

ABSTRACT
In vitro, infection of polarized human intestinal epithelial cells by coxsackievirus B3 (CVB3) depends on virus interaction with decay-accelerating factor (DAF), a receptor expressed on the apical cell surface. Although mice are highly susceptible to CVB3 infection when virus is delivered by intraperitoneal injection, infection by the enteral route is very inefficient. Murine DAF, unlike human DAF, does not bind virus, and we hypothesized that the absence of an accessible receptor on the intestinal surface is an important barrier to infection by the oral route. We generated transgenic mice that express human DAF specifically on intestinal epithelium and measured their susceptibility to infection by a DAF-binding CVB3 isolate. Human DAF permitted CVB3 to bind to the intestinal surface ex vivo and to infect polarized monolayers of small-intestinal epithelial cells derived from DAF transgenic mice. However, expression of human DAF did not facilitate infection by the enteral route either in immunocompetent animals or in animals deficient in the interferon alpha/beta receptor. These results indicate that the absence of an apical receptor on intestinal epithelium is not the major barrier to infection of mice by the oral route.
IMPORTANCE CVB3 infection of human intestinal epithelial cells depends on DAF at the apical cell surface, and expression of human DAF on murine intestinal epithelial cells permits their infection in vitro. However, expression of human DAF on the intestinal surface of transgenic mice did not facilitate infection by the oral route. Although the role of intestinal DAF in human infection has not been directly examined, these results suggest that DAF is not the critical factor in mice.
INTRODUCTION
Coxsackieviruses belong to the genus Enterovirus of the family Picornaviridae, a group of small, nonenveloped viruses with a single-strand, positive-sense RNA genome (1). Group B coxsackieviruses (CVBs) are important causes of viral myocarditis and meningitis, and they have been proposed to be a possible triggering agent for juvenile diabetes (1).
CVBs, like other enteroviruses, are transmitted by the fecal-oral route and are believed to initiate infection by crossing the intestinal mucosa, which is lined by polarized epithelial cells with distinct apical and basolateral surfaces. In polarized epithelial cells, the primary CVB receptor, the coxsackievirus and adenovirus receptor (CAR) (2–4), is not expressed on the apical cell surface but is instead confined to the basolateral surface, where it functions as part of the intercellular tight junction (5); CAR is thus inaccessible to virus approaching the apical cell surface from the intestinal lumen. Some CVB isolates bind to a second receptor, decay-accelerating factor (DAF) (6–8). Unlike CAR, DAF is highly expressed on the apical surface of polarized epithelial cells, and infection of polarized cells depends on virus attachment to DAF (9). In addition to providing a docking site for virus, DAF mediates a series of intracellular signals that permit virus to move across the cell surface, interact with CAR in the tight junction, and then enter the cell (10).
CVBs, when delivered by intraperitoneal (i.p.) injection, readily infect mice, causing myocarditis (11) and pancreatitis (12); infection of the heart and pancreas is mediated by murine CAR (13, 14). Although infection can be established by the enteral (oral [p.o.]) route in some murine models (15, 16), the gastrointestinal tract has been reported to act as a barrier to infection, particularly beyond the neonatal period (17). Consistent with the idea of an intestinal barrier, our own experience has been that infection by the enteral route requires virus doses much higher (10,000-fold or more) than those needed to infect mice by intraperitoneal injection (unpublished data; see Fig. 2 and 3).
FIG 2.
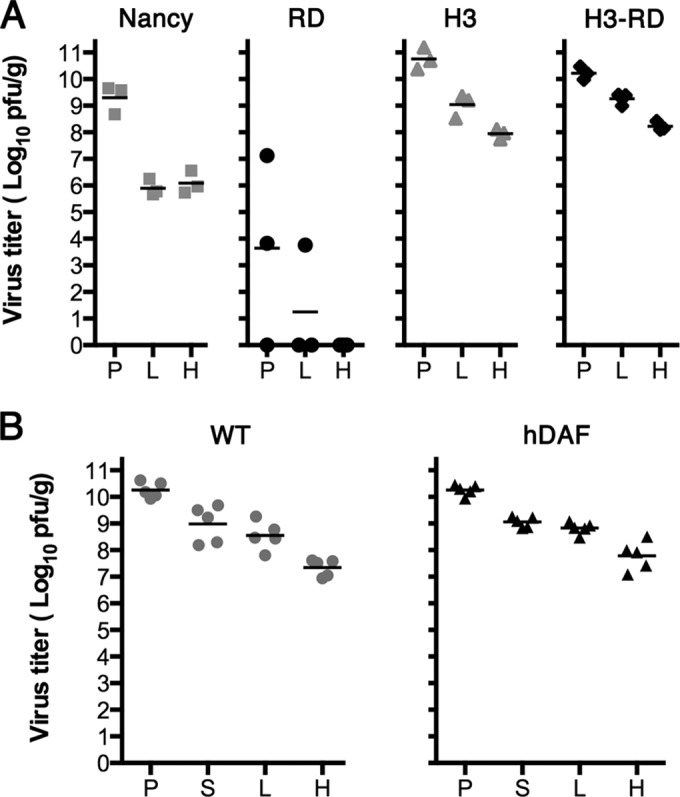
Wild-type and hDAF transgenic mice are equally susceptible to intraperitoneal infection by a DAF-binding virus isolate, CVB3 H3-RD. (A) Wild-type C57BL/6 mice were injected intraperitoneally with 105 PFU of CVB3 Nancy, RD, H3, and H3-RD, and virus titers in tissues were measured after 2 days. Titers obtained in individual animals are shown, with mean titers indicated as bars. (B) Wild-type (WT) and hDAF transgenic mice were injected intraperitoneally with 105 PFU of CVB3 H3-RD, and virus titers in tissues were measured as described above. P, pancreas; S, spleen; L, liver; H, heart.
