

ABSTRACT
Respiratory syncytial virus (RSV) infects epithelial cells of the respiratory tract and is a major cause of bronchiolitis and pneumonia in children and the elderly. The virus assembles and buds through the plasma membrane, forming elongated membrane filaments, but details of how this happens remain obscure. Oligomerization of the matrix protein (M) is a key step in the process of assembly and infectious virus production. In addition, it was suggested to affect the conformation of the fusion protein, the major current target for RSV antivirals, in the mature virus. The structure and assembly of M are thus key parameters in the RSV antiviral development strategy. The structure of RSV M was previously published as a monomer. Other paramyxovirus M proteins have been shown to dimerize, and biochemical data suggest that RSV M also dimerizes. Here, using size exclusion chromatography-multiangle laser light scattering, we show that the protein is dimeric in solution. We also crystallized M in two crystal forms and show that it assembles into equivalent dimers in both lattices. Dimerization interface mutations destabilize the M dimer in vitro. To assess the biological relevance of dimerization, we used confocal imaging to show that dimerization interface mutants of M fail to assemble into viral filaments on the plasma membrane. Additionally, budding and release of virus-like particles are prevented in M mutants that fail to form filaments. Importantly, we show that M is biologically active as a dimer and that the switch from M dimers to higher-order oligomers triggers viral filament assembly and virus production.
IMPORTANCE Human respiratory syncytial virus (RSV) is the most frequent cause of infantile bronchiolitis and pneumonia. The enormous burden of RSV makes it a major unmet target for a vaccine and antiviral drug therapy. Oligomerization of the matrix protein is a key step in the process of assembly and production of infectious virus, but the molecular mechanism of RSV assembly is still poorly understood. Here we show that the RSV matrix protein forms dimers in solution and in crystals; the dimer is essential for formation of higher-order oligomers. Destabilizing the dimer interface resulted in the loss of RSV filament formation and a lack of budding of virus-like particles. Importantly, our findings can potentially lead to new structure-based RSV inhibitors targeting the assembly process.
INTRODUCTION
Human respiratory syncytial virus (RSV), an enveloped, nonsegmented negative-strand RNA paramyxovirus, is the major worldwide cause of bronchiolitis and pneumonia in infants and of morbidity and mortality in the elderly. Despite the large global impact of RSV infection, no specific antivirals or licensed vaccine is yet available (1, 2). Designing new antivirals requires detailed molecular and structural understanding of key steps in RSV replication.
RSV encodes 11 proteins, with the 3 glycoproteins, the fusion protein (F), glycoprotein (G), and small hydrophobic protein (SH), being present in the viral envelope. The virion itself contains an internal ribonucleoprotein (RNP) complex comprising the negative-sense genome encapsidated within the nucleoprotein (N), the phosphoprotein (P), and large (L) RNA-directed RNA polymerase. Viral proteins N, P, L, matrix protein (M) 2-1 (M2-1), and M were shown to localize to cytoplasmic inclusion bodies (IBs) of various sizes (3–7). On the basis of localization of viral RNA and RNP proteins into small IBs, it is assumed that these are sites of viral transcription and replication (8), whereas big IBs were shown to contain only small amounts of viral RNA and were suggested to antagonize the innate immune response (9). RSV assembles and buds through the plasma membrane, forming elongated membrane filaments (10). The minimal RSV protein requirement for filament formation and budding of virus-like particles (VLPs) is F, N, P, and M (11, 12). M, a key structural protein, directs assembly and budding on the plasma membrane, presumably by interacting with the cytoplasmic tails of the glycoproteins and with the RNP complex in the cytoplasm (7, 13–15). At the start of RSV assembly, M localizes into viral IBs (5, 16). It is thought that M triggers viral filament formation by interacting with F (17). At the final budding step, M is crucial for viral filament maturation and elongation (15). An intact M layer is also thought to be critical for the infectivity of the virus particle. It was suggested that the M layer interacts with the F cytoplasmic tail when F is in its prefusion conformation. This interaction is lost upon triggering of F, resulting in M lattice disassembly and gradual transition to a spherical noninfectious virus particle (18).
Higher-order assembly is an important aspect of M function through formation of a two-dimensional lattice, but the structural and mechanistic details of the process are not fully understood. M has previously been shown to form ordered oligomers in vitro when incubated with specific sets of lipids (19). We have previously shown that the role of RSV M in virus assembly/release is strongly dependent on the threonine 205 (Thr 205) residue. This threonine is located within a consensus site for casein kinase 2, which appears to play a key regulatory role in modulation of M oligomerization and the association with virus filaments (20), probably defining filamentous morphology (18). However, it is not clear what the biologically relevant assembly unit of M is. RSV belongs to the order Mononegavirales. Crystal structures have been solved for a number of M proteins from the Mononegavirales (21–24), all of which form dimers. Although the monomeric unit of RSV M protein is structurally closely related to that of other paramyxovirus M proteins that exist as dimers, M was crystallized as a monomer (24, 25), which sparked a debate in the field whether RSV M may use a different mechanism for its oligomerization.
In this study, we show that RSV M forms dimers in solution and in crystals. We characterized the dimerization interface from the crystal structure and used dimerization interface mutants to show that dimeric M is the biologically relevant assembly unit required for viral filament formation and particle release.
MATERIALS AND METHODS
M protein expression and purification.
Wild-type (WT) M and mutant derivatives were expressed as His6 fusion proteins in a modified pCDF-Duet1 vector (20) in Escherichia coli Rosetta 2. Cells were grown from fresh starter cultures in 0.25 liter of 6× Luria-Bertani broth for 5 h at 30°C, followed by induction with 0.4 mM isopropylthio-β-galactoside (IPTG) for 4 h at 25°C. All following procedures were done on ice or at 4°C. Cells were lysed by sonication (4 times for 20 s each time) and lysozyme (1 mg/ml; Sigma) in 50 mM NaH2PO4-Na2HPO4, 300 mM NaCl, pH 7.4, plus protease inhibitors (Roche Diagnostics), RNase (12 μg/ml, Sigma), and 0.25% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}. Lysates were clarified by centrifugation (23,425 × g, 30 min, 4°C), and the soluble His6-M protein was purified on an Ni-nitrilotriacetic acid column (His Trap HP; Qiagen). The bound protein was washed extensively with loading buffer plus 20 mM imidazole and eluted with a 20 to 250 mM imidazole gradient. The eluted protein was incubated with 3C protease (20 μg per 1 mg M) in the presence of 1 mM dithiothreitol (DTT) overnight at 4°C to remove the His6 tag. The processed protein was diluted 5 times to reduce the DTT concentration and applied to HIS-Select HF nickel affinity beads (Sigma) to separate it from the 3C protease and uncleaved protein. Untagged M was concentrated to 2 ml using Vivaspin 20 columns (Sartorius Stedim Biotech) and purified on a HiLoad 10/600 Superdex S200 column (GE Healthcare) in 50 mM NaH2PO4-Na2HPO4, 300 mM NaCl, pH 7.4. The M peak was concentrated to 9 mg/ml using Vivaspin 4 columns and subjected to crystallization or to size exclusion chromatography (SEC)-multiangle laser light scattering (MALS).
