRNA minikit (Qiagen) according to the manual. The reverse transcriptase reaction with oligo(dT) (to amplify the C-end coding region) or random primers (to amplify all other parts of GBF1 coding region) was performed with a MonsterScript First-Strand cDNA synthesis kit (Epicentre Biotechnologies). A full list of GBF1-specific primers used for generation of overlapping PCR fragments and sequencing is available upon request.
Transfection of Vero cells by electroporation.
For the expression of exogenous GBF1, Vero cells were transfected with pVenos-GBF1 plasmid using Amaxa Nucleofector according to the manufacturer's protocol. Briefly, 106 cells per sample were resuspended in 100 μl of Ingenio electroporation solution (Mirus) at room temperature and mixed with 2 μg of the plasmid. Immediately after electroporation using the device program I-013, 500 μl of culture medium was added to the cuvette, and the cells were gently transferred into cell culture plates prefilled with complete cell culture medium.
Selection of poliovirus mutants resistant to BFA.
Selection was performed by serial passages of the virus in the presence of BFA. For the first passage, a cell monolayer grown on a 6-cm plate was infected with poliovirus in a cell culture adjusted to 50 PFU/cell, and growth medium containing the indicated amount of BFA was added after virus attachment. The total material was harvested at 6 h postinfection. After three cycles of freeze-thawing, half of the total virus-containing material was used for the next passage. At the indicated passages, the virus yield was determined by a standard plaque assay on a HeLa cell monolayer. Statistical analysis was performed using a GraphPad Prism package unpaired t test module.
Identification of resistant mutations.
Individual plaques from titrations of resistant virus populations were collected, and viral RNA was isolated using a QIAamp viral RNA minikit (Qiagen) directly from the plaque material without additional viral propagation. RNA was reverse transcribed using a MonsterScript First-Strand cDNA synthesis kit (Epicentre Biotechnologies) with random primers. A PCR fragment covering 2C-3A genomic region was amplified using a Phusion PCR kit (New England BioLabs) and sequenced using a commercial sequencing service (Genewiz, Inc.).
Polio replicon assays.
Polio replicon assays were performed essentially as described previously (27), with minor modifications depending on the cell culture used. Briefly, cells grown on 96-well plates were transfected with purified replicon RNA (10 ng/well for HeLa, 293 and SH-S5Y5 cells, and 30 ng/well for Vero cells) with Trans-it mRNA transfection reagent (Mirus) according to the manufacturer's recommendations. A transfection mix was added to the incubation medium containing 60 μM live-cell Renilla Endu-Ren (Promega) substrate and the indicated amount of BFA (or the corresponding amount of dimethyl sulfoxide [DMSO] solvent in control samples). The cells were incubated at 37°C in an Infinite M1000 (Tecan) plate reader, and the luciferase signal was measured every hour for 16 h. The signal from at least eight wells was averaged for each sample. All data were processed using GraphPad Prism statistical software. Error bars on graphs show the standard deviations, regression curves, and cell culture-specific 50% inhibitory concentrations (IC50) of BFA were calculated using the exponential decay analysis module provided in the GraphPad Prism package. Total replication was calculated as an integrated luciferase signal (area under the curve). An unpaired t test was performed to generate P values where indicated.
In vitro translation and replication.
HeLa S10 extracts for translation reactions were prepared as described previously (28) with minor modifications. Translation reaction mixtures of 50 μl included 2.5 μg of mRNA transcripts coding for polio proteins. A 9-μl aliquot from each reaction mixture was mixed with 1 μl of 35S-labeled methionine-cysteine mix (Perkin-Elmer), followed by incubation for 3.5 h at 34°C, and an aliquot of the labeled material was resolved on a 12% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel to visualize the translation products. The remaining 40 μl was also incubated for 3.5 h at 34°C and then centrifuged for 20 min at 16,000 × g at 4°C to collect the membranous pellets with associated viral and cellular proteins. These pellets were either resuspended in 50 μl of guanidine-HCl-free replication buffer containing [32P]αCTP (Perkin-Elmer), or the total pellet material was resolved on SDS-PAGE gel and subjected to Western blot analysis. After the replication reaction, the RNA was extracted with an RNeasy kit (Invitrogen) and resolved on a denaturing agarose gel. Staining of the gel with ethidium bromide was used to visualize rRNA to ensure the equal recovery of RNA from the samples. The agarose gel was dried and exposed for autoradiography. The intensity of the replication signal was analyzed using ImageJ software (NIH).
Cell lysates.
All lysis buffers were supplemented with protease inhibitors cocktail (Sigma-Aldrich). For preparation of the cytoplasmic lysates, the cell monolayer was treated with mild lysis buffer (0.1 M Tris-HCl [pH 7.8], 0.5% Triton X-100) for 10 min on a shaker platform; the lysate was clarified by centrifugation and used for Western blot analysis. For preparation of total cell lysates, the cells were collected in an Eppendorf tube and lysed directly with 1× Laemmli sample buffer (2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.002% bromphenol blue, 0.625 M Tris-HCl [pH ∼7.0]), followed by sonication to fragment the nuclear DNA. The lysate was used for Western blotting without further purifications. A total of 60 μg of total protein per lane was loaded for the Western blot analyses, a portion which lies within the linear range of signal as determined by serial dilutions. For coimmunoprecipitation (co-IP) experiments, the cells were lysed with a buffer recommended for use with anti-FLAG affinity gel (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) and processed according to the protocol provided by the manufacturer of the affinity resin.
Poly(I·C) treatment.
HeLa cells grown on 96-well plates were treated with 100 μg of poly(I·C) (Sigma-Aldrich)/ml in serum-free Dulbecco modified Eagle medium. After 3 h of treatment, the cells were used for a poliovirus replicon assay.
Immunofluorescence microscopy.
Cells grown on glass coverslips were fixed with 4% formaldehyde in Dulbecco phosphate-buffered saline (PBS) for 20 min, washed three times with PBS, and then permeabilized with 0.2% Triton X-100 in PBS for 5 min. After three washes with PBS, the cells were incubated in 3% blocking reagent (GE Healthcare) containing 10 ng of Hoechst 33342 (DNA stain)/ml for 1 h, followed by 1-h incubations with primary and secondary antibodies diluted in the same blocking buffer, with 3× PBS washes between incubations. The processed slides were mounted with Fluoromount-G anti-fade medium (Electron Microscopy Sciences). Digital images were taken with Zeiss Axiovert 200M fluorescence microscope equipped with AxioCam MRm monochrome digital camera; confocal images were obtained with a Zeiss ApoTome AxioImager M2 microscope. Images were processed with Adobe Photoshop or ImageJ (NIH) software. Colors were artificially assigned to monochrome images according to the channel where they were taken. Software processing was applied universally to the whole image, and images from different samples were taken and processed under identical conditions.
Secretion assay.
HeLa cells were transfected with pCMV-GLUC plasmid using Mirus 2020 transfection reagent (Mirus Bio) and grown overnight on a 96-well plate. The next day, the cells were thoroughly washed with serum-free medium and incubated with BFA (or an equivalent amount of DMSO in the control) for 4 h in normal growth medium (75 μl/well). A 20-μl aliquot of the medium was collected, and the secreted luciferase signal was assayed using Gaussia luciferase assay solution (New England BioLabs). The cells were then washed with serum-free medium, lysed, and assessed for the intracellular Gaussia luciferase signal.
RESULTS
Cell-specific establishment of drug resistance by poliovirus.