FIG 3.

Human DAF permits infection of primary murine intestinal cells in vitro, but hDAF transgenic mice resist infection by the oral route, despite virus attachment to hDAF on intestinal epithelium. (A) Primary intestinal epithelial cells from hDAF transgenic or wild-type (WT) mice were cultured as polarized monolayers on Transwell filters and stained with anti-DAF (green) and anti-CAR (red) antibodies, as described in Materials and Methods. DAF is present on the apical surface of monolayers from hDAF but not WT mice. (B) Polarized monolayers were exposed to CVB3 H3-RD (5 PFU per cell) for 1 h at 16°C, and then monolayers were washed and incubated at 37°C to permit infection to proceed. At 8 h, monolayers were stained to detect newly synthesized viral capsid protein VP1; input virus (0 h) was not detectable. (C) Intestinal segments (1 cm long) were treated with medium containing anti-hDAF MAb IF7 or with medium alone (Control) and then exposed to 35S-labeled CVB3 H3-RD, and binding was determined as described in Materials and Methods. Results are indicated as the mean cpm bound + SD for triplicate samples. (D) Wild-type mice (n = 4) and hDAF transgenic mice (n = 5) were exposed to 1 × 109 PFU of CVB3 H3-RD by gavage, and virus titers in tissues were determined at 3 days postinfection. SI, small intestine; P, pancreas; L, liver; H, heart; LI, large intestine.
Several elements are likely to contribute to the barrier, including both physical factors—such as gastric acidity and the impermeability of the intestinal epithelium—and the host's innate immune responses. Another factor may be the lack of an accessible CVB receptor on the intestinal mucosa: although murine CAR binds virus (3, 18), murine DAF does not (19). As is true of CAR expression in human intestinal epithelium, murine CAR is confined to intercellular junctions (20) and is absent from the apical surface of mouse intestinal cells (our unpublished data). To test the idea that access to a receptor on intestinal epithelium might facilitate infection by the enteral route, we generated transgenic mice that express human DAF (hDAF) on intestinal epithelium and measured their susceptibility to infection by a DAF-binding coxsackievirus B3 (CVB3) isolate. Although intestinal epithelial cells isolated from the transgenic mice were susceptible to infection in vitro, expression of human DAF on intestinal epithelium did not facilitate infection by the enteral route.
MATERIALS AND METHODS
Viruses.
Coxsackievirus B3 strains Nancy (21), RD (21), H3 (22), and H3-RD (H3-VP2-N138D) (21) were derived from full-length viral cDNA clones. Nancy and RD were produced by in vitro transcription with T7 RNA polymerase (RiboMax; Promega) and transfection of RNA into HeLa cells using DMRIE-C reagent (Invitrogen). H3 and H3-RD were generated by transfection of HeLa cells directly with plasmid DNA, using Lipofectamine 2000 (Invitrogen). Virus was harvested 4 days later.
Intestine-specific hDAF transgenic mice.
All experiments with animals were performed under protocols approved by the Institutional Animal Care and Use Committee of the Children's Hospital of Philadelphia Research Institute. cDNA encoding full-length human DAF was cloned downstream of a 9-kb regulatory region of the mouse villin gene previously shown to direct gene expression within intestinal epithelium (23) and upstream of the bovine growth hormone polyadenylation signal (Fig. 1A). After linearization with the restriction enzyme AclI, the construct was injected into C57BL/6 murine embryos to generate transgenic mice in the Children's Hospital of Philadelphia Transgenic Mouse Core Facility. hDAF transgenic mice were maintained in a C57BL/6 background.
FIG 1.

hDAF expressed on the epithelium of transgenic mice. (A) hDAF RNA and protein expression. (Top) RNA was measured by real-time PCR to assess hDAF RNA expression in the intestines of wild-type (WT) and transgenic mice (lines 11, 24, and 27). Data are shown as the ratio of the hDAF RNA copy number to the copy number for endogenous murine DAF RNA. (Bottom) Immunoblot to measure hDAF protein expression in intestines of wild-type and transgenic (tg) animals (line 24). (B) Immunofluorescence detection of hDAF in the intestines of wild-type and transgenic animals. In the small intestine (SI), hDAF (green) was detected both within cells (SI-1, arrow) and on the apical cell surface (SI-2, arrowheads), marked by phalloidin staining of the cortical actin layer (red). In the large intestine (LI), hDAF was expressed on the apical cell surface. (C) Immunogold electron microcopy to detect DAF in the small intestine. Gold label (arrows) is associated with the microvilli in transgenic but not wild-type mice. (D) Black-and-white image of DAPI-stained distal small intestine, showing a lymphoid follicle (LF) with overlying epithelium (ep) and adjacent microvilli (mv). The expanded color panel shows follicle-associated epithelium stained for hDAF (green); arrowheads indicate DAF at the apical epithelial cell surface. (E) Small intestine stained for hDAF (green) and an M cell marker (red). Some M cells appear to stain for surface DAF (inset a); however, the red M cell staining is associated with a neighboring cell in a different focal plane (inset b, arrow).
To detect the hDAF transgene, PCR analysis of tail DNA was performed with primers hDAF-transF (5′-GATAGAAGACGGGTAGTACC-3′) and hDAF-transR (5′-GATAGAAGACGGGTAGTACC-3′) and internal control primers specific for the intestinal fatty acid binding protein gene, primers Fabpi-500-F (5′-CCTCCGGAGAGCAGCGATTAAAAGTGTCAG-3′) and Fabpi-500-R (5′-TAGAGCTTTGCCACATCACAGGTCATTCAG-3′) (http://mgc.wustl.edu/Protocols/PCRGenotypingPrimerPairs/tabid/154/Default.aspx). Wild-type animals show a single control band (466 bp), and transgenic animals show a 229-bp hDAF band as well as the control band.