In vitro dimerization experiments.
SEC was combined with online detection by MALS and refractometry to determine the absolute molecular mass of M protein mutants independently of dimensions and shape. SEC was performed with a Tricorn Superdex 75 10/30 column (GE Healthcare) equilibrated overnight with 50 mM NaH2PO4-Na2HPO4, 300 mM NaCl, pH 7.4. M was purified as described above for the crystallization experiments and concentrated to 9 mg/ml. Separations were performed at 20°C at a flow rate of 0.4 ml/min. MALS detection was performed with a miniDAWN Treos detector (Wyatt Technology) using a laser emitting at 657.3 nm. The refractive index of the solution was measured with an Optilab T-rEX detector (Wyatt Technology), with the refractive index increment (dn/dc) being set to 0.185 ml/g. Weight-averaged molar masses were calculated with ASTRA software (Wyatt Technology).
For dimerization assays on size exclusion chromatography, M proteins were expressed and lysed as described above; the soluble His6-M protein was purified using HIS-Select HF nickel affinity beads (Sigma) and eluted with 250 mM imidazole. Protein-containing fractions were collected, and the protein was concentrated up to ∼3 mg/ml using Vivaspin 4 columns (Sartorius Stedim Biotech) and analyzed by size exclusion chromatography (Superdex-200 10/300 GL; GE Healthcare) in 50 mM NaH2PO4-Na2HPO4, 300 mM NaCl, pH 7.4, 1 day later.
Crystallization and structure determination.
M was crystallized by vapor diffusion at 20°C. Two crystal forms (CFs) appeared overnight. Crystal form 1 (CF 1) grew in 10% (wt/vol) polyethylene glycol 8000; 20% (vol/vol) ethylene glycol; 0.02 M (each) sodium formate, ammonium acetate, trisodium citrate, and sodium potassium tartrate; and 0.1 M MOPS (morpholinepropanesulfonic acid)–HEPES-Na (7.5). Crystal form 2 (CF 2) grew in 30% glycerol, 0.1 M MES (morpholineethanesulfonic acid; 6.5), and 1.8 M Am2SO4. Crystals were flash-frozen by dipping into liquid nitrogen without additional cryoprotection.
Two CF 1 data sets, a highly redundant data set for phasing and a high-resolution data set for refinement, were collected on a Rigaku MicroMax 007-HF X-ray generator. Data were processed in the HKL-3000 program. The structure was solved in the SHELX program (26) by single anomalous dispersion on the basis of the sulfur signal. CF 2 data were collected at beamline I24 of the Diamond Light Source and showed quick radiation damage. To obtain a full data set, the first five degrees of data from nine crystals were combined in the Blend program (27) after processing with the XDS package (28). The structure was solved in the Phaser program (29) with a monomer of M, as solved previously (25), as the search model (PDB accession no. 2VQP). Both structures were refined in the Refmac5 program (30) and the phenix.refine program (31) and rebuilt in the Coot program (32) until convergence. Crystallographic statistics are summarized in Table 1. Interface analysis was performed on the Proteins, Interfaces, Structures, and Assemblies (PISA) server (33). Molecular graphics figures were prepared with PyMOL (Schrödinger, LLC).
TABLE 1.
Data collection and refinement statistics
| Parametera | Valueb |
||
|---|---|---|---|
| Anomalous CF 1 | Native CF 1 | CF 2 | |
| Data collection statistics | |||
| Space group | C 2 | C 2 | P65 2 2 |
| Unit cell dimensions | |||
| a, b, c (Å) | 52.3, 79.2, 66.0 | 52.3, 79.2, 65.9 | 87.1, 87.1, 144.7 |
| α, β, γ (°) | 90, 96.2, 90 | 90, 96.2, 90 | 90, 90, 120 |
| Resolution (Å) | 23.5–1.9 (1.94–1.90) | 23.5–1.70 (1.73–1.70) | 33.5–1.90 (1.94–1.90) |
| Rmerge | 0.057 (0.190) | 0.050 (0.292) | 0.101 (0.753) |
| I/σ〈I〉 | 41.2 (15.6) | 16.1 (2.9) | 13.5 (3.2) |
| Completeness (%) | 99.7 (100) | 99.8 (99.1) | 99.1 (99.6) |
| Redundancy | 23.0 (19.9) | 4.9 (2.8) | 5.5 (5.5) |
| Refinement statistics | |||
| No. of reflections | 29,389 (2,463) | 25,938 (1,643) | |
| Rwork/Rfree | 0.175/0.211 | 0.183/0.206 | |
| No. of atoms | |||
| Protein | 2,077 | 2,023 | |
| Ligand/ion | 1 | 12 | |
| Water | 183 | 119 | |
| B factors | |||
| Protein | 22.1 | 28.3 | |
| Ligand/ion | 16.5 | 33.06 | |
| Water | 30.2 | 37.4 | |
| RMSD | |||
| Bond length (Å) | 0.017 | 0.016 | |
| Bond angle (°) | 1.32 | 1.04 | |
| PDB accession no. | 4V23 | 4D4T | |
Rmerge = Σ|I – 〈I〉 |/ΣI × 100, where I is the intensity of a reflection and 〈I〉 is the average intensity.
Values in parentheses are for the highest-resolution shell.
Cell culture and RSV preparation.
HEp-2 (ATCC CCL-23) and Vero cells (provided by W. Barclay, Imperial College London) were maintained in Dulbecco modified Eagle medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco), 1% l-glutamine, and 1% penicillin-streptomycin. Transfections were performed using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's specifications. Plaque-purified human RSV (A2 strain from ATCC) was cultured in Vero cells. In the case of cell-associated virus, the culture supernatant was removed when extensive cytopathic effects were observed, and the cells were resuspended in serum-free SPGA medium (218 mM sucrose, 7.1 mM K2HPO4, 4.9 mM sodium glutamate, 1% bovine serum albumin [BSA]), followed by centrifugation (1,300 × g, 15 min, 4°C) and storage at −80°C. RSV titers were determined by an immune plaque assay in triplicate on HEp-2 cells (20).
Virus and virus-like filament/particle formation.
Overnight cultures of HEp-2 cells seeded at 4 × 105 cells/well in 6-well plates (on a 16-mm micro-cover glass for immunostaining) were transfected with pcDNA3.1 codon-optimized plasmids (0.4 μg each) carrying the RSV A2 WT or the mutant M protein along with pcDNA3.1 codon-optimized plasmids carrying RSV A2 N, P, and F (gifts from Marty Moore, Emory University, Atlanta, GA, USA) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations. Cells were fixed at 24 h posttransfection, immunostained, and imaged as described below. For VLP formation, released VLPs were harvested from the supernatant; the supernatant was clarified of cell debris by centrifugation (1,300 × g, 10 min, 4°C) and pelleted through a 20% sucrose cushion. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer. Cellular lysates and VLP pellets were dissolved in Laemmli buffer and subjected to Western analysis. For virus particle formation, HEp-2 cells were infected with RSV A2 at a multiplicity of infection (MOI) of 3, and cells and released virus were harvested at 24 h postinfection. Cells were lysed in RIPA buffer, and released virus was harvested from the supernatant as described above for VLPs and subjected to Western analysis.