To investigate the development of poliovirus resistance to BFA inhibition in cells of different origin, we chose cell lines that robustly support poliovirus infection. HeLa (human cervical carcinoma) cell line is a standard laboratory model for growing poliovirus. Vero (African green monkey kidney) cell line is also widely used in poliovirus research and is an approved cell line for high-yield commercial poliovirus propagation for vaccine production (29). First, we characterized poliovirus growth in both cell lines. We observed that the standard plaque assay performed with the same preparation of poliovirus shows significantly different titers on HeLa and Vero monolayers (Table 1); on average, the titers obtained on Vero were ∼1.5 to 2 logs lower than the same viral stock would produce on HeLa cells. This phenomenon was observed previously by other researchers and is believed to be associated with lower penetration/uncoating efficiency of poliovirus in Vero cells (E. Ehrenfeld, unpublished data). For further experiments, we infected HeLa and Vero cells with the same multiplicity of infection (MOI) calculated according to the virus titer for each culture, i.e., the amount of the viral suspension used per cell was higher for Vero than for HeLa because the titer of the viral preparation on Vero was lower. To assess the kinetics of poliovirus propagation on HeLa and Vero, the cells were infected with a cell type-adjusted MOI of 10 PFU/cell, the samples were collected at different times postinfection, and the virus yield from both cultures was assessed on HeLa monolayers. Both cultures demonstrated similar kinetics of poliovirus propagation and virus yield in the time course experiment (Fig. 1A). The higher value for the 2-h time point for the Vero sample reflects the excess of the original infectious material added to Vero cells compared to that added to HeLa. To determine whether the adjustment of the poliovirus infectious dose according to the cell-specific infectivity really compensated for lower infectivity of poliovirus in Vero cells, we incubated the infected cells in the presence of 2 mM guanidine HCl, a strong specific inhibitor of poliovirus RNA replication (30). Accumulation of a viral antigen over the first 3 h of infection in the context of inhibited replication (Fig. 1B, Gua +) showed that the penetration/uncoating and translation steps of the poliovirus infection were similar in both cultures under these conditions.
TABLE 1.
Poliovirus pools examined in this study
| Poliovirus pool | Titration (PFU/ml) |
|
|---|---|---|
| HeLa cells | Vero cells | |
| 1 | 2.00E + 09 | 6.00E + 07 |
| 2 | 4.00E + 09 | 4.00E + 07 |
| 3 | 3.00E + 09 | 1.20E + 08 |
FIG 1.
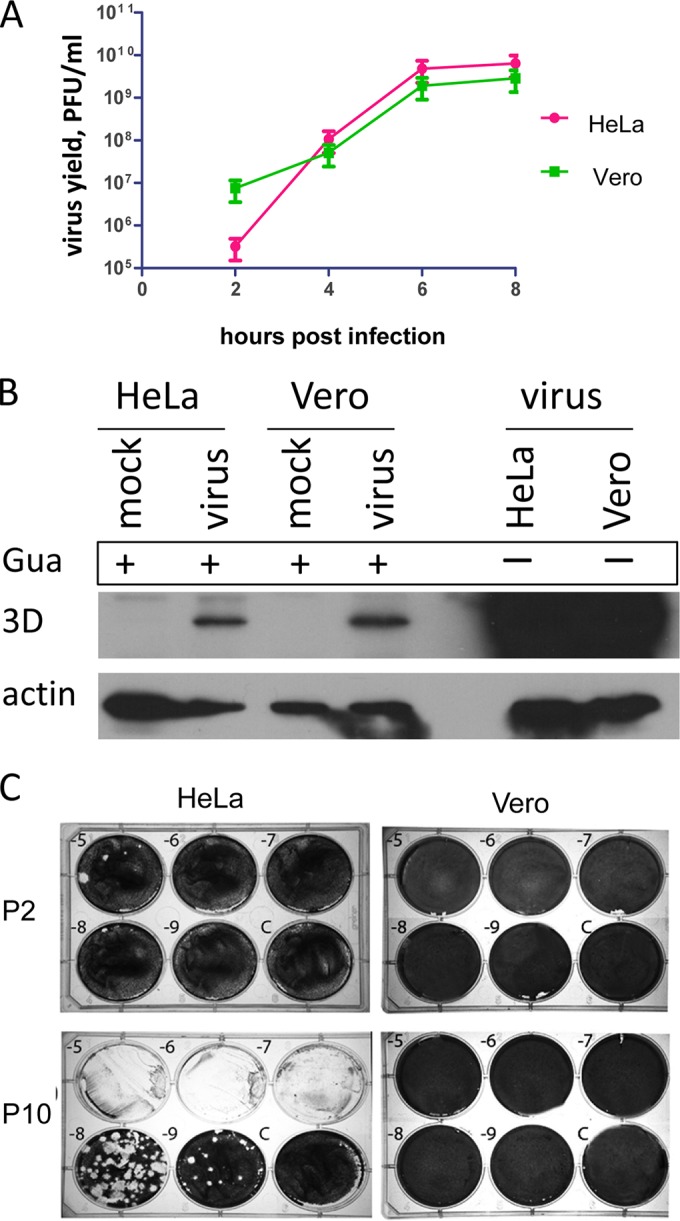
BFA-resistant polioviruses can be selected in HeLa cells but not in Vero cells. (A) Poliovirus replication in HeLa and Vero cells. The cells were infected with a cell culture adjusted to 50 PFU/cell. At the indicated time points, the samples were collected, and the total virus yield was determined by a standard plaque assay on a HeLa cell monolayer. (B) Cell culture adjustment of the infectious dose provides a similar initiation of poliovirus infection in both HeLa and Vero cells. The cells were infected with a cell culture adjusted to 50 PFU of poliovirus/ml, followed by incubation in the presence of 2 mM guanidine-HCl to inhibit replication (Gua +). Control replication was performed in the absence of the inhibitor (Gua −). The cells were collected at 3 h postinfection and analyzed in a Western blot with anti-3D antibodies. (C) Population of BFA-resistant viruses that emerged after 10 passages of poliovirus in the presence of 2 μg of BFA/ml in HeLa cells but not in Vero cells. Titrations of the viral stocks on HeLa cells monolayer is shown after passages 2 (P2) and 10 (P10) for both cultures. Dilutions are indicated; C, mock-infected control wells.
For the selection of BFA-resistant mutants, we first performed 10 serial passages in the presence of 2 μg of BFA/ml. The first infection was performed with cell culture adjusted to an MOI of 50 PFU/cell to ensure similar starting conditions in both cell cultures. In HeLa cells this scheme resulted in selection of a resistant population that grew similar to the wild-type (wt) virus propagated without the drug (Fig. 1C, compare HeLa cells, passages 2 and 10). Selection of the BFA-resistant variants by passaging the same virus on Vero cells was unsuccessful (Fig. 1C, compare Vero cells, passages 2 and 10). We repeated serial passaging on Vero cells starting with the lower concentrations of the drug (0.5 and 0.1 μg/ml). However, even under these conditions we could not select BFA-resistant poliovirus mutants in Vero cells (not shown). Thus, while both HeLa and Vero cells support robust replication of poliovirus, mutants resistant to BFA can be readily selected on HeLa but not on Vero cells.
BFA-resistant mutants selected in HeLa cells are BFA sensitive in Vero cells.