Wild-type and IFNAR-KO mice.
Wild-type C57BL/6 mice were obtained from The Jackson Laboratory. Interferon alpha/beta receptor1 (IFNAR1)-knockout (IFNAR-KO) mice (C57BL/6 background) were provided by Stefania Gallucci of Temple University. Genotyping of IFNAR-KO mice was performed by PCR with primers IFNAR1-F (5′-AAAAGACGAGGCGAAGTGG-3′), IFNAR1-R (5′-CATTCCACGAAGATGTGCTG-3′), and neo-R (5′-AATTCGCCAATGACAAGACGC-3′) as described previously (24). Wild-type animals show a 149-bp band (IFNAR1-F to IFNAR1-R), and knockout animals show a 249-bp band (IFNAR1-F to neo-R).
Immunoblots.
Immunoblots to detect DAF protein were performed with rabbit anti-CD55 (H-319) antibody (Santa Cruz Biotechnology) and horseradish peroxidase-conjugated anti-rabbit IgG (GE Healthcare).
Immunofluorescence.
Tissue samples were fixed overnight in 4% paraformaldehyde (PFA) at 4°C and then dehydrated in 30% sucrose overnight at 4°C; samples were embedded in OCT medium and frozen in a dry ice-ethanol solution, and frozen tissue blocks were used to prepare cryosections 6 to 8 μm thick. Sections were fixed with 4% PFA (10 min at room temperature), washed with phosphate-buffered saline (PBS), and then stained with fluorescein isothiocyanate (FITC)-labeled anti-human CD55 (clone IA10; BD Pharmingen) and AF594 phalloidin (catalog number A12381; Life Technologies) for 1 h at room temperature. Samples were washed, mounted in Vectashield mounting medium containing DAPI (4′,6-diamidino-2-phenylindole), and examined in an Olympus BX51 fluorescence microscope. For detection of M cells, sections were stained with anti-CD55 and with a rat monoclonal antibody that recognizes an M cell-specific α-(1,2)-fucose-containing carbohydrate (25) (clone NKM 16-2-4; MBL International). Sections were examined with a Zeiss LSM 510 confocal microscope, and z-stack images were obtained at 0.3-μm intervals.
Immunogold electron microscopy.
Tissue was prepared for immune detection as follows: high-pressure freezing in an Abra HPM010 machine, freeze substitution in 100% acetone, 0.1% uranyl acetate, 0.1% glutaraldehyde for 3 days at −90°C, embedding in HM20 medium at −50°C, and polymerization under a 360-nm light for 48 h. Sections of HM20-embedded mouse intestinal tissue cut at a 60-nm thickness were labeled with anti-CD55 monoclonal antibody (MAb; MAb GTX113170; Genetex) and then secondarily detected with 15-nm gold-conjugated protein G (Electron Microscopy Science, Hatfield, PA). Images of labeled tissues were collected at 80 keV on an FEI Tecnai 12 microscope equipped with a Gatan US1000 2K charge-coupled-device camera.
Real-time RT-PCR.
Mouse tissues were homogenized, and total RNA was isolated with TRIzol reagent (Invitrogen) and purified using a Qiagen RNeasy minikit. cDNA was produced with murine leukemia virus reverse transcriptase (RT; Promega), and real-time PCR was performed using Power SYBR green PCR master mix (Applied Biosystems) and the following primers: hDAF-transF and hDAF-transR (described above for detection of the hDAF transgene) to measure hDAF RNA; mDAF-F (5′-GTTGCTCCAGAAAGACTGAG-3′) and mDAF-R (5′-ATAATATGCCGGTTGGTATG-3′) to measure mouse DAF (mDAF); and mGapdhF (5′-AAATGGTGAAGGTCGGTGTG-3′) and mGapdhR (5′-CATGTAGACCATGTAGTTGAG-3′) to measure GAPDH (glyceraldehyde-3-phosphate dehydrogenase). For each sample, copy numbers of mDAF and hDAF RNA were determined relative to the copy number for the GAPDH control.
Virus infection.
For parenteral (i.p.) infection, virus (100 μl, diluted in PBS) was injected into the peritoneum of 8- to 10-week-old mice; for enteral (p.o.) infection, an animal feeding needle (20 gauge, 1.5 inch; Fisher) was used to administer virus by gavage (200 μl diluted in PBS buffered with 3% NaHCO3). Preliminary experiments indicated that peak tissue titers were achieved at 2 days after i.p. infection, and we expected that p.o. infection would be somewhat slower. Mice were therefore euthanized at 2 days after i.p. infection and 3 days after p.o. infection, and tissues were collected and stored at −80°C. The tissues were homogenized in PBS, homogenates were centrifuged to remove any debris, and the supernatant containing the virus was subjected to a plaque assay. Results were analyzed by 2-tailed Student's t test for unpaired samples.
Virus binding to mouse intestine ex vivo.
Freshly harvested intestines were fixed in 4% PFA for 1 h at 4°C and then washed with binding buffer (minimal essential medium, 20 mM HEPES buffer). Intestines were cut into 1-cm-long segments, slit lengthwise to expose the mucosal surface, and then incubated in binding buffer with or without anti-DAF MAb IF7 (ascitic fluid diluted 1:50) for 1 h at room temperature. Intestinal segments were then incubated with 35S-labeled CVB3 RD-H3 (40,000 cpm, approximately 550 PFU/cpm) in binding buffer for 1 h at room temperature. After three washes in binding buffer, the intestine-bound radioactivity was measured in a scintillation counter. Results were analyzed by 2-tailed Student's t test for unpaired samples.
Polarized intestinal epithelial cell monolayers from transgenic mice.