Immunostaining and imaging.
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, blocked with 3% BSA in 0.2% Triton X-100–PBS for 10 min, and immunostained with monoclonal anti-M (1:200; a gift from Mariethe Ehnlund, Karolinska Institute, Sweden), monoclonal anti-N (1:300; Abcam), or monoclonal anti-F (1:500; Millipore) antibodies, followed by species-specific secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 568 (1:1,000; Invitrogen). Images were obtained on a Zeiss 5 Pascal confocal laser scanning microscope (LSM) using a 63× (numerical aperture, 1.4) Plan-Apochromat oil lens (averaging 4 determinations). Images were acquired using Zeiss LSM Image Browser software (v4.2.0.121; Zeiss).
SDS-PAGE and Western analysis.
Protein samples were separated by electrophoresis on 12% polyacrylamide gels in Tris-glycine buffer. All samples were boiled for 3 min prior to electrophoresis. Proteins were then transferred onto a polyvinylidene difluoride membrane (Roche Diagnostics). The blots were blocked with 5% nonfat milk in Tris-buffered saline (pH 7.4), followed by incubation with polyclonal anti-RSV (1:1,000; Abcam) and horseradish peroxidase (HRP)-conjugated donkey antigoat (1:5,000, Santa Cruz Biotechnology) antibodies. Western blots were developed using freshly prepared chemiluminescent substrate (100 mM Tris-HCl, pH 8.8, 1.25 mM luminol, 0.2 mM p-coumaric acid, 0.05% H2O2) and exposed to Fuji autoradiography films.
Protein structure accession numbers.
The protein structures of native CF 1 and CF 2 have been deposited in the Protein Data Bank (PDB) under accession numbers 4V23 and 4D4T, respectively.
RESULTS
RSV M dimerizes similarly to other paramyxovirus M proteins.
Controversy in the literature regarding the oligomeric state of M arose from a crystal structure reporting that it is a monomer (25). More recently, analytical size exclusion chromatography showed M to be dimeric (20). To resolve this, we first purified recombinant M protein by modifying the previously published protocol. We repeated the size exclusion experiments but analyzed eluting protein by multiangle light scattering (SEC-MALS) to determine the absolute molecular mass of M protein independently of dimensions and shape. M eluted in one peak with a calculated molecular mass of 60.0 kDa, which corresponds to a dimer (Fig. 1A). We then crystallized M in two different crystal forms that diffracted to 1.7 and 1.9 Å, respectively. We solved the structure from crystal form 1 (CF 1) by single anomalous dispersion based on the sulfur signal and that from CF 2 by molecular replacement with the existing M monomeric structure (PDB accession no. 2VQP) (25) as a search model (Fig. 1B). Crystallographic statistics are summarized in Table 1.
FIG 1.

RSV M forms dimers. (A) SEC-MALS chromatogram (loading concentration, 9 mg/ml) for bacterially expressed wild-type M protein. The differential refractive index signal (left axis) is shown in blue, and the MALS-derived apparent mass (right axis) is shown in green. The calculated weight-average molar mass of 60.0 kDa indicates that M protein is dimeric in solution. RIU, refractive index units; Ve, volume of elution. (B) Dimer of RSV M protein generated by application of crystal symmetry to the protomer present in the asymmetric unit. (Left) The structure obtained from our CF 1; (middle) the structure obtained from our CF 2; (right) the previously solved structure (PDB accession no. 2VQP) (25). In each case, the protomers that comprise the dimer are colored differently. The NTD and CTD of an M protomer are indicated in the left panel. The potassium ions observed in our structures are shown as purple spheres. (C) The RSV M dimer (left) is very similar to the dimeric matrix proteins from hMPV (middle) (PDB accession no. 4LP7) (24) and NDV (right) (PDB accession no. 4G1G) (23). NDV M possesses a number of extensions (shown in red) not present in either RSV M or hMPV M.
Both structures are highly similar, with a root mean square (RMS) deviation (RMSD) of 0.9 Å between 235 C-α atoms. The main difference is a peripheral segment (residues 170 to 178 inclusive) which forms a short helix in CF 1 but is disordered in CF 2. In either CF, the asymmetric unit contains 1 molecule of M. However, interface analysis suggests that the likely stable assembly is a dimer in either case (Table 2), which is consistent not only with our chromatography results but also with crystal structures of homologous M proteins. The RSV M dimerization interface is the same as that observed in the dimeric M structures from Newcastle disease virus (NDV) (23) and human metapneumovirus (hMPV) (24), and dimeric RSV M can be superposed onto the homologous M dimers with matching quaternary structure (Fig. 1C).
TABLE 2.
PISA analysisa
| Parameter | Value |
||
|---|---|---|---|
| Previous structure | CF 1 | CF 2 | |
| PDB accession no. | 2VQP | 4V23 | 4D4T |
| Accessible surface area (Å2) | 23,310 | 22,040 | |
| Excluded area (Å2) | 2,660 | 3,830 | |
| Solvation free energy gain (kcal/mol) | −221.9 | −245.6 | |
| Stable | No | Yes | Yes |
| Range of residues | 60, 63–68, 71, 93, 96–98, 104, 105, 129, 130, 133, 142, 144, 159, 161, 163, 228, 229, 231, 232, 252, 255b | 60, 62–68, 71, 93, 96–100, 104, 105, 129, 130, 133, 144, 159, 161, 163, 225, 228, 229, 231, 232, 252, 255 | 63–68, 93, 96–101, 104, 105, 129, 133, 142, 144, 146, 156, 159–163, 228, 229, 231–233, 252 |
| No. of salt bridges (salt bridges) | 2 (Lys 252-Asp 105) | 6 (Lys 60-Glu 255 [n = 2], Lys 252-Asp 105) | 8 (Lys 101-Glu 233 [n = 2], Lys 101-Glu 98, Lys 252-Asp 105) |
| No. of hydrogen bonds (hydrogen bonds) | 6 (Ser 63-Pro 161, Lys 232-Asn 93, Lys 252-Asp 105) | 8 (Ser 63-Pro 161, Asn 93-Ala 228, Lys 232-Leu 96, Lys 232-Asp 97) | 20 (Ser 63-Tyr 229, Thr 64-Tyr 229 [n = 2], Asn 93-Ala 228, Leu 96-Lys 232, Lys 101-Glu 98, Lys 101-Glu 233, Asp 105-Lys 252, Tyr 163-Ser 63, Lys 232-Glu 231) |
PISA analysis was performed with the PDBe PISA program (v1.51).
The numbers in bold are residues involved in the dimerization interface in all three structures.