We sequenced several plaque-purified BFA-resistant viruses from the passage 10 population selected on HeLa cells and identified two amino acid substitutions, one in 2C (C4200T), resulting in substitution of S26 to L and another in 3A (G5211A) resulting in substitution of R34 to K (nucleotide positions correspond to the full-length poliovirus RNA, and amino acids are numbered according to their positions in individual proteins) (Fig. 2A). The majority of the sequenced plaque-purified viruses contained both mutations together; this genotype will be referred to as DM1 (double mutant 1), and one contained only the mutation in 2C. Among the isolated plaques there were no viruses with only a single mutation in 3A. Interestingly, these mutations are different from the ones previously identified by Crotty et al. (26). To confirm that these mutations were responsible for the BFA-resistant phenotype, we introduced them into the poliovirus replicon encoding Renilla luciferase (Fig. 2A). In HeLa cells in the absence of the drug, all constructs replicated similarly to the wt control, and all displayed a BFA-resistant phenotype. The G5211A mutant with the substitution in 3A protein replicated better in the presence of the drug than did the 2C mutant (C4200T), and the mutations provided synergistic effect when combined together in one genome (Fig. 2B), similarly to the previously described DM mutant with mutations G4361A and C5190T (26). Both double mutants (DM and DM1) showed very similar levels of BFA resistance and were used interchangeably in further experiments.
FIG 2.

Poliovirus mutants resistant to BFA in HeLa cells are sensitive to the drug in Vero cells. (A) Scheme of poliovirus genome and poliovirus replicon. The BFA-resistant mutations in 2C and 3A are shown. The ones reported previously by Crotty et al. (26) are indicated in black; the ones identified in the present study are indicated in red. (B) Replication of the replicons bearing BFA-resistant mutations in HeLa cells in the presence of 1 μg of BFA/ml (a corresponding amount of DMSO solvent was added to the control samples). (C) Replication of the replicons bearing BFA-resistant mutations in Vero cells in the presence of 1 μg of BFA/ml (a corresponding amount of DMSO solvent was added to the control samples). (D) Replication of the DM BFA-resistant mutant replicon (G4361A + C5190T) in HeLa and Vero cells at different concentrations of BFA. The total replication is calculated as an integrated luciferase signal (area under the curve). Statistically significant differences are indicated by asterisks (****, P < 0.0001).
When we investigated the BFA-resistant phenotype of the replicons bearing single C4200T and G5211A BFA-resistant mutations, as well as the DM mutant (G4361A and C5190T) in Vero cells, none of the mutants could replicate in Vero cells at 1 μg of BFA/ml, in contrast to HeLa cells (compare Fig. 2B and C). We compared the BFA sensitivity range of the DM mutant replicon in HeLa and Vero cells. The replication of this mutant showed much stronger resistance to BFA in HeLa cells than in Vero cells (Fig. 2D). At lower concentrations of the drug, its replication in HeLa cells was even stimulated (up to ∼1.5 times at 125 ng of BFA/ml) over replication under control conditions (Fig. 2D, total replication). In Vero cells the replication of the same DM mutant was somewhat more resistant to the inhibitor than the replication of the wt (not shown), but it was strongly suppressed compared to the replication in HeLa. At 0.5 and 1 μg of BFA/ml, the replication of DM in Vero cells was down to ca. 20 and 5% of control, respectively, while in HeLa cells at the same drug concentrations its replication was very robust, i.e., ∼115 and 85% of the control, respectively. (Fig. 2D). These data show that the mutations sufficient to bypass the inhibition in HeLa cells are not effective in Vero cells.
Poliovirus replication is less sensitive to inhibition by BFA than the cellular secretory pathway.
BFA inhibits the cellular secretory pathway by blocking the recycling of small cellular GTPases Arf by guanine nucleotide exchange factor GBF1 (21). The targeting of GBF1 also explains the inhibitory effect of the drug on poliovirus and CVB3 replication, although the mechanistic role of GBF1 in the viral life cycle is not well understood (11, 12). We looked at whether the functioning of the cellular secretory pathway or the viral replication is more sensitive to BFA. We assessed the response of the cellular secretory pathway to BFA by measuring the secretion of Gaussia luciferase (GLUC), which has a natural secretion signal. HeLa and Vero cells transfected with a plasmid expressing GLUC were incubated with different concentrations of BFA for 4 h, and the activity of the luciferase secreted in the extracellular medium was measured. The inhibition of the secretory pathway by BFA was practically identical in both cultures, although the signal in HeLa in the absence of the drug was much stronger, likely reflecting higher transfection efficiency of these cells (Fig. 3A). The inhibition of secretion was accompanied by the corresponding increase of the intracellular GLUC activity, showing that the decrease of the signal from the media was indeed from the inhibition of secretion and not due to the cell toxicity or inhibition of translation (not shown). At 125 ng of BFA/ml in both HeLa and Vero cells, the inhibition of secretion reached the plateau level at ∼17 and 34% of control, respectively (Fig. 3A, normalized graph). Inhibition of poliovirus replication, on the other hand, required significantly higher concentration of BFA than inhibition of secretion. In HeLa cells IC50 of BFA for replication was ∼80 ng/ml, while that for secretion was ∼15 ng/ml (Fig. 3B). In Vero cells poliovirus replication was much more sensitive to the drug than in HeLa cells. Concentrations of BFA from 50 ng/ml and up almost completely blocked replication of the wt replicon, whereas the effect of BFA at 50 ng/ml was barely noticeable in HeLa cells (compare Fig. 3B and C). The replication in Vero cells started to recover, especially in samples with lower concentrations of BFA, after ca. 6 to 8 h posttransfection, likely reflecting the degradation of BFA due to the cellular metabolism (Fig. 3C, replication kinetics). At the lower range of drug concentrations in Vero cells, the poliovirus replicon replication was much more resistant to BFA than the secretory pathway, an observation similar to the phenomenon noted in HeLa cells. The IC50 of BFA for secretion in Vero cells was ∼2 ng/ml, while that for poliovirus replication was at least 10 times higher at ∼27 ng/ml (Fig. 3D). It should be noted that even at the lowest concentrations of BFA tested (5 ng/ml for Vero cells and 50 ng/ml for HeLa cells), the inhibition of secretion was almost complete, and thus the IC50s for secretion derived from regression analysis represent the upper limit and will likely be even lower. Thus, poliovirus replication is much more sensitive to BFA inhibition in Vero cells than in HeLa cells, and the viral replication can withstand significantly higher concentrations of the drug than those required for virtually complete shutdown of the cellular secretory pathway.
FIG 3.

Poliovirus replication is much more resilient to BFA inhibition than the cellular secretory pathway. (A) HeLa and Vero cells were transfected with a plasmid expressing secreted Gaussia luciferase. Before the experiment, the cells were washed and incubated in the presence of indicated amount of BFA (DMSO solvent in control) for 4 h. The secreted luciferase was then measured in the cell culture medium. The data on the normalized secretion graph were calculated relative to the control secretion with no BFA in corresponding cell lines. (B and C) Replication of the wt poliovirus replicon in HeLa (B) and Vero (C) cells at different concentrations of BFA. The total replication was calculated as an integrated luciferase signal (area under the curve) and normalized to the replication of the control sample without BFA for normalized secretion and the replication graph. Normalized secretion curves are the same as in panel A. (D) Replication of the wt poliovirus replicon (left) and secretion of Gaussia luciferase (center) in Vero cells at a lower range of BFA concentrations. The total replication was calculated as an integrated luciferase signal (area under the curve). The total replication and secretion signals are normalized to the control samples without BFA (right).
Intracellular distribution of GBF1 and its recruitment to the viral replication complexes is different in HeLa and Vero cells.