Intestinal crypts were isolated from the duodenum as described previously (26) and suspended in a 24-well plate in 50 μl of Matrigel containing recombinant murine epidermal growth factor (EGF; 50 ng/ml; PeproTech), murine Noggin (100 ng/ml; PeproTech), human R-spondin-1 (250 ng/ml); and Y27632 (10 μM; R&D Systems), an inhibitor of Rho-associated protein kinase. Matrigel was overlaid with 50% WRN medium (conditioned medium from L cells expressing Wnt, R-spondin, and Noggin, as described in reference 27). The medium was replaced on the next day with 50% WRN medium supplemented with 10 μM Y27632 (50% WRN-Y) and then daily with 50% WRN-Y to permit growth of stem cell-enriched spheroids. Spheroids were trypsinized, split 1:3, and resuspended in fresh Matrigel approximately every 3 days.
To establish polarized epithelial cell monolayers, spheroids were treated with TrypLE enzyme (Life Technologies) to obtain a single-cell suspension, and cells were plated in 50% WRN-Y on Transwell filters precoated with placental type IV collagen; medium was replaced daily with 50% WRN (without Y27632) until transepithelial electrical resistance (measured with an epithelial volt ohmmeter [World Precision Instruments]) was appreciable (typically, >500 Ω cm2).
To examine DAF and CAR expression, polarized monolayers were fixed with 4% PFA, excised, stained with FITC-labeled anti-DAF IA10, and then permeabilized and stained with CAR-specific rabbit antiserum (28) and AF594-labeled antirabbit secondary antibody. To measure susceptibility to infection, polarized monolayers were exposed to CVB3 H3-RD (5 PFU/cell) for 1 h at 16°C and then cultured for 8 h at 37°C. Infected cells were identified by staining with antibody specific for enteroviral VP1 (NCL-Entero; Leica), as described previously (10).
RESULTS
Transgenic mice expressing hDAF on intestinal epithelium.
We generated mice expressing human DAF under the control of the murine villin promoter, which drives gene expression specifically in intestinal epithelium. Three transgenic lines were established. Real-time RT-PCR analysis, using endogenous murine DAF as the internal standard (Fig. 1A), and immunoblot analysis of intestinal tissue demonstrated that the highest levels of hDAF RNA and protein were expressed in mice of line 24, and all further experiments were performed with these mice. Immunofluorescence staining of sections of small and large intestine with a monoclonal anti-hDAF antibody demonstrated that hDAF was expressed in intestinal epithelial cells (Fig. 1B); only faint background staining was detected in wild-type mice.
Throughout the large intestine, intense staining was evident on the apical cell surface, just above the cortical actin layer that directly underlies the plasma membrane. In the small intestine, two interspersed patterns of staining were observed. In approximately two-thirds of the small-intestinal epithelium, the predominant staining was intracellular, below the cortical actin. In patches comprising approximately one-third of the epithelial cell surface, strong expression was observed on the epithelial cell surface, with hDAF being detectable on intestinal microvilli by immunogold electron microscopy (Fig. 1C). Human DAF was also present on the follicle-associated epithelium overlying lymphoid follicles in the small intestine (Fig. 1D); as expected with the villin promoter, transgenic hDAF was not expressed on lymphoid cells.
Intestinal M cells are specialized epithelial cells that transport antigens and microorganisms from the intestinal lumen to the gut-associated lymphoid tissue (29). Because M cells have been implicated in the transcytosis of viruses across the intestinal mucosa (30–34), we determined whether hDAF was expressed on M cells by costaining with antibody specific for an M cell marker (25). Although there appeared to be some overlap of hDAF and the M cell marker in some sections (Fig. 1E, inset a), careful analysis of serial z-sections revealed that hDAF was expressed on the surface of neighboring cells rather than on the M cells themselves (Fig. 1E, inset b).
DAF-binding CVB3 RD is infectious in the mouse model.
The prototype DAF-binding CVB3 isolate is CVB3 RD, a derivative of CVB3 Nancy isolated after passage in RD rhabdomyosarcoma cells (35). In comparison to CVB3 Nancy, CVB3 RD was reported to be avirulent in mice (35), and we found that it replicated poorly in mice after intraperitoneal injection (Fig. 2A). For experiments with DAF transgenic mice, we wanted to use a DAF-binding isolate that replicated efficiently in mice. We therefore tested CVB3 H3-RD, a DAF-binding derivative of the cardiovirulent isolate CVB3 H3 which differs from CVB3 H3 in only a single capsid residue (21). After inoculation with 105 PFU by the intraperitoneal route, CVB3 H3-RD replicated to high titers in the pancreas, liver, and heart (Fig. 2A). Virus titers were the same in wild-type mice and in DAF transgenic mice (Fig. 2B; P > 0.1), indicating that expression of hDAF on intestinal epithelium did not prevent or enhance virus replication in other tissues.
Polarized epithelial cell monolayers from hDAF transgenic mice are susceptible to infection in vitro.
Because a complex set of DAF-dependent intracellular signals and cytoskeletal rearrangements is required for CVB3 infection of polarized human intestinal epithelium (10, 36), we wanted to be sure that human DAF functions to permit infection of murine cells. To test this, we made use of recent technical advances in the culture and maintenance of nontransformed primary epithelial cells (27, 37, 38). We isolated intestinal stem cells from the duodenum of a DAF transgenic mouse and from a wild-type littermate and used these to produce polarized epithelial cell monolayers. Monolayers from the DAF transgenic (but not the wild-type) mouse expressed human DAF on the apical surface (Fig. 3A); monolayers from both the transgenic and wild-type mice expressed endogenous CAR, primarily within tight junctions and along the lateral cell membrane. When exposed to CVB3 H3-RD, the DAF transgenic monolayer, but not the wild-type monolayer, became infected, as demonstrated by expression of newly synthesized viral capsid protein (seen at 8 h but not at 0 h postinfection; Fig. 3B). These results demonstrate that human DAF expressed on the surface of primary murine intestinal epithelial cells facilitates infection by CVB3.