In light of our biochemical analysis (20) and the fact that we crystallized M as a dimer, we reanalyzed the previously published structure of RSV M (25), which was reported to be monomeric. This older structure is similar to both structures reported here. Its CF corresponds to our CF 1 (Fig. 1B), and the old structure and our CF 1 structure superpose with an RMSD of 0.4 Å over 234 C-α atoms. Application of crystal symmetry to the old structure creates a dimer similar to the one observed in our structures and in other paramyxovirus M proteins. The previously suggested hypothesis that RSV M crystallized as a monomer because it is missing residues at the C terminus that, in NDV, form a helix involved in mediating monomer-to-monomer contacts (23) is thus incorrect. Clearly, similar to other published paramyxovirus M proteins, RSV M crystallizes as a dimer, suggesting that the biologically relevant assembly unit of RSV M is dimeric.
Ion binding sites in Paramyxovirinae M proteins.
In both our CFs, we observed a potassium ion at the interface between the N-terminal domain (NTD) and the C-terminal domain (CTD), near the dimerization interface. This ion is coordinated by 6 carbonyl oxygens (Asn 93, Val 94, Leu 96, Leu 230, Glu 231, and Glu 233) and 1 water molecule and was identified unambiguously by its strong anomalous signal in data collected at 1.54 Å (Fig. 2A) and by its coordination geometry. The ion is absent from the earlier structure, where an equivalent density was modeled as a water molecule.
FIG 2.
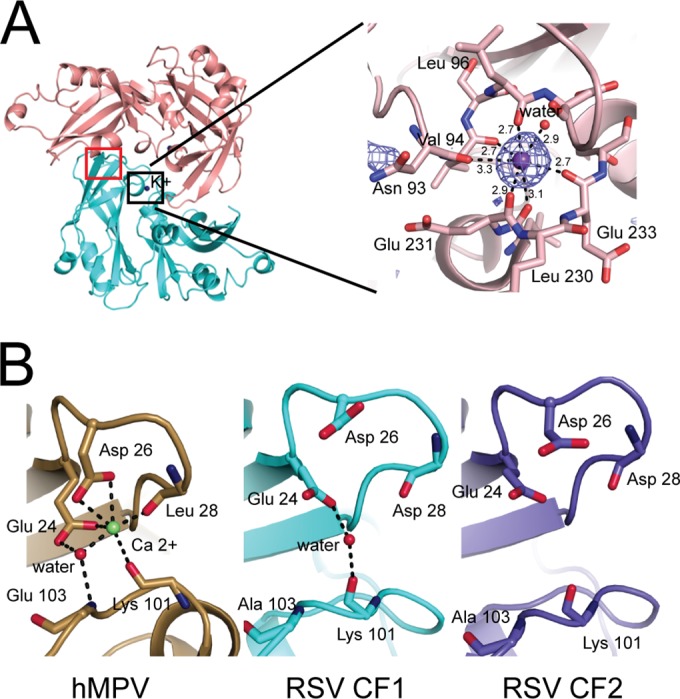
Ion binding sites in Paramyxovirinae M proteins. (A) A potassium ion (purple sphere) is coordinated by the main-chain carbonyl oxygens of 6 side chains and by 1 water molecule, as indicated. The experimental anomalous density is shown at the 3σ contour level. Atomic distances between potassium and its coordinating atoms are shown in angstroms. (B) Closeup view of the calcium ion binding site in hMPV M (left) and the comparable site in RSV M CF 1 (middle; marked with a red square in panel A) and RSV M CF 2 (right). The Ca2+ ion is represented as a green sphere in the hMPV structure. Ordered water molecules are shown as red spheres.
The recently published structure of hMPV M reported that a calcium ion is part of the dimeric structure and showed that it is important for M stability (24). However, comparison of the Ca2+-binding site in hMPV with corresponding regions in CF 1 and CF 2 showed clear differences (Fig. 2B). The site is much more open in RSV M, and its geometry is incompatible with Ca binding. In RSV M, Asp 26 points away from the site instead of into it, and the distance between the two loops forming the site is larger. The distance between the proximal carboxylic oxygen of Asp 26 and the main-chain nitrogen of residue 103 is 5.5 Å in hMPV and 8.8 Å and 8.6 Å in RSV M proteins of CF 1 and CF 2, respectively.
Dimerization interface of RSV M.
The dimerization interface observed in RSV M crystals, shown in Fig. 3A, comprises (sequentially numbered) loop L1 (residues 63 to 68), helix H2 and the loop immediately following it (residues 92 to 105), helix H3 (residues 129 to 134), sheet S4 (residues 144 and 163), and loop L5 (residues 225 to 235). All residues involved in dimerization are listed in Table 2. The interface is large, burying a total of 2,660 Å2 in CF 1 and 3,830 Å2 in CF 2. In the region around the symmetry axis, the residues involved in the dimerization interface are polar or charged, e.g., Asn 93 and Glu 231 (Fig. 3A, interface 1). Distal from the symmetry axis of the dimerization interface, the CTD of one protomer contacts the NTD of the other protomer. Contacts at this interface are largely hydrophobic, but residues Ser 63 and Tyr 229 are part of a hydrogen-bonding network (Fig. 3A, interface 2).
FIG 3.

Dimerization interface of RSV M. (A) (Left) A magnified view of the dimerization interface in CF 1. The black ellipse indicates the 2-fold axis, which is perpendicular to the page. The structural features participating in the interaction are loop L1, helix H2, helix H3, sheet S4, and loop L5. Only one protomer is labeled for clarity. Hydrogen bonds are shown for interactions highlighted in more detail in the two insets. (Top right) Interface 1 comprising residues Asn 93, Ala 228, and Glu 231. The potassium ion is indicated. (Bottom right) Interface 2 comprising residues Ser 63, Thr 64, Pro 161, and Tyr 229. The figure is colored and oriented as in Fig. 1B, left. (B) Superposition of the RSV M dimers from CF 1 and CF 2 onto one protomer (left) shows plasticity in the dimerization interface. The nonsuperposed protomers assume positions with a relative rational difference of nearly 20° (right). Coloring is as in Fig. 1B. The view is perpendicular to the 2-fold axis.
The two structures determined here have the same quaternary structure, but the details of dimerization vary. The dimer seen in CF 2 is narrower than that seen in CF 1, and the central portion is much more compact (Fig. 1B). Both dimers can be superimposed with an RMSD of 2.2 Å over equivalent C-α atoms. In contrast, the dimer assembled from the previously determined RSV M crystal structure superposes onto the CF 1 dimer with an RMSD of 0.4 Å. However, when only one chain is considered, the RMS difference is reduced to 0.9 Å for the CF 1-CF 2 pair, which indicates that the relative orientation of the protomers in the dimer differs between CF 1 and CF 2. Superposition of the one set of protomers in the dimers makes this difference visible (Fig. 3B). The nonsuperposed CF 2 protomer appears to be rotated by nearly 20° with respect to its CF 1 counterpart.
Mutations in the dimerization interface affect M oligomerization.