Inhibition of poliovirus replication by BFA in HeLa cells was shown to depend on GBF1 (11); thus, the inability to select BFA-resistant mutants in Vero cells suggests that interaction of GBF1 with the viral replication complexes may be significantly different in HeLa and Vero cells. First, we compared the GBF1 content and distribution in both cell types. The GBF1 signal on Western blots prepared with cytoplasmic lysates obtained with a mild lysis buffer that does not destroy nuclei was significantly lower in Vero cells than that in HeLa cells (Fig. 4A, cytoplasmic). To compare the total content of GBF1, we prepared unfractionated lysates by treating the cells directly with SDS-containing Laemmli sample buffer, followed by ultrasound homogenization. The total GBF1 content was still lower in Vero cells than in HeLa cells, but the difference was not as striking as the one observed in cytoplasmic lysates (Fig. 4A, total). Thus, the results suggest that the amount of this protein and/or its interaction with the cellular structures differs in these cell types. The samples in this experiment were adjusted by the total amount of protein; virtually the same results were obtained when the samples were adjusted by the number of cells (data not shown). Immunostaining of GBF1 in mock-infected cells revealed that in HeLa the protein is localized in a compact perinuclear pattern, while in Vero cells the distribution of GBF1 is scattered in many isolated spots occupying much bigger area than in HeLa cells (Fig. 4B, mock). GBF1 is mostly localized to cis-Golgi membranes (31), and the observed distribution likely reflects differences in organization of this organelle in HeLa and Vero cells. Overall, the intensity of the GBF1 immunostaining in both cultures was similar, strengthening the conclusion that the significantly lower signal observed in the Western blot with cytoplasmic lysates is mostly due to the differences in intracellular GBF1 distribution. In cells of both types infected with poliovirus carrying FLAG-Y epitope insert in the 3A protein, we observed similar redistribution of the protein to the perinuclear pattern characteristic of the distribution of the viral replication complexes consistent with the previous data (11). We used this virus for compatibility of the available anti-FLAG and anti-GBF1 antibodies suitable for immunostaining. This virus replicates with wt phenotype in cell culture and recapitulates strong 3A-GBF1 interaction (32). Poliovirus infection rapidly shuts off host protein synthesis and, accordingly, the level of GBF1 did not increase in either HeLa or Vero cells during the time course of infection (not shown); however, the immunofluorescent signal of GBF1 in infected cells was always stronger than that in mock-infected controls (compare Fig. 4B mock and infected). This suggests that the epitope recognized by monoclonal antibody becomes more accessible, likely reflecting significant changes in the GBF1 interactions upon poliovirus infection. GBF1 is recruited to the poliovirus and CVB3 replication complexes through interaction with the viral protein 3A (33, 34). To assess the level of recruitment of GBF1 to poliovirus replication complexes formed in HeLa and in Vero cells, we also used the same poliovirus carrying FLAG-Y epitope insert in the 3A protein (32). The replication complexes were pulled down with the anti-FLAG antibodies conjugated to a resin and GBF1 was detected in the coimmunoprecipitated material. The lysis buffer in the co-IP experiment was similar to the mild lysis buffer used for the preparation of cytoplasmic lysates shown on the Fig. 4A and, accordingly, in the aliquot of the input lysates loaded on the gel, the signal for GBF1 was detectable only in HeLa cells (Fig. 4C, input). The co-IP signal for GBF1, on the other hand, was strong in both HeLa and Vero cells, showing that GBF1 is recruited to the replication complexes in both cultures, although the ratio of the amount of GBF1 to that of 3A and 3AB recovered from infected Vero cells was noticeably lower than that from HeLa cells (Fig. 4C, co-IP). Thus, the amount and/or intracellular distribution of GBF1, the target of BFA inhibition, is different in HeLa and Vero cells, likely resulting in the lower level of recruitment of this protein to the poliovirus replication complexes formed in Vero cells.
FIG 4.
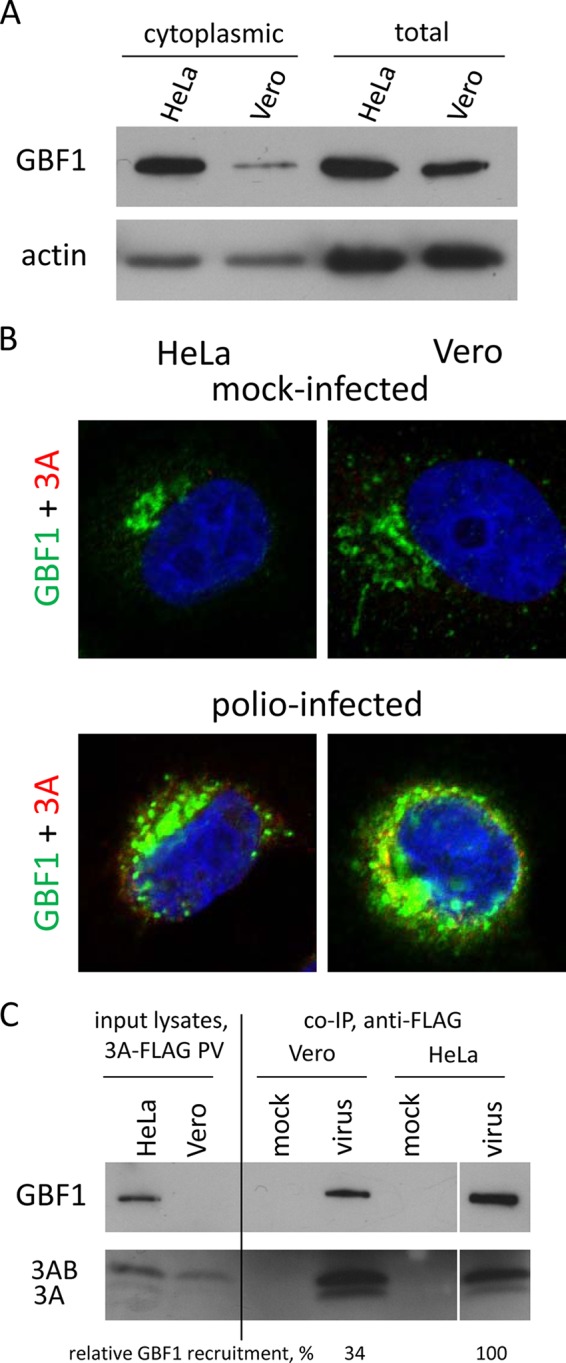
GBF1 is less accessible for poliovirus replication complexes in Vero cells than in HeLa cells. (A) Western blot showing GBF1 in cytoplasmic and total lysates from HeLa and Vero cells. (B) Confocal images showing GBF1 distribution in mock-infected (top) and poliovirus-infected (bottom) HeLa and Vero cells. The cells were infected with poliovirus with a FLAG-Y insert in the 3A sequence at a culture-adjusted dose of 50 PFU/cell and incubated for 6 h postinfection. The GBF1 signal is in the green channel, poliovirus 3A (anti-FLAG antibody) is indicated in red. (C) Co-IP of 3A and GBF1 from Vero and HeLa cells. The cells were infected (mock infected) with poliovirus with FLAG-Y insert in the 3A sequence at a culture-adjusted dose of 50 PFU/cell and incubated for 6 h postinfection. The input lysate samples represent 4% of the amount of the lysates from infected cells taken for immunoprecipitation with anti-FLAG resin. The relative GBF1 recruitment was calculated as a ratio of GBF1 signal to the combined 3A and 3AB signal. The relative GBF1 recruitment in HeLa cells is set as 100%. The results from a representative experiment are shown.
Vero cell-specific GBF1 is fully functional in supporting poliovirus replication and is the limiting factor for replication of a BFA-resistant mutant in Vero cells.