Intestinal hDAF binds virus ex vivo but is not sufficient for infection by the enteral route.
To determine whether hDAF was expressed on intestine at levels sufficient to permit virus attachment, we incubated sections of small and large intestine with radiolabeled CVB3 H3-RD and measured DAF-specific binding. Significantly more virus bound to sections of both the small and large intestines from transgenic mice than to sections from wild-type mice (Fig. 3C; P < 0.005). Virus attachment to transgenic mouse samples was inhibited by the anti-DAF monoclonal antibody IF7, which blocks CVB3 attachment to human DAF (7), indicating that the binding was DAF dependent (P < 0.001); attachment to wild-type samples was not significantly inhibited by IF7 (P > 0.1). Significantly more virus bound to sections of large intestine than to sections of small intestine (P < 0.005), consistent with the more extensive expression of hDAF on the surface of the large intestine (Fig. 1B). Nonetheless, virus binding to the small intestine was substantial. Given the specific radioactivity of the virus used in this experiment (550 PFU/cpm), approximately 8 × 105 PFU bound per cm of intestinal segment, suggesting that the small intestine (which is 30 to 40 cm long [39]) had the capacity to bind more than 2 × 107 PFU, at least under ex vivo conditions.
However, when hDAF transgenic mice were exposed to CVB3 H3-RD delivered by gavage, little or no systemic infection was noted. Results for hDAF transgenic mice were the same as those obtained for wild-type mice: virus replication at 72 h was detected in only a few animals exposed by gavage to 109 PFU of CVB3 H3-RD (Fig. 3D); virus titers in the small intestine, heart, lung, and pancreas were not significantly different in transgenic versus wild-type mice (P > 0.1 for all comparisons). Similar results were obtained whether or not virus was administered in bicarbonate buffer (data not shown), which has been reported to protect poliovirus from neutralization by gastric acid (40). We also conducted experiments in suckling mice, in which intestinal barriers to infection are reportedly less effective (17); however, we observed no differences in the susceptibility of wild-type and transgenic animals to virus delivered by gavage (not shown). Taken together, the results indicated that although human DAF permits infection of murine polarized intestinal epithelial cells in vitro, and although virus binds to human DAF on the surface of the intestinal epithelium of DAF transgenic mice, the mice were nonetheless not susceptible to oral infection.
Interferon type I signaling inhibits infection by the enteral route, but intestinal hDAF does not enhance susceptibility in interferon receptor-knockout mice.
Work with poliovirus receptor (PVR) transgenic mice has demonstrated that the innate immune system is a powerful barrier to infection by the enteral route; infection does not occur, even when the receptor is expressed on intestinal epithelium (41), unless the type I interferon response has been ablated by deletion of interferon alpha/beta receptor 1 (40). We therefore tested whether expression of hDAF on intestinal epithelium would permit enteral infection in the absence of an effective interferon response by crossing hDAF transgenic mice with mice deficient in IFNAR1 (IFNAR-KO mice). IFNAR-KO mice were much more susceptible to infection than were wild-type mice, with robust virus replication being found in animals exposed to oral doses of 107 and 108 PFU (Fig. 4B and C). However, expression of hDAF in these immunodeficient mice—which we confirmed by immunostaining, as in Fig. 1B (data not shown)—did not enhance their susceptibility to enteral infection across a range of virus doses (Fig. 4A to C). A lower virus titer was detected in the small intestines of hDAF transgenic mice exposed to 108 PFU (P < 0.05), but in no case was significantly more virus detected in DAF transgenic mouse tissues (P > 0.1 for all other comparisons).
FIG 4.
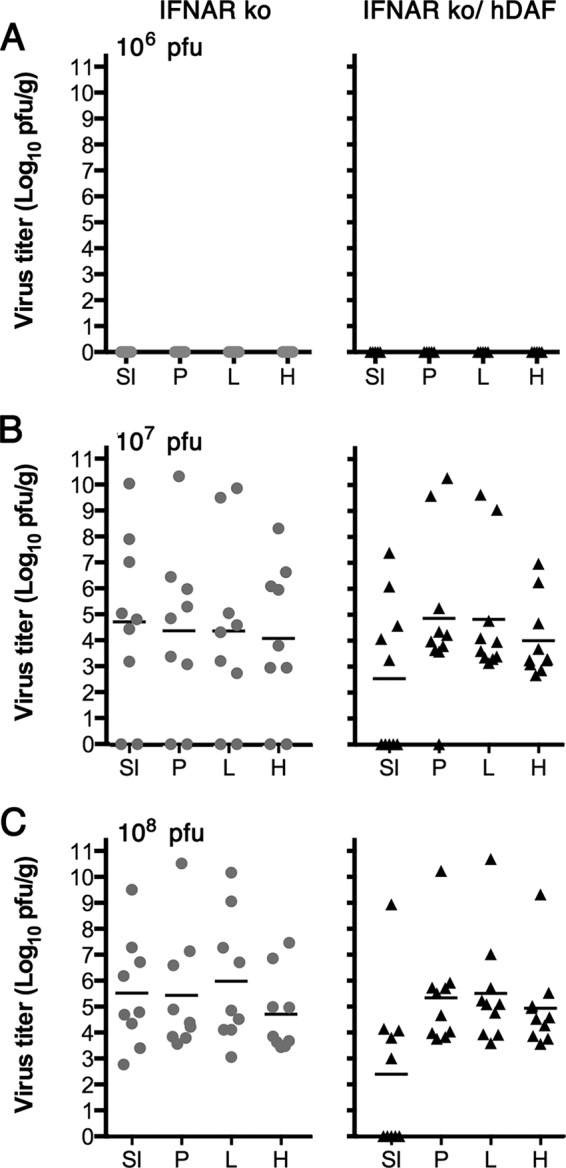
Human DAF on intestinal epithelium does not enhance the susceptibility of IFNAR-KO mice to oral infection. IFNAR-KO mice and IFNAR-KO mice expressing the hDAF transgene were exposed to CVB3 H3-RD by gavage (106 to 108 PFU), and virus titers in tissues were determined at 3 days postinfection. (A) 1 × 106 PFU; (B) 1 × 107 PFU; (C) 1 × 108 PFU. Titers obtained in individual animals are shown, with mean titers indicated as bars. SI, small intestine; P, pancreas; L, liver; H, heart.