To understand the significance of dimerization of M protein for the life cycle of RSV, we designed a set of mutations to destabilize the dimer interface. Because of the size of the dimerization interface and its ambiguous character, we not only mutated residues to alanine but also introduced large charged residues, predicted to be the most disruptive. We chose Ser 63, Asn 93, Tyr 229, and Glu 231 as targets (Fig. 3A), with Lys 50 on the opposite of the dimerization interface serving as a negative control.
To compare the ability of WT and mutant M proteins to form dimers/higher-order oligomers in solution, recombinant RSV M WT, K50A, S63A, S63E, N93A, N93E, Y229A, and E231A proteins were expressed in bacteria and purified by nickel affinity chromatography under conditions similar to those used for RSV M crystallization. After concentration to 3 mg/ml, incubation for 24 h at 4°C, and centrifugation to pellet aggregated protein, the extent of oligomerization was analyzed by assessing the distribution of M protein between the insoluble and soluble fractions. As seen in Fig. 4A, all proteins were observed in both fractions. The pellet contained insoluble protein, presumably largely comprising higher-order oligomers which tend to aggregate in the absence of stabilizing lipids (19), and the supernatant contained soluble protein. While WT M and the K50A, E231A, and N93E mutants were nearly equally distributed between the pellet (50 to 60%) and supernatant (40 to 50%) fractions, the S63A, N93A, and Y229A mutants were found enriched in the supernatant fraction, suggesting decreased oligomerization. The percentage of each protein found in the pellet and supernatant fractions is shown in Fig. 4A, and data represent the means. The S63E mutant M protein could not be concentrated to comparable levels and was found predominantly in the pellet fraction, suggesting increased aggregation (data not shown). As this mutant could not be concentrated to more than to 0.5 mg/ml, we could not perform analysis of the secondary structure by circular dichroism.
FIG 4.

Dimerization mutants fail to form stable dimers in solution. (A) Bacterially expressed His6-tagged WT M and the indicated mutant derivative proteins were purified using nickel affinity chromatography. Proteins were concentrated to 3 mg/ml, incubated for 24 h at 4°C, and pelleted to separate higher-order oligomers in the pellet (P) fraction from the supernatant (S) fraction. Band intensity, representing the protein distribution in the pellet (black) and supernatant (gray) fractions, was calculated with ImageJ software. Data represents the means ± SDs (n = 2). (B) Analytical size exclusion chromatography was performed on the soluble fraction of the WT, K50A, S63A, S63E, N93A, N93E, Y229A, and E231A M proteins. (Top) M proteins showing an elution pattern similar to that of WT M; (middle) WT, N93A and N93E M proteins; (bottom) results of a separate analytical size exclusion chromatography experiment performed on WT M and S63E mutant M concentrated to 0.5 mg/ml. mAU, milli-absorbance units. (C) Molecular masses were estimated by comparing the gel-phase distribution coefficient (Kav) of the M protein peaks with the values obtained for known calibration standards (GE Healthcare). The estimated molecular mass of M peaks was determined by fitting to the calibration curve (plot of Kav versus log Mr); volumes of elution (Ves) of 16.5 ml (estimated molecular mass, 32 kDa), 15 ml (estimated molecular mass, 65 kDa), and 13.7 ml (estimated molecular mass, 133 kDa) are indicated by arrows.
Next, the oligomerization state of the protein in the soluble fraction was analyzed by analytical size exclusion chromatography (Fig. 4B). WT M (molecular mass of the monomer, 29 kDa) and the K50A and E231A mutants all eluted as single dimeric species (Fig. 4B, top) with an estimated molecular mass of 65 kDa (Fig. 4C). The S63A and Y229A mutants, which appeared to be more soluble than WT M (Fig. 4A), also eluted as dimers. This suggests that the S63A and Y229A mutations do not prevent dimerization but influence the higher-order oligomerization of M. The N93A and N93E mutants eluted as intermediate species in addition to a dimer, implying that mutation of Asn 93 results in an assembly defect (Fig. 4B, middle). The S63E mutant showed the most severe defect, with the S63E mutation resulting in reduced protein levels in the soluble fraction when concentrated (data not shown). We thus performed the same analysis but used a protein concentration of only 0.5 mg/ml. The S63E mutant eluted as three species corresponding to monomeric, dimeric, and higher-molecular-mass intermediate forms (Fig. 4B, bottom). In summary, M dimerization interface mutations S63A, S63E, N93A, N93E, and Y229A all resulted in unstable dimers and/or oligomerization defects compared to the stability and oligomerization capacity of WT M.
M dimerization interface mutants cannot form virus-like filaments.
To assess the biological relevance of dimerization, we analyzed M mutants in cells. Our hypothesis was that M mutants that fail to form stable dimers will not form higher-order oligomeric structures at the sites of budding and as a result will fail to produce virus-like filaments. RSV VLPs can be generated independently of viral infection by transfecting cells with plasmids encoding the RSV M, N, P, and F proteins (12, 20). We used this transfection-based assay to study M dimerization interface mutants. HEp-2 cells were transfected to express WT F, N, P, and various M constructs, and the intracellular localization of RSV proteins and the formation of RSV virus-like filaments were determined by confocal imaging after staining with monoclonal anti-M, anti-N, or anti-F antibodies (Fig. 5).
FIG 5.

M dimerization mutants abolish virus-like filament formation. (A) HEp-2 cells were cotransfected with pcDNA3.1 plasmids expressing RSV P, N, and F proteins and WT M (row 1, positive control), no M (row 2, negative control), or the indicated RSV M mutant constructs (rows 3 to 8). Cells were fixed, permeabilized at 24 h posttransfection, immunostained with anti-M, anti-N, or anti-F as described in Materials and Methods, and analyzed by confocal microscopy. The primary antibodies used are indicated at the top. BF, bright field. Bars, 10 μm. (B) The percentage of virus-like filament-forming cells was calculated for cells expressing each of the M constructs (n = 100 for each construct) and stained with anti-M, anti-N, or anti-F antibody.
In the presence of F, N, P, and WT M, the formation of virus-like filaments was detected by staining for M (Fig. 5A, left, row 1, positive control). No filaments were formed at 24 h after transfection in the absence of M (Fig. 5A, left, row 2, negative control), in agreement with previous reports (12, 15). M containing the K50A mutation, not located at the dimerization interface, formed virus-like filaments similar to those formed by the WT (Fig. 5A, left, row 3). One of the dimer interface mutants (E231A) showed filament formation that appeared to be slightly more robust than that of WT (row 8). In contrast, each of the S63A, N93A, and Y229A mutations in M prevented virus-like filament formation (Fig. 5A, left, rows 4, 6, and 7, respectively) to various extents. In cells cotransfected with the S63A, N93A, or Y229A mutant, M formed small protrusions coincident with the plasma membrane, and these were especially evident in cells expressing the Y229A M mutant. The S63E mutant showed the most severe phenotype (Fig. 5A, left, row 5), consistent with oligomerization defects in vitro (Fig. 4); M protein localized into big IBs and the nucleus.