The inefficient replication of the BFA-resistant mutants in Vero cells in the presence of the drug may indicate that human and African green monkey-specific GBF1s are different in their ability to support polio replication. The full sequence of GBF1 from the African green monkey is not available in the databases; thus, we sequenced GBF1 mRNA expressed in Vero cells. We found eight single amino acid mutations in the Vero GBF1 coding sequence compared to the human GBF1 variant used in the GBF1 expression plasmid (23). None of them were localized in the Sec7 domain which is directly responsible for Arf activation function of GBF1 and which is the target of BFA inhibition (24). Three amino acid substitutions were localized at the N terminus (G44S, S52N, and E58G), one was in the HDS2 domain (A1219G), and the others were concentrated at the C-terminal part of the protein (Q1728H, Q1730H, S1770G, and R1808Q) (Fig. 4A). The sequencing was performed by generating overlapping PCR fragments from cellular mRNA; interestingly, not all of the substitutions were present together, a finding consistent with existence of multiple splice variants and/or allelic differences of GBF1 transcripts in diploid cells (35). We tested whether the Vero cell-specific GBF1 sequence may be compromised in its functioning in the viral replication complexes by assessing its ability to support poliovirus replicon replication. HeLa cells transiently expressing GBF1 with a BFA-resistant mutation in the Sec7 domain and with Vero cell-specific substitutions in the other parts of the protein were transfected with the poliovirus replicon, and the replication was assessed in the presence of BFA. In these settings the endogenous GBF1 in HeLa cells is inhibited by the drug, and the replication can be supported only by the BFA-resistant exogenously expressed GBF1 protein. As can be seen in Fig. 5B, GBF1 containing all mutations found in the Vero sequence was as efficient in supporting poliovirus replication as was the control human protein.
FIG 5.

Replication of a BFA-resistant virus can be rescued in Vero cells by expression of exogenous GBF1. (A) Scheme of GBF1 protein with Vero cell-specific amino acid substitutions indicated. (B) GBF1 with Vero cell-specific mutations is fully functional in supporting poliovirus replication. HeLa cells were transfected with plasmids expressing either human or Vero cell-specific GBF1s with BFA-resistant mutation A795E in the Sec7 domain. Replication of the wt poliovirus replicon was assessed in these cells in the presence or the absence of BFA. The expression of the exogenous GBF1 species is shown. (C) Replication of the DM BFA-resistant mutant poliovirus (G4361A + C5190T) is rescued in Vero cells expressing exogenous GBF1 (arrow). A low level of replication was observed in nontransfected cells (arrowheads). (D) Selection of BFA-resistant mutants in Vero cells starting from the DM BFA-resistant mutant poliovirus (G4361A + C5190T) was not successful. The virus yield was determined by titration on the HeLa cell monolayer of the material from passages 2 (P2) and 10 (P10). The control shows replication of the DM mutant on Vero cells in the absence of BFA. The P value is shown; the difference between titers from passages 2 and 10 was not statistically significant (NS).
To determine whether GBF1 was the limiting factor preventing replication of BFA resistant mutants in Vero cells, we transfected these cells with a plasmid expressing wt, BFA-sensitive GBF1 with N-terminally fused fluorescent protein. When the cells were infected with the DM BFA-resistant virus, the replication in the presence of the inhibitor was rescued in cells expressing exogenous GBF1 (Fig. 5C, arrow). We also noticed that in nontransfected cells there was a detectable positive signal for a viral antigen indicative of a low-level replication of this mutant (Fig. 5C, arrowheads). This prompted us to repeat the selection of Vero cell-specific BFA-resistant mutants starting with DM mutant rather than with the wt virus. However, after 10 passages the selected population demonstrated the same low level of replication in the presence of BFA in Vero cells as at the beginning of the selection process (Fig. 5D). Thus, Vero cell-specific GBF1 is fully functional in the viral replication complexes, and the inefficient replication of the BFA-resistant poliovirus mutant in Vero cells is likely related to the smaller amount and/or the accessibility of the protein to the virus.
Strong 3A-GBF1 interaction is dispensable for replication in normal conditions but is important for BFA resistance.
In Vero cells the BFA-resistant mutants selected in HeLa cells lost their ability to efficiently replicate in the presence of the drug, and this lack of BFA resistance correlated with the diminished recruitment of GBF1 to the replication complexes. The N-terminal part of GBF1 strongly interacts with the N terminus of the poliovirus protein 3A (33). To understand whether 3A-mediated recruitment of GBF1 is required for BFA-resistant phenotype in HeLa cells, we took advantage of poliovirus mutants with strongly impaired 3A-GBF1 interaction. 3A-2 mutant has an insert of one extra serine in the N-terminal part of 3A and was originally characterized as having a cold-sensitive growth phenotype (36) (Fig. 6A). It was shown that this mutation strongly interferes with 3A-GBF1 interaction in a yeast two-hybrid assay, in mammalian cells, and in an in vitro translation system based on HeLa cell extract (11, 25, 34). The other mutation affecting 3A-GBF1 interaction is an insertion of the HA epitope in the 3A sequence (Fig. 6A). In the co-IP experiments with the virus bearing this mutation the recruitment of GBF1 to the replication complexes was not detectable, while the virus with the FLAG-Y epitope inserted in the same position showed strong recruitment of this host factor (32). First, we compared the level of GBF1 recruitment by wt 3A, 3A-2, 3A-HA, and 3A-FLAG-Y in the in vitro translation system based on HeLa cell extract (Fig. 6B). Consistent with the previously reported data, expression of both wt 3A and 3A-FLAG-Y induced strong binding of GBF1 to membranes, while 3A-2 and 3A-HA showed a much lower level of GBF1 recruitment, with 3A-2 being the weakest. It is important, however, that even in the case of 3A-2 the level of GBF1 recruitment was still higher than that of the control background (Fig. 6C). We introduced mutations conferring BFA resistance in the context of 3A-2, 3A-HA, and 3A-FLAG-Y poliovirus replicons. In the absence of BFA, all constructs replicated with similar efficiency showing that replication in the normal conditions can tolerate significant variability in the level of 3A-dependent GBF1 recruitment (Fig. 6D, controls). In the presence of the inhibitor, however, 3A-2 mutation strongly compromised the BFA resistance conferred by mutations in 2C (C4200T), 3A (G5211A), and both of them together (DM1) (Fig. 6C, 3A-2). Inhibition of the BFA-resistant phenotype conferred by mutation G5211A was also observed in the context of HA insert but was usually less pronounced than that in the context of 3A-2, and no inhibition was observed when this mutation was placed in the context of the FLAG-Y insert (Fig. 6D, 3A-HA and 3A-FLAG-Y). Thus, the BFA-resistant phenotype of the same mutations strongly correlates with the efficiency of recruitment of GBF1 by the 3A protein.
FIG 6.

A BFA-resistant phenotype correlates with recruitment of GBF1. (A) Sequences of poliovirus 3As with mutations affecting recruitment of GBF1. An R34 amino acid mutated to K in BFA-resistant mutants G5211A is marked in red in the wt 3A sequence. (B) Scheme of the experiment for assessment of recruitment of GBF1 to membranes upon translation of 3A-coding RNAs. (C) Western blot of the material associated with the membranous pellets upon translation of 3A-specific RNAs in HeLa S10 extracts. 35S-labeled translation products show equal expression of 3A species. (D) Replication of replicons bearing BFA-resistant mutations in the context of different 3As in HeLa cells in the presence or in the absence of BFA. Replication of the C4200A mutant in the 2C protein was assessed at 0.5 μg of BFA/ml because this mutation has a weak BFA-resistant phenotype. All other experiments in this panel were performed with a drug concentration of 1 μg/ml.