DISCUSSION
Because murine DAF does not bind CVB3 (19) and because attachment to DAF on the apical cell surface is important for infection of polarized human epithelial cells in vitro (9, 10), we hypothesized that the lack of an accessible apical receptor on intestinal epithelium might be a major barrier to infection of mice by the oral route, and that expression of human DAF on murine intestinal epithelium would enhance the susceptibility of transgenic mice to oral infection. We found that expression of human DAF permitted virus to bind to the intestinal mucosa of DAF transgenic mice ex vivo and permitted infection in vitro of polarized intestinal epithelium derived from these mice. Nonetheless, expression of human DAF on intestinal epithelium did not permit infection by the enteral route.
We cannot exclude the possibility that, despite the results of in vitro and ex vivo control experiments, the levels of DAF expression on the apical surface were inadequate to permit virus to attach to intestinal epithelium in living animals, in which factors such as the mucus layer or rapid peristalsis may have interfered with virus access to DAF. We do not know why, in some areas of the small intestine, DAF was retained within cells rather than expressed at the apical surface; because apical sorting depends on DAF's glycosylphosphatidylinositol (GPI)-linked membrane anchor (42), one possible explanation is that some small-intestinal cells may lack the machinery required for synthesis and attachment of the GPI moiety, as has been reported for some murine cell lines (43).
In a common view of enterovirus pathogenesis—largely based on studies of poliovirus infection in nonhuman primates—ingested virus first replicates in the alimentary tract, moves to draining lymph nodes, and spreads through the bloodstream (or by neuronal transport) to other organs (44). There has been controversy about whether primary poliovirus replication occurs in mucosal epithelium itself (45) or whether virus moves directly to mucosal lymphoid tissues (46) by transcytosis through overlying M cells (31, 32, 34). In humans, the poliovirus receptor (PVR) is expressed on epithelial cells, on M cells, and in mucosal lymphoid follicles, and it has been suggested that infection by the oral route may involve expression of the receptor on all these cell types within the intestine (47). Although we consider it unlikely, it remains possible that expression of human DAF on M cells or intestinal lymphoid cells might have permitted establishment of an oral model of CVB3 infection.
Studies of poliovirus infection in mouse models indicate that multiple factors influence infection by the enteral route, including PVR expression, innate immunity, mucosal integrity, and interactions with intestinal flora (40, 48, 49). A major difference between the mouse models of poliovirus and CVB3 infection is that, unlike poliovirus, CVB3 has a natural receptor in mice, murine CAR; once CVB3 enters the bloodstream, infection can spread readily in the absence of any transgenic receptor. We found that IFNAR-knockout mice were susceptible to relatively low oral doses of CVB3, whether or not they expressed human DAF on the intestinal epithelium.
Poliovirus replicates efficiently in the intestines of PVR transgenic mice (49), presumably either in epithelium or in mucosal lymphoid tissue, but systemic spread occurs only when IFNAR1 is deleted (40); these results suggest that interferon signaling is a major barrier to viral dissemination from the intestine rather than to local replication. We did not directly trace the passage of CVB3 from the intestinal lumen to target organs, and we do not know specifically where infection is blocked in wild-type and hDAF transgenic mice, or how the block(s) is overcome in IFNAR1-knockout mice. How IFNAR1 controls poliovirus or CVB3 spread from the intestine is not well understood. The simplest explanation is that type I interferon, acting through IFNAR1, suppresses viral replication. However, it is conceivable that indirect effects of IFNAR deletion—changes in intestinal flora, or alterations in mucosal structure or permeability that permit virus access to mouse CAR—may also be important.
The results presented here do not support our initial idea that the presence or absence of a receptor on intestinal epithelium determines whether mice are susceptible to oral infection. They suggest that, at least in mice, infection of polarized epithelium may be neither necessary nor sufficient for infection to occur. However, we believe that the results leave open the question of whether intestinal DAF is important for the pathogenesis of CVB3 infection in humans. CVB3 and other enteroviruses have evolved to infect humans by the enteral route, and although infection in mice depends on IFNAR1 deletion, immunocompetent humans are highly susceptible. Given that the barriers to infection may be very different in humans and mice, we do not believe that the current data exclude a possible role for DAF in the transmission of DAF-binding CVB3 across human intestinal mucosa.
ACKNOWLEDGMENTS
We thank Dewight Williams of the University of Pennsylvania Electron Microscopy Resource Laboratory for performing immunogold electron microscopy. We are grateful to Carolyn Coyne for helpful comments on the manuscript.
This work was supported by grants from the National Institutes of Health (R01AI052281, R01AI072490, R01DKO61931, R01DK068271, T32HD007009, and F30DK103511) and by the Plotkin Endowed Chair at the Children's Hospital of Philadelphia.