Additionally, transfected cells were stained with monoclonal anti-N antibody (Fig. 5A, middle). In the presence of F, N, P, and WT M, virus-like filaments were formed, and N was detected in IBs and on virus-like filaments (Fig. 5A, middle, row 1). No filaments were formed at 24 h after transfection in the absence of M, and N was detected in cytoplasmic IBs only (Fig. 5A, middle, row 2). M containing the K50A or the E231A mutation formed virus-like filaments (Fig. 5A, middle, rows 3 and 8, respectively) and N localized to cytoplasmic IBs and to virus-like filaments. In contrast, in cells expressing M with the S63A, S63E, N93A, or Y229A mutation, N localized into cytoplasmic IBs only (Fig. 5A, middle, rows 4 to 7, respectively). In summary, all M dimerization interface mutations except the E231A mutation prevented virus-like filament formation.
Staining with monoclonal anti-F antibody (Fig. 5A, right) confirmed these results. In the presence of F, N, P, and WT M, virus-like filaments were formed (Fig. 5A, right, row 1). No filaments were formed at 24 h after transfection in the absence of M (Fig. 5A, right, row 2). M containing the K50A or the E231A mutation formed virus-like filaments similar to those formed by WT M (Fig. 5A, right, rows 3 and 8, respectively). In contrast, the S63A, S63E, N93A, and Y229A M mutations all prevented virus-like filament formation (Fig. 5A, right, rows 4 to 7, respectively). In summary, all M dimerization interface mutations except the E231A mutation prevented virus-like filament formation. The E231A M mutant appeared to form more robust virus-like filaments than WT M, as it was stained with anti-M antibody, and the S63E M mutant showed the most severe phenotype, being localized into the IBs and the nucleus.
To analyze the robustness of the phenotypes, the percentage of virus-like filament-forming cells was calculated for cells expressing each of the M constructs (Fig. 5B). Clearly, only cells expressing WT, K50A, or E231A M formed virus-like filaments. Staining with anti-M antibody showed filaments in 66% and 47% of the cells for WT and K50A mutant M, respectively, and up to 88% for E231A mutant M. Staining with anti-N or anti-F antibody showed up to 50% and 61% virus-like filament-forming cells, respectively, with the levels being comparable for WT, K50A, and E231A M constructs. Virus-like filaments could not be detected on cells expressing S63A, S63E, N93A, or Y229A M.
M dimerization interface mutations abolish VLP release.
Next, we asked whether M dimerization interface mutations affect VLP release. HEp-2 cells were transfected to express WT F, N, P, and various M constructs; cell lysates (soluble fraction) and the VLPs released into the soluble fraction were analyzed by Western blotting using an anti-RSV antibody (Fig. 6A). All M proteins except the S63E and N93E mutant proteins were expressed in cells at comparable levels (Fig. 6A, bottom), demonstrating that most of the M dimerization interface mutations have no impact on M stability. The undetectable level of the S63E mutant possibly reflects the low level of stability of monomeric M (Fig. 4) and its localization into the nucleus and the IBs (Fig. 5A), which are not soluble under the lysis conditions used. In the presence of F, N, P, and WT M, RSV VLPs were formed (Fig. 6A, top, lane 1, positive control), whereas no VLPs could be detected at 24 h posttransfection in the absence of M (Fig. 6A, top, lane 2, negative control). Cells expressing the M K50A or E231A mutant released VLPs, with the M E231A mutation resulting in slightly increased VLP budding levels compared to those for WT M (Fig. 6A, top; compare lane 9 to lane 1), consistent with more robust filament formation, as determined by staining with anti-M antibody (Fig. 5). In contrast, each M construct containing the S63A, S63E, N93A, N93E, or Y229A mutation prevented VLP release (Fig. 6A, top, lanes 4 to 8, respectively), consistent with a lack of virus-like filaments (Fig. 5). The clear implication is that proper M dimerization and oligomerization are critical for viral budding and release.
FIG 6.
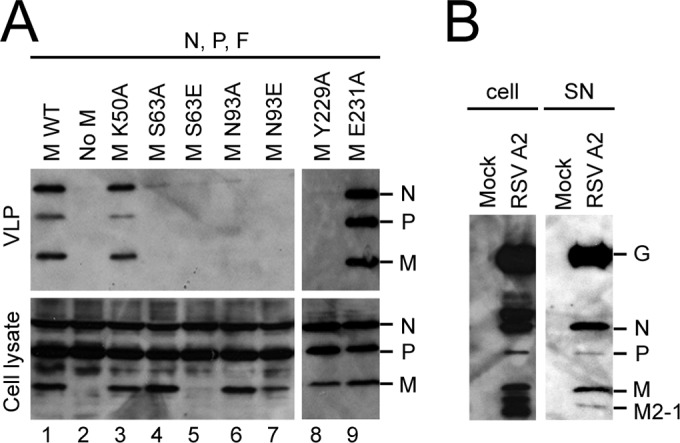
M dimerization mutants abolish VLP release. (A) HEp-2 cells were cotransfected with pcDNA3.1 plasmids expressing RSV P, N, and F plus pcDNA3.1 RSV WT M (lane 1, positive control) or pcDNA3.1 plasmids carrying RSV P, N, and F plus an empty pcDNA3.1 vector (lane 2, negative control) or with the indicated RSV M mutant constructs (lanes 3 to 9). At 48 h posttransfection, VLPs (top) were isolated from the supernatant by centrifugation (1,300 × g, 10 min, 4°C) to get rid of cell debris and pelleting of the clean supernatant through a sucrose cushion. Cell lysates (bottom) were generated using RIPA buffer. VLPs and cell lysates were then subjected to Western analysis using anti-RSV polyclonal antibody. The detected proteins are indicated to the right of the blot. (B) HEp-2 cells were infected without (mock) or with the RSV A2 strain at an MOI of 3. At 24 h postinfection, virus (right) was isolated from the supernatant (SN) after centrifugation (1,300 × g, 10 min, 4°C) to remove cell debris and pelleted through a sucrose cushion. Cell lysates (left) were generated using RIPA buffer. Virus and cell lysates were then subjected to Western analysis using anti-RSV polyclonal antibody. The detected proteins are indicated to the right of the blot.
Since staining of released VLPs formed in the presence of the E231A M mutant suggested a slightly different ratio of N, P, and M proteins, we determined whether the relative abundance of N, P, and M differs between VLPs and released virus. We infected HEp-2 cells with RSV A2 strain at an MOI of 3 and analyzed both cell-associated and supernatant (released) virus by Western blotting using the polyclonal anti-RSV antibody (Fig. 6B). Staining of the infected cell lysate (Fig. 6B, left) detected the G, N, P, M, and, presumably (on the basis of the protein size), M2-1 proteins (20, 34). In both the infected cell lysate and the released virus fraction, G and N were the main proteins detected with the polyclonal anti-RSV antibody, while P and M protein levels were lower. These were comparable to the N, P, and M levels in VLPs formed in the presence of WT or K50A mutant M but differed from those in VLPs formed in the presence of E231A mutant M, where higher levels of P were detected (Fig. 6A).