It is possible that since the BFA-resistant mutations were selected starting with wt virus, they could not function in the context of 3A-2 or 3A-HA background, but other mutations could potentially exist that may provide BFA resistance in the context of diminished 3A-GBF1 interaction. We repeated the selection of a BFA-resistant population in HeLa cells starting with the 3A-2 mutant. We plaque purified several viruses selected after 15 passages in the presence of 2 μg of BFA/ml and sequenced the 2C-3A area of the genome where all previously identified BFA-resistant mutations were located. All of the sequenced viruses had a C4200A mutation in 2C that was identified previously. Two of them also had an additional new mutation, C5197A (D29E), in the 3A region. Importantly, none of the sequenced viruses retained the original 3A-2 serine insertion, showing a strong correlation between the restoration of 3A-GBF1 interaction and the establishment of BFA resistance. These data show that while poliovirus could achieve BFA resistance in HeLa cells through many different mutations, the acquisition and expression of BFA resistance is restricted by the interaction between 3A and GBF1, which has much less stringent constraints for replication without the drug.
BFA-resistant mutations incur a fitness cost associated with a reduced efficiency of viral RNA replication.
Closer examination of the replication of poliovirus replicons bearing BFA-resistant mutations revealed that in the absence of the drug they demonstrated significantly impaired replication kinetics. Although eventually the replication signal of the mutant replicons reached essentially the same level as the wt control construct, the replication of all BFA-resistant mutants tested was considerably delayed during the exponential phase of replication throughout the first 6 h posttransfection (Fig. 7A). To understand the step in the replication cycle affected by the mutations, we used an in vitro polio translation/replication system. In this system, poliovirus-specific RNA is first translated in the postnuclear crude HeLa cells extract in the presence of 2 mM guanidine-HCl, which does not interfere with translation of the RNA and polyprotein processing but strongly inhibits RNA replication. During the second step, membranes with replication complexes formed during the translation step are collected by centrifugation and resuspended in a guanidine-HCl-free buffer supplemented with a labeled nucleotide, thus allowing a synchronous start of the replication reaction and visualization of newly synthesized poliovirus RNA (37, 38). The translation efficiency and polyprotein processing of both single mutants, C4200T and G5211A, as well as the double mutant DM1 (C4200T + G5211A), was indistinguishable from that of the control wt construct (Fig. 7B, translation panel). RNA replication of all BFA-resistant mutants, however, was significantly compromised, with the double mutant showing the strongest defect (Fig. 7B, replication panel). In this experiment, we used the same preparations of poliovirus replicon RNAs that were used in the in vivo replication kinetics experiments; thus, the in vitro translation data also provided an additional control that the replication kinetics defects observed in vivo were not due to different quality of the replicon RNA preparations. The impaired RNA replication capacity of the BFA-resistant mutants apparently resulted in their higher sensitivity to treatment of cells with poly(I·C), a synthetic analog of double-stranded RNA and a potent inducer of innate antiviral response, and to guanidine-HCl, an inhibitor of poliovirus RNA replication (not shown). Thus, the establishment of BFA resistance reduces the efficiency of poliovirus RNA replication and results in generally increased susceptibility of the infection to at least some antiviral agents.
FIG 7.

BFA-resistant mutants demonstrate replication defect in cell culture and in an in vitro system. (A) Replication of the replicons bearing BFA-resistant mutations in HeLa cells (no BFA is added). Statistically significant differences in the luciferase signal are indicated by asterisks (**, P < 0.004; ***, P < 0.0005; ****, P < 0.0001). (B) Translation and replication of the poliovirus replicon RNAs bearing BFA-resistant mutations in a two-step in vitro system. An aliquot of translation reaction was labeled with a [35S]methionine-cysteine mix (translation panel). Polyprotein processing products are indicated. Replication was performed in the presence of [32P]αCTP to label the newly synthesized RNA (replication panel). The replication signal was quantified using ImageJ software, and the results of a representative experiment are shown.
The level of BFA resistance of the same mutant is different in human cells of different origins.
The development of paralysis associated with poliovirus infection requires replication of the virus in diverse cell types—at least in the gut epithelium and neurons and likely many others involved in the dissemination of the virus to the central neural system. Our data show that poliovirus demonstrates significantly different levels of sensitivity to BFA in HeLa and Vero cells. However, HeLa and Vero cells originate from different species of primates and thus may not be a representative model for replication of the virus in a natural host. To determine whether cells of human origin may differ in their ability to support the replication of BFA-resistant mutants, we compared the replication of DM mutant in 293 (human embryonic kidney) and SH-S5Y5 (human neuroblastoma) cell lines. In 293 cells, this mutant replicated with practically the same efficiency in the absence and in the presence of the drug, similarly to the replication in HeLa cells (Fig. 8, 293). In the neuroblastoma cells, however, its replication was much more sensitive to BFA, reaching only ∼10% of control with the same concentration of the drug (Fig. 8). Thus, the antipoliovirus efficiency of BFA and the likely antiviral activity of other drugs targeting cellular factors may be different in different cell types, affecting the possibility of the emergence and spread of resistant mutants.
FIG 8.

The BFA-resistant phenotype is cell type dependent in cells of human origin. A replicon assay with the wt and the DM BFA-resistant mutant (G4361A + C5190T) in 293 (human embryonic kidney) and SH-S5Y5 (human neuroblastoma) cell lines was performed. The total replication (area under the curve) was normalized to the replication of each replicon in the control samples containing equivalent amounts of DMSO solvent. P values are shown. ns, not significant.
DISCUSSION
Targeting the cellular factors involved in the infectious cycle instead of virus-specific proteins holds the promise of creating broad-spectrum antivirals and raising the barrier for the emergence of resistant mutants, which is especially relevant for (+)RNA viruses known for their rapid adaptation rates. In the present study, we investigated the establishment of poliovirus resistance to a compound targeting a cellular factor required for viral replication on a model of BFA, an inhibitor of the cellular protein GBF1 and a potent suppressor of replication of poliovirus and other related picornaviruses (11–13). We showed that while poliovirus mutants resistant to the drug can be easily selected in HeLa cells, the selection process was inefficient in Vero cells. Accordingly, the BFA-resistant mutants selected in HeLa could not replicate in Vero in the presence of the drug, while both cultures can support effective poliovirus replication without the inhibitor. The lower availability of GBF1 for the replication complexes was the main limiting factor in Vero cells since replication of a BFA-resistant poliovirus could be rescued in the cells expressing exogenous GBF1 protein.
The role of GBF1 in the poliovirus replication process is not clear. GBF1 is a guanine nucleotide exchange factor for several small cellular GTPases of the Arf family, and in uninfected cells the protein organizes the formation of COPI transport vesicles transferring cargo between cis-Golgi membranes and the endoplasmic reticulum. It also coordinates recruitment of other proteins to the Golgi membranes, and its activity is important for maintaining the overall organization of the Golgi structure (39, 40). It was proposed that Arf-activating function of GBF1 was important for enterovirus replication and that enrichment of activated Arf on the replication membranes may be required to attract other cellular factors supporting replication such as the phosphatidylinositol kinase PI4KIIIβ (41). However, although the knockdown of GBF1 expression severely compromises poliovirus and CVB3 replication, showing that it is an essential cellular factor for the viruses (11, 12), the importance of its Arf activating property is uncertain. It has been shown that GBF1 variants lacking Arf activation capacity can partially rescue poliovirus replication from BFA inhibition and that the replication of a BFA-resistant poliovirus mutant in the presence of the drug does not induce Arf activation (27). Here, we observed that the cellular function of GBF1, as measured by the functioning of the cellular secretory pathway, was inhibited at a much lower concentration of BFA than was poliovirus replication. Moreover, replication of a BFA-resistant mutant in HeLa cells was even visibly stimulated at BFA concentrations sufficient to completely block the secretory activity. Collectively, these data strongly suggest that the replication requires GBF1 protein but is not dependent on its Arf-GTP regeneration activity. Thus, BFA itself or BFA-like compounds targeting the Arf activation function of GBF1 cannot serve as antiviral therapeutics since they would inhibit the cellular function of GBF1 at much lower concentrations than those required for suppressing viral replication, but the replication-specific role of GBF1 could be an attractive antiviral target.