REFERENCES
- 1.Pallansch M, Roos R. 2007. Enteroviruses: poliovirus, coxsackieviruses, echoviruses, and newer enteroviruses, p 839–894. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 3.Tomko RP, Xu R, Philipson L. 1997. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci U S A 94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martino TA, Petric M, Weingartl H, Bergelson JM, Opavsky MA, Richardson CD, Modlin JF, Finberg RW, Kain KC, Willis N, Gauntt CJ, Liu PP. 2000. The coxsackie-adenovirus receptor (CAR) is used by reference strains and clinical isolates representing all six serotypes of coxsackievirus group B, and by swine vesicular disease virus. Virology 271:99–108. doi: 10.1006/viro.2000.0324. [DOI] [PubMed] [Google Scholar]
- 5.Cohen CJ, Shieh JT-C, Pickles RJ, Okegawa T, Hsieh J-T, Bergelson JM. 2001. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc Natl Acad Sci U S A 98:15191–15196. doi: 10.1073/pnas.261452898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergelson JM, Modlin JF, Wieland-Alter W, Cunningham JA, Crowell RL, Finberg RW. 1997. Clinical coxsackievirus B isolates differ from laboratory strains in their interaction with two cell surface receptors. J Infect Dis 175:697–700. doi: 10.1093/infdis/175.3.697. [DOI] [PubMed] [Google Scholar]
- 7.Bergelson JM, Mohanty JG, Crowell RL, St John NF, Lublin DM, Finberg RW. 1995. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J Virol 69:1903–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. 1995. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J Virol 69:3873–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shieh JTC, Bergelson JM. 2002. Interaction with decay-accelerating factor facilitates coxsackievirus B infection of polarized epithelial cells. J Virol 76:9474–9480. doi: 10.1128/JVI.76.18.9474-9480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 11.Esfandiarei M, McManus BM. 2008. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol 3:127–155. doi: 10.1146/annurev.pathmechdis.3.121806.151534. [DOI] [PubMed] [Google Scholar]
- 12.Huber S, Ramsingh AI. 2004. Coxsackievirus-induced pancreatitis. Viral Immunol 17:358–369. doi: 10.1089/vim.2004.17.358. [DOI] [PubMed] [Google Scholar]
- 13.Kallewaard NL, Zhang L, Chen JW, Guttenberg M, Sanchez MD, Bergelson JM. 2009. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe 6:91–98. doi: 10.1016/j.chom.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi Y, Chen C, Lisewski U, Wrackmeyer U, Radke M, Westermann D, Sauter M, Tschope C, Poller W, Klingel K, Gotthardt M. 2009. Cardiac deletion of the coxsackievirus-adenovirus receptor abolishes coxsackievirus B3 infection and prevents myocarditis in vivo. J Am Coll Cardiol 53:1219–1226. doi: 10.1016/j.jacc.2008.10.064. [DOI] [PubMed] [Google Scholar]
- 15.Bopegamage S, Borsanyiova M, Vargova A, Petrovicova A, Benkovicova M, Gomolcak P. 2003. Coxsackievirus infection of mice. I. Viral kinetics and histopathological changes in mice experimentally infected with coxsackieviruses B3 and B4 by oral route. Acta Virol 47:245–251. [PubMed] [Google Scholar]
- 16.Bopegamage S, Kovacova J, Vargova A, Motusova J, Petrovicova A, Benkovicova M, Gomolcak P, Bakkers J, van Kuppeveld F, Melchers WJ, Galama JM. 2005. Coxsackie B virus infection of mice: inoculation by the oral route protects the pancreas from damage, but not from infection. J Gen Virol 86:3271–3280. doi: 10.1099/vir.0.81249-0. [DOI] [PubMed] [Google Scholar]
- 17.Loria RM, Shadoff N, Kibrick S, Broitman S. 1976. Maturation of intestinal defenses against peroral infection with group B coxsackievirus in mice. Infect Immun 13:1397–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergelson JM, Krithivas A, Celi L, Droguett G, Horwitz MS, Wickham T, Crowell RL, Finberg RW. 1998. The murine CAR homologue (mCAR) is a receptor for coxsackie B viruses and adenoviruses. J Virol 72:415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiller OB, Goodefellow IG, Evans DJ, Almond JW, Morgan BB. 2000. Echoviruses and coxsackie B viruses that use human decay-accelerating factor (DAF) as a receptor do not bind the rodent analogues of DAF. J Infect Dis 181:340–343. doi: 10.1086/315210. [DOI] [PubMed] [Google Scholar]
- 20.Raschperger E, Thyberg J, Pettersson S, Philipson L, Fuxe J, Pettersson RF. 2006. The coxsackie- and adenovirus receptor (CAR) is an in vivo marker for epithelial tight junctions, with a potential role in regulating permeability and tissue homeostasis. Exp Cell Res 312:1566–1580. doi: 10.1016/j.yexcr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 21.Pan J, Narayanan B, Shah S, Yoder JD, Cifuente JO, Hafenstein S, Bergelson JM. 2011. Single amino acid changes in the virus capsid permit coxsackievirus B3 to bind decay-accelerating factor. J Virol 85:7436–7443. doi: 10.1128/JVI.00503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber S. 1996. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol 70:7811–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinto D, Robine S, Jaisser F, El Marjou FE, Louvard D. 1999. Regulatory sequences of the mouse villin gene that efficiently drive transgenic expression in immature and differentiated epithelial cells of small and large intestines. J Biol Chem 274:6476–6482. doi: 10.1074/jbc.274.10.6476. [DOI] [PubMed] [Google Scholar]
- 24.Roundy KM, Spangrude G, Weis JJ, Weis JH. 2005. Partial rescue of B cells in microphthalmic osteopetrotic marrow by loss of response to type I IFNs. Int Immunol 17:1495–1503. doi: 10.1093/intimm/dxh327. [DOI] [PubMed] [Google Scholar]