DISCUSSION
Viral filament formation and particle release are essential steps in the life cycle of RSV that require the assembly of a regular layer of M proteins. Here we show that RSV M forms dimers in solution and in crystals and that proper M dimerization and oligomerization are critical for viral budding and release. Mutations in the dimerization interface both disrupt the formation of virus-like filaments on the surface of transfected cells and abolish the release of VLPs.
Our two crystal structures show that RSV M assembles into dimers much like the M proteins of other paramyxoviruses. The previously solved crystal structure of RSV M was reported to be a monomer, but interface analysis highlights the true dimerization interface to be the strongest in the crystal. It was probably missed in the earlier analysis because it is less extensive than the interfaces in our structures (Table 2) and the interactions do not meet the criteria for a stable quaternary structure. While most identified interface residues are shared among the old structure and our structures, residue Arg 99 was not modeled in the earlier structure and residue Ser 100 was missing atoms. We suspect that the absence of this structural information from the dimerization interface and the attendant reduction in the calculated solvation free energy gain thwarted the recognition of the dimeric nature of the old M structure.
The significance of the potassium ion identified in both of our structures is unclear, but it is unlikely to be an artifact, as the protein was purified in the absence of potassium and one of the crystallization conditions (those for CF 2) did not contain potassium. The recently published structure of hMPV M reported that a calcium ion is part of the dimeric structure and showed that it is important for M stability (24). Our detailed analysis of the potential binding site of Ca2+ in RSV M, based on similarity to hMPV M, ruled out the presence of ordered Ca2+ in both RSV M CFs, but it is possible that in the presence of high concentrations of Ca2+, the site is rearranged in RSV to become competent for Ca2+ binding. Regulation of RSV function by calcium might thus function through M, as has been proposed for hMPV (24). Indeed, a negative effect of calcium depletion on RSV replication has been reported in the past (35). On the other hand, as the potassium ion found in our RSV M structures is located near the dimerization interface and at the interface between the NTD and the CTD, it is tempting to speculate that it has a function similar to that of calcium, but experimental evidence for this is currently lacking.
It is known that the contacts and, thus, the relative angles between dimeric matrix proteins determine the curvature of the matrix protein layer that forms underneath the cell membrane during budding (23). Our two crystal structures show that the dimerization interface itself also shows plasticity. The slightly different opening angle of the dimers, if borne out in vivo, would result in different degrees of curvature in the assembled virus and help explain the structural heterogeneity observed in RSV virions (14, 18). Atomic-resolution structural information is needed to propose a model of the fully assembled virion.
All of the mutants with dimerization interface mutations that we studied in this work exist as dimeric species under some conditions and to some degree, but they vary in their propensity to aggregate over time. The S63E mutant was largely insoluble, and the little that could be purified eluted as a mixture of molecular mass intermediates, dimers, and monomers. Most likely, the S63E mutant aggregates either because it is not folded properly or because severe disruption of the dimerization interface causes it to be monomeric and unstable. The N93A and N93E mutants eluted in two peaks, with the second one corresponding to the WT (dimeric). The first peak corresponds to a molecular mass of 133 kDa, which suggests a tetramer, possibly the result of abortive oligomerization due to unstable dimers caused by the N93 mutation. Although both N93 M mutants formed intermediate species as soluble proteins, the M distribution between the pellet and supernatant fractions differed, and the N93A mutant formed lower levels of higher-order oligomers than the N93E mutant, as was reflected in the enriched pellet fraction of the recombinant M N93E mutant. Based on the lower expression levels in transfected cells, we can speculate that N93E mutant aggregates which are not extracted under the conditions used are also formed in the cell.
We have found no evidence for the existence of stable monomeric M, and it is not clear whether it has any biological function in the infected cell at all. However, we cannot exclude the possibility that RSV M exists as a monomer at early stages of infection, when M is found in the nucleus and in IBs before the assembly process starts. The M S63E mutant as a recombinant protein formed monomers and localized to the IBs and the nucleus in transfected cells. RSV M protein was previously reported to contain two nuclear export signals (NESs), nNES (amino acids 47 to 60) and cNES (amino acids 194 to 206), with cNES being the key signal for nuclear export (36). However, our structural analysis shows that S63 does not directly affect either of the two NES domains. Additional experiments are required to determine whether M dimerization affects NES accessibility and regulates nuclear trafficking. M was shown to be phosphorylated (34), and our earlier studies reported phosphorylation on Thr 205 by the host casein kinase 2 to be critical for RSV M assembly into higher-order oligomers (20). Future studies are required to show whether there is a switch between monomeric and dimeric M and whether phosphorylation regulates this process.
The M E231A mutant as a recombinant protein showed enrichment in the pellet fraction; robust virus-like filament formation, as determined by staining with anti-M antibody; and altered ratios of RSV proteins in VLPs released from transfected cells. Being part of the dimerization interface, the E231 residue could presumably have a positive effect on the dimerization of M and result in more stable dimers, leading to increased M oligomerization, virus-like filament formation, and VLP budding. Future analyses of recombinant RSV containing the M E231A mutation will determine whether this also affects virus release and/or infectivity.
All dimerization interface mutants except the E231A mutant showed differences in M cellular localization and defects in filament formation, but some degree of filament formation in the cell was observed with some mutants (e.g., the Y229A mutant). As M is required for recruiting glycoproteins into the virus (37), we speculate that the failure to extend these filaments is caused by a lack of interaction with other viral proteins, such as F. The C-terminal tail of F (FCT) is necessary for RSV filament formation and budding (12). While direct binding between M and FCT has not been shown, possibly because the interaction is weak and transient in the absence of a lipid membrane or because it requires additional viral and/or cellular factors, an interaction between M and FCT was demonstrated by cellular colocalization and functional studies (17). Therefore, it is possible that some M mutants, while still forming dimers, are unable to bind F and that, conversely, F interacts with and possibly disrupts the dimerization interface of M during budding.
In conclusion, we have shown here using structural, biochemical, and functional analyses that the biologically relevant assembly unit of RSV M is dimeric. This brings RSV in line with other paramyxoviruses, where the switch from M dimers to higher-order oligomers triggers virus filament assembly and virus production. The RSV assembly process is most likely driven by interaction of the four viral proteins M, F, N, and P with each other and possibly with additional cellular factors. This leads to the oligomerization of M and the formation of a curved two-dimensional lattice that is eventually located beneath the membrane and bridges the glycoproteins and the nucleocapsid of the virion. Currently, it is not clear whether some of the M dimerization interface mutants fail to trigger virus filament formation due to their inability to oligomerize or to bind FCT. Future studies are necessary to fully elucidate the specific viral and cellular protein network during RSV assembly, but it is clear that oligomerization of M is a key step in the process of assembly and infectious virus production. This clearer understanding of the mechanism that drives RSV budding should aid with the identification of new structure-based antivirals aimed at destabilizing essential protein-protein interactions.
ACKNOWLEDGMENTS
We thank Peter Openshaw for helpful discussions.
This work was supported by institutional grant (number 707999) from Imperial College London and by an FP7 Marie Curie Career Integration Grant (number 321931) from the EU (to M.B.).