In this study, we identified different point mutations conferring BFA resistance to poliovirus replication. Similar to the ones reported previously (26), they localized in the 2C and 3A proteins. The mutations in 3A are responsible for most of the BFA-resistant phenotype, while the ones in 2C seem to have an auxiliary role. There is strong evidence that the functions of 2C and 3A proteins in the replication complex are interdependent, since mutations in one protein can be compensated for by changes in the other (42). Thus, it is likely that BFA-resistant mutations in these proteins affect the same process rather than that changes in 2C and 3A provide separate mechanisms of resistance. The dependence of 2C-mediated BFA resistance on 3A is supported by the observation that a mutation in 3A disrupting 3A-GBF1 interaction reduced the BFA resistance of the construct with the BFA-resistant mutation in 2C. Interestingly, the BFA-resistant mutations in 3A (R34K and D29E identified here and A27V described previously [26]) are substitutions for amino acids with properties very similar to the ones in the parental sequence, suggesting that they could not dramatically change the protein properties but likely modulate its interaction with membranes and/or protein partners, which is apparently enough to escape the BFA inhibition. It could also suggest that this region of 3A is under a strong selective pressure that cannot tolerate significant changes. Indeed, the inability to develop of BFA-resistant mutations in Vero cells under conditions of the limited GBF1 supply may reflect the limited capacity of this segment of 3A to accommodate more extensive modifications than those sufficient for resistance in HeLa cells.
The interaction between 3A and GBF1 was critical for BFA resistance since the resistant phenotype was lost in the context of mutations that disrupt 3A-mediated recruitment of GBF1 to membranes. At the same time, none of these mutants showed significant defects in replication in the absence of the drug. This apparent excess of the strength of 3A-GBF1 interaction in the context of the wt poliovirus may suggest that the virus is actually adapted to replication under the conditions of a very limited GBF1 supply that probably may exist in the natural sites of infection in the human host. More likely, however, is that strong GBF1-3A interaction is required for rapid inhibition of the cellular secretory pathway preventing the secretion of cytokines and suppressing the presentation of antigens on the surfaces of infected cells (43, 44), which is likely important for replication in a host with a fully functional immune system and is mostly dispensable in cell culture. Indeed, in a mouse model of myocarditis, CVB3 with a mutation in the 3A protein disrupting 3A-GBF1 interaction was significantly attenuated, while showing minimal defects in replication in cell culture (25).
The original expectations that targeting a cellular factor which cannot change during infection process rather than a highly adaptable virus protein will prevent emergence of resistant mutants seem to hold true in certain cell types but not the in others, at least in the case of BFA. Importantly, different cell lines of human origin also differ in their ability to support the growth of already selected BFA-resistant mutants; these viruses could bypass BFA inhibition in HeLa and 293 epithelial cells but not in the neuroblastoma cell line SH-S5Y5. This is to be expected for other prospective inhibitors targeting cellular factors recruited for viral replication as well, since the level of expression and/or availability of these proteins for viral processes would likely be different in different tissues. In this respect for the future development of antivirals it will be important to take into consideration the contribution of tissue-specific replication for the actual development of the disease. For example, an agent restricting replication in neurons would work as an antipoliomyelitis drug, whereas the emergence of resistant mutants replicating in epithelial cells can be tolerated as long as they cannot bypass the inhibition in neuronal cells, such as BFA-resistant viruses. Another desirable property of an antiviral compound would be that, even if the virus could develop resistance, there should be a trade-off between resistance to the inhibitor and fitness, such as susceptibility to the immune defenses, as we observed in the case of BFA-resistant mutants. BFA-resistant mutations are associated with a reduced efficiency of poliovirus RNA replication without the drug, resulting in slower replication kinetics and rendering the replication more sensitive to induction of the cellular innate immune response and other antiviral agents.
Currently, there is practically no rational approach to the selection of cellular targets for development of antiviral therapeutics, for the most part due to our very limited knowledge of any host factors required to support viral infection in any viral system. Practically any protein identified is considered a prospective target. However, with the advancement of future research, the situation might change, and facing the choice among different targets the lessons learned using the model of inhibition of GBF1 by BFA may provide important guidance. First, we need to know the specific role of the cellular protein in the viral life cycle and not just try to inhibit its known cellular function; second, the goal of complete prevention of the emergence of resistant mutants is probably unrealistic. Instead, the focus should be on choosing a target that minimizes the replication potential of resistant mutants in the cell types of importance for the development of the disease. Finally, the emergence of resistant mutants can be tolerated as long as they lead to a fitness trade-off.
ACKNOWLEDGMENTS
This study was supported in part by University of Maryland funds (G.A.B.) and a National Institutes of Health Virology training grant (L.A.F.-S.).
REFERENCES
- 1.Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA, Fraser-Liggett CM, Liggett SB. 2009. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 324:55–59. doi: 10.1126/science.1165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papi A, Contoli M. 2011. Rhinovirus vaccination: the case against. Eur Respir J 37:5–7. doi: 10.1183/09031936.00145710. [DOI] [PubMed] [Google Scholar]
- 3.Manolea F, Claude A, Chun J, Rosas J, Melancon P. 2008. Distinct functions for Arf guanine nucleotide exchange factors at the Golgi complex: GBF1 and BIGs are required for assembly and maintenance of the Golgi stack and trans-Golgi network, respectively. Mol Biol Cell 19:523–535. doi: 10.1091/mbc.E07-04-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borderia AV, Stapleford KA, Vignuzzi M. 2011. RNA virus population diversity: implications for inter-species transmission. Curr Opin Virol 1:643–648. doi: 10.1016/j.coviro.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 5.Acevedo A, Brodsky L, Andino R. 2014. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vignuzzi M, Stone JK, Andino R. 2005. Ribavirin and lethal mutagenesis of poliovirus: molecular mechanisms, resistance, and biological implications. Virus Res 107:173–181. doi: 10.1016/j.virusres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Thibaut HJ, De Palma AM, Neyts J. 2012. Combating enterovirus replication: state-of-the-art on antiviral research. Biochem Pharmacol 83:185–192. doi: 10.1016/j.bcp.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Geller R, Vignuzzi M, Andino R, Frydman J. 2007. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev 21:195–205. doi: 10.1101/gad.1505307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Schaar HM, van der Linden L, Lanke KH, Strating JR, Purstinger G, de Vries E, de Haan CA, Neyts J, van Kuppeveld FJ. 2012. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res 22:1576–1592. doi: 10.1038/cr.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2011. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol 85:2364–2372. doi: 10.1128/JVI.02249-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E. 2008. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog 4:e1000216. doi: 10.1371/journal.ppat.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanke KH, van der Schaar HM, Belov GA, Feng Q, Duijsings D, Jackson CL, Ehrenfeld E, van Kuppeveld FJ. 2009. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J Virol 83:11940–11949. doi: 10.1128/JVI.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gazina EV, Mackenzie JM, Gorrell RJ, Anderson DA. 2002. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J Virol 76:11113–11122. doi: 10.1128/JVI.76.21.11113-11122.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irurzun A, Perez L, Carrasco L. 1992. Involvement of membrane traffic in the replication of poliovirus genomes: effects of brefeldin A Virology 191:166–175. [DOI] [PubMed] [Google Scholar]