- 25.Nochi T, Yuki Y, Matsumura A, Mejima M, Terahara K, Kim DY, Fukuyama S, Iwatsuki-Horimoto K, Kawaoka Y, Kohda T, Kozaki S, Igarashi O, Kiyono H. 2007. A novel M cell-specific carbohydrate-targeted mucosal vaccine effectively induces antigen-specific immune responses. J Exp Med 204:2789–2796. doi: 10.1084/jem.20070607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato T, Clevers H. 2013. Primary mouse small intestinal epithelial cell cultures. Methods Mol Biol 945:319–328. doi: 10.1007/978-1-62703-125-7_19. [DOI] [PubMed] [Google Scholar]
- 27.Miyoshi H, Stappenbeck TS. 2013. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc 8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen CJ, Xiang ZQ, Gao G-P, Ertl HCJ, Wilson JM, Bergelson JM. 2002. Chimpanzee adenovirus CV-68 adapted as a gene delivery vector interacts with the coxsackievirus and adenovirus receptor (CAR). J Gen Virol 83:151–155. [DOI] [PubMed] [Google Scholar]
- 29.Kraehenbuhl J-P, Neutra MR. 2000. Epithelial M cells: differentiation and function. Annu Rev Cell Dev Biol 16:301–332. doi: 10.1146/annurev.cellbio.16.1.301. [DOI] [PubMed] [Google Scholar]
- 30.Wolf JL, Rubin DH, Finberg R, Kauffman RS, Sharpe AH, Trier JS, Fields BN. 1981. Intestinal M cells: a pathway for entry of reovirus into the host. Science 212:471–472. doi: 10.1126/science.6259737. [DOI] [PubMed] [Google Scholar]
- 31.Sicinski P, Rowinski J, Warchol JB, Jarzabek Z, Gut W, Szczygiel B, Bielecki K, Koch G. 1990. Poliovirus type 1 enters the human host through intestinal M cells. Gastroenterology 98:56–58. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi Y, Misumi S, Muneoka A, Masuyama M, Tokado H, Fukuzaki K, Takamune N, Shoji S. 2008. Nonhuman primate intestinal villous M-like cells: an effective poliovirus entry site. Biochem Biophys Res Commun 368:501–507. doi: 10.1016/j.bbrc.2008.01.120. [DOI] [PubMed] [Google Scholar]
- 33.Fotopoulos G, Harari A, Michetti P, Trono D, Pantaleo G, Kraehenbuhl J-P. 2002. Transepithelial transport of HIV-1 by M cells is receptor-mediated. Proc Natl Acad Sci U S A 99:9410–9414. doi: 10.1073/pnas.142586899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouzilou L, Caliot E, Pelletier I, Prévost M-C, Pringault E, Colbere-Garapin F. 2002. Poliovirus transcytosis through M-like cells. J Gen Virol 83:2177–2182. [DOI] [PubMed] [Google Scholar]
- 35.Reagan KJ, Goldberg B, Crowell RL. 1984. Altered receptor specificity of coxsackie B3 after growth in rhabdomyosarcoma cells. J Virol 49:635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coyne CB, Shen L, Turner JR, Bergelson JM. 2007. Coxsackievirus entry from epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2:181–192. doi: 10.1016/j.chom.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 38.Moon C, VanDussen KL, Miyoshi H, Stappenbeck TS. 2014. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunol 7:818–828. doi: 10.1038/mi.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aust G, Kerner C, Gonsior S, Sittig D, Schneider H, Buske P, Scholz M, Dietrich N, Oldenburg S, Karpus ON, Galle J, Amasheh S, Hamann J. 2013. Mice overexpressing CD97 in intestinal epithelial cells provide a unique model for mammalian postnatal intestinal cylindrical growth. Mol Biol Cell 24:2256–2268. doi: 10.1091/mbc.E13-04-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohka S, Igarashi H, Nagata N, Sakai M, Koike S, Nochi T, Kiyono H, Nomoto A. 2007. Establishment of a poliovirus oral infection system in human poliovirus receptor-expressing transgenic mice that are deficient in alpha/beta interferon receptor. J Virol 81:7902–7912. doi: 10.1128/JVI.02675-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang S, Racaniello VR. 1997. Expression of the poliovirus receptor in intestinal epithelial cells is not sufficient to permit poliovirus replication in the mouse gut. J Virol 71:4915–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujita M, Kinoshita T. 2012. GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta 1821:1050–1058. doi: 10.1016/j.bbalip.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Kamitani T, Chang HM, Rollins C, Waneck GL, Yeh ET. 1993. Correction of the class H defect in glycosylphosphatidylinositol anchor biosynthesis in Ltk− cells by a human cDNA clone. J Biol Chem 268:20733–20736. [PubMed] [Google Scholar]
- 44.Racaniello VR. 2006. One hundred years of poliovirus pathogenesis. Virology 344:9–16. doi: 10.1016/j.virol.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 45.Sabin AB. 1956. Pathogenesis of poliomyelitis: reappraisal in the light of new data. Science 123:1151–1157. doi: 10.1126/science.123.3209.1151. [DOI] [PubMed] [Google Scholar]
- 46.Bodian D. 1955. Emerging concept of poliomyelitis infection. Science 122:105–108. doi: 10.1126/science.122.3159.105. [DOI] [PubMed] [Google Scholar]
- 47.Iwasaki A, Welker R, Mueller S, Linehan M, Nomoto A, Wimmer E. 2002. Immunofluorescence analysis of poliovirus receptor expression in Peyer's patches of humans, primates and CD155 transgenic mice: implications for poliovirus infection. J Infect Dis 186:585–592. doi: 10.1086/342682. [DOI] [PubMed] [Google Scholar]
- 48.Kuss SK, Etheredge CA, Pfeiffer JK. 2008. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog 4:e1000082. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. 2011. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334:249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]