REFERENCES
- 1.Guvenel AK, Chiu C, Openshaw PJ. 2014. Current concepts and progress in RSV vaccine development. Expert Rev Vaccines 13:333–344. doi: 10.1586/14760584.2014.878653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins PL, Graham BS. 2008. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol 82:2040–2055. doi: 10.1128/JVI.01625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia J, Garcia-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247. doi: 10.1006/viro.1993.1366. [DOI] [PubMed] [Google Scholar]
- 4.Carromeu C, Simabuco FM, Tamura RE, Farinha Arcieri LE, Ventura AM. 2007. Intracellular localization of human respiratory syncytial virus L protein. Arch Virol 152:2259–2263. doi: 10.1007/s00705-007-1048-4. [DOI] [PubMed] [Google Scholar]
- 5.Ghildyal R, Mills J, Murray M, Vardaxis N, Meanger J. 2002. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J Gen Virol 83:753–757. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Barreno B, Delgado T, Melero JA. 1996. Identification of protein regions involved in the interaction of human respiratory syncytial virus phosphoprotein and nucleoprotein: significance for nucleocapsid assembly and formation of cytoplasmic inclusions. J Virol 70:801–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Jans DA, Bardin PG, Meanger J, Mills J, Ghildyal R. 2008. Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J Virol 82:8863–8870. doi: 10.1128/JVI.00343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindquist ME, Lifland AW, Utley TJ, Santangelo PJ, Crowe JE Jr. 2010. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J Virol 84:12274–12284. doi: 10.1128/JVI.00260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE Jr, Santangelo PJ. 2012. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts SR, Compans RW, Wertz GW. 1995. Respiratory syncytial virus matures at the apical surfaces of polarized epithelial cells. J Virol 69:2667–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teng MN, Collins PL. 1998. Identification of the respiratory syncytial virus proteins required for formation and passage of helper-dependent infectious particles. J Virol 72:5707–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaikh FY, Cox RG, Lifland AW, Hotard AL, Williams JV, Moore ML, Santangelo PJ, Crowe JE Jr. 2012. A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles. mBio 3(1):e00270-11. doi: 10.1128/mBio.00270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghildyal R, Ho A, Jans DA. 2006. Central role of the respiratory syncytial virus matrix protein in infection. FEMS Microbiol Rev 30:692–705. doi: 10.1111/j.1574-6976.2006.00025.x. [DOI] [PubMed] [Google Scholar]
- 14.Kiss G, Holl JM, Williams GM, Alonas E, Vanover D, Lifland AW, Gudheti M, Guerrero-Ferreira RC, Nair V, Yi H, Graham BS, Santangelo PJ, Wright ER. 2014. Structural analysis of respiratory syncytial virus reveals the position of M2-1 between the matrix protein and the ribonucleoprotein complex. J Virol 88:7602–7617. doi: 10.1128/JVI.00256-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitra R, Baviskar P, Duncan-Decocq RR, Patel D, Oomens AG. 2012. The human respiratory syncytial virus matrix protein is required for maturation of viral filaments. J Virol 86:4432–4443. doi: 10.1128/JVI.06744-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghildyal R, Baulch-Brown C, Mills J, Meanger J. 2003. The matrix protein of human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch Virol 148:1419–1429. [DOI] [PubMed] [Google Scholar]
- 17.Baviskar PS, Hotard AL, Moore ML, Oomens AG. 2013. The respiratory syncytial virus fusion protein targets to the perimeter of inclusion bodies and facilitates filament formation by a cytoplasmic tail-dependent mechanism. J Virol 87:10730–10741. doi: 10.1128/JVI.03086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liljeroos L, Krzyzaniak MA, Helenius A, Butcher SJ. 2013. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc Natl Acad Sci U S A 110:11133–11138. doi: 10.1073/pnas.1309070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPhee HK, Carlisle JL, Beeby A, Money VA, Watson SM, Yeo RP, Sanderson JM. 2011. Influence of lipids on the interfacial disposition of respiratory syncytical virus matrix protein. Langmuir 27:304–311. doi: 10.1021/la104041n. [DOI] [PubMed] [Google Scholar]
- 20.Bajorek M, Caly L, Tran KC, Maertens GN, Tripp RA, Bacharach E, Teng MN, Ghildyal R, Jans DA. 2014. The Thr205 phosphorylation site within respiratory syncytial virus matrix (M) protein modulates M oligomerization and virus production. J Virol 88:6380–6393. doi: 10.1128/JVI.03856-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dessen A, Volchkov V, Dolnik O, Klenk HD, Weissenhorn W. 2000. Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J 19:4228–4236. doi: 10.1093/emboj/19.16.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann P, Lieber D, Meyer S, Dautel P, Kerth A, Kraus I, Garten W, Stubbs MT. 2009. Crystal structure of the Borna disease virus matrix protein (BDV-M) reveals ssRNA binding properties. Proc Natl Acad Sci U S A 106:3710–3715. doi: 10.1073/pnas.0808101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Battisti AJ, Meng G, Winkler DC, McGinnes LW, Plevka P, Steven AC, Morrison TG, Rossmann MG. 2012. Structure and assembly of a paramyxovirus matrix protein. Proc Natl Acad Sci U S A 109:13996–14000. doi: 10.1073/pnas.1210275109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leyrat C, Renner M, Harlos K, Huiskonen JT, Grimes JM. 2014. Structure and self-assembly of the calcium binding matrix protein of human metapneumovirus. Structure 22:136–148. doi: 10.1016/j.str.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Money VA, McPhee HK, Mosely JA, Sanderson JM, Yeo RP. 2009. Surface features of a Mononegavirales matrix protein indicate sites of membrane interaction. Proc Natl Acad Sci U S A 106:4441–4446. doi: 10.1073/pnas.0805740106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr A 64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 27.Foadi J, Aller P, Alguel Y, Cameron A, Axford D, Owen RL, Armour W, Waterman DG, Iwata S, Evans G. 2013. Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 69:1617–1632. doi: 10.1107/S0907444913012274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W. 2010. Xds. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 34.Lambert DM, Hambor J, Diebold M, Galinski B. 1988. Kinetics of synthesis and phosphorylation of respiratory syncytial virus polypeptides. J Gen Virol 69(Pt 2):313–323. doi: 10.1099/0022-1317-69-2-313. [DOI] [PubMed] [Google Scholar]
- 35.Shahrabadi MS, Lee PW. 1988. Calcium requirement for syncytium formation in HEp-2 cells by respiratory syncytial virus. J Clin Microbiol 26:139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghildyal R, Ho A, Dias M, Soegiyono L, Bardin PG, Tran KC, Teng MN, Jans DA. 2009. The respiratory syncytial virus matrix protein possesses a Crm1-mediated nuclear export mechanism. J Virol 83:5353–5362. doi: 10.1128/JVI.02374-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison MS, Sakaguchi T, Schmitt AP. 2010. Paramyxovirus assembly and budding: building particles that transmit infections. Int J Biochem Cell Biol 42:1416–1429. doi: 10.1016/j.biocel.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]