- 15.Maynell LA, Kirkegaard K, Klymkowsky MW. 1992. Inhibition of poliovirus RNA synthesis by brefeldin A J Virol 66:1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doedens J, Maynell LA, Klymkowsky MW, Kirkegaard K. 1994. Secretory pathway function, but not cytoskeletal integrity, is required in poliovirus infection. Arch Virol 9(Suppl):159–172. [DOI] [PubMed] [Google Scholar]
- 17.Monaghan P, Cook H, Jackson T, Ryan M, Wileman T. 2004. The ultrastructure of the developing replication site in foot-and-mouth disease virus-infected BHK-38 cells. J Gen Virol 85:933–946. doi: 10.1099/vir.0.19408-0. [DOI] [PubMed] [Google Scholar]
- 18.Ehrenfeld E, Domingo E, Roos RP. 2010. The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 19.Lowery J, Szul T, Styers M, Holloway Z, Oorschot V, Klumperman J, Sztul E. 2013. The Sec7 guanine nucleotide exchange factor GBF1 regulates membrane recruitment of BIG1 and BIG2 guanine nucleotide exchange factors to the trans-Golgi network (TGN). J Biol Chem 288:11532–11545. doi: 10.1074/jbc.M112.438481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takashima K, Saitoh A, Hirose S, Nakai W, Kondo Y, Takasu Y, Kakeya H, Shin HW, Nakayama K. 2011. GBF1-Arf-COPI-ArfGAP-mediated Golgi-to-ER transport involved in regulation of lipid homeostasis. Cell Struct Funct 36:223–235. doi: 10.1247/csf.11035. [DOI] [PubMed] [Google Scholar]
- 21.Zhao XH, Claude A, Chun J, Shields DJ, Presley JF, Melancon P. 2006. GBF1, a cis-Golgi and VTCs-localized ARF-GEF, is implicated in ER-to-Golgi protein traffic. J Cell Sci 119:3743–3753. doi: 10.1242/jcs.03173. [DOI] [PubMed] [Google Scholar]
- 22.Szul T, Grabski R, Lyons S, Morohashi Y, Shestopal S, Lowe M, Sztul E. 2007. Dissecting the role of the ARF guanine nucleotide exchange factor GBF1 in Golgi biogenesis and protein trafficking. J Cell Sci 120:3929–3940. doi: 10.1242/jcs.010769. [DOI] [PubMed] [Google Scholar]
- 23.Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. 2005. Dynamics of GBF1, a brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol Biol Cell 16:1213–1222. doi: 10.1091/mbc.E04-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mossessova E, Corpina RA, Goldberg J. 2003. Crystal structure of ARF1*Sec7 complexed with brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol Cell 12:1403–1411. doi: 10.1016/S1097-2765(03)00475-1. [DOI] [PubMed] [Google Scholar]
- 25.Wessels E, Duijsings D, Niu TK, Neumann S, Oorschot VM, de Lange F, Lanke KHW, Klumperman J, Henke A, Jackson CL, Melchers WJG, van Kuppeveld FJM. 2006. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev Cell 11:191–201. doi: 10.1016/j.devcel.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Crotty S, Saleh MC, Gitlin L, Beske O, Andino R. 2004. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J Virol 78:3378–3386. doi: 10.1128/JVI.78.7.3378-3386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belov GA, Kovtunovych G, Jackson CL, Ehrenfeld E. 2010. Poliovirus replication requires the N terminus but not the catalytic Sec7 domain of ArfGEF GBF1. Cell Microbiol 12:1463–1479. doi: 10.1111/j.1462-5822.2010.01482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fogg MH, Teterina NL, Ehrenfeld E. 2003. Membrane requirements for uridylylation of the poliovirus VPg protein and viral RNA synthesis in vitro. J Virol 77:11408–11416. doi: 10.1128/JVI.77.21.11408-11416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duchene M. 2006. Production, testing and perspectives of IPV and IPV combination vaccines: GSK biologicals' view. Biologicals 34:163–166. doi: 10.1016/j.biologicals.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 30.Caliguiri LA, Tamm I. 1968. Action of guanidine on the replication of poliovirus RNA. Virology 35:408–417. doi: 10.1016/0042-6822(68)90219-5. [DOI] [PubMed] [Google Scholar]
- 31.Jackson CL, Casanova JE. 2000. Turning on ARF: the Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol 10:60–67. doi: 10.1016/S0962-8924(99)01699-2. [DOI] [PubMed] [Google Scholar]
- 32.Teterina NL, Pinto Y, Weaver JD, Jensen KS, Ehrenfeld E. 2011. Analysis of poliovirus protein 3A interactions with viral and cellular proteins in infected cells. J Virol 85:4284–4296. doi: 10.1128/JVI.02398-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wessels E, Duijsings D, Lanke KH, van Dooren SH, Jackson CL, Melchers WJ, van Kuppeveld FJ. 2006. Effects of picornavirus 3A proteins on protein transport and GBF1-dependent COP-I recruitment. J Virol 80:11852–11860. doi: 10.1128/JVI.01225-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wessels E, Duijsings D, Lanke KH, Melchers WJ, Jackson CL, van Kuppeveld FJ. 2007. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J Virol 81:5238–5245. doi: 10.1128/JVI.02680-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Claude A, Zhao BP, Melancon P. 2003. Characterization of alternatively spliced and truncated forms of the Arf guanine nucleotide exchange factor GBF1 defines regions important for activity. Biochem Biophys Res Commun 303:160–169. doi: 10.1016/S0006-291X(03)00316-4. [DOI] [PubMed] [Google Scholar]
- 36.Bernstein HD, Baltimore D. 1988. Poliovirus mutant that contains a cold-sensitive defect in viral-RNA synthesis. J Virol 62:2922–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton DJ, Black EP, Flanegan JB. 1995. Complete replication of poliovirus in vitro: preinitiation RNA replication complexes require soluble cellular factors for the synthesis of VPg-linked RNA. J Virol 69:5516–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molla A, Paul AV, Wimmer E. 1993. In vitro synthesis of poliovirus. Dev Biol Stand 78:39–53. [PubMed] [Google Scholar]
- 39.Bui QT, Golinelli-Cohen MP, Jackson CL. 2009. Large Arf1 guanine nucleotide exchange factors: evolution, domain structure, and roles in membrane trafficking and human disease. Mol Genet Genomics 282:329–350. doi: 10.1007/s00438-009-0473-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright J, Kahn RA, Sztul E. 2014. Regulating the large Sec7 ARF guanine nucleotide exchange factors: the when, where and how of activation. Cell Mol Life Sci 71:3419–3438. doi: 10.1007/s00018-014-1602-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teterina NL, Levenson E, Rinaudo MS, Egger D, Bienz K, Gorbalenya AE, Ehrenfeld E. 2006. Evidence for functional protein interactions required for poliovirus RNA replication. J Virol 80:5327–5337. doi: 10.1128/JVI.02684-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dodd DA, Giddings TH Jr, Kirkegaard K. 2001. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J Virol 75:8158–8165. doi: 10.1128/JVI.75.17.8158-8165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deitz SB, Dodd DA, Cooper S, Parham P, Kirkegaard K. 2000. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci U S A 97:13790–13795. doi: 10.1073/pnas.250483097. [DOI] [PMC free article] [PubMed] [Google Scholar]