

ABSTRACT
The type I interferon (IFN) system, including IFN induction and signaling, is the critical component of the host defense line against viral infection, which, in turn, is also a vulnerable target for viral immune evasion. Severe fever with thrombocytopenia syndrome virus (SFTSV) is an emerging bunyavirus. Previous data have shown that SFTSV can interfere with the early induction of type I IFNs through targeting host kinases TBK1/IKKε. In this study, we demonstrated that SFTSV also can suppress type I IFN-triggered signaling and interferon-stimulated gene (ISG) expression. Interestingly, we observed the significant inhibition of IFN signaling in cells transfected with the plasmids encoding the nonstructural protein (NSs) but not the nucleocapsid protein (NP), indicating the role of NSs as an antagonist of IFN signaling. Furthermore, coimmunoprecipitation (Co-IP) and pulldown assays indicated that NSs interacts with the cellular signal transducer and activator of transcription 2 (STAT2), and the DNA-binding domain of STAT2 may contribute to the NSs-STAT2 interaction. Combined with confocal microscopy analyses, we demonstrated that NSs sequesters STAT2 and STAT1 into viral inclusion bodies (IBs) and impairs IFN-induced STAT2 phosphorylation and nuclear translocation of both STATs, resulting in the inhibition of IFN signaling and ISG expression. SFTSV NSs-mediated hijacking of STATs in IBs represents a novel mechanism of viral suppression of IFN signaling, highlighting the role of viral IBs as the virus-built “jail” sequestering some crucial host factors and interfering with the corresponding cellular processes.
IMPORTANCE SFTSV is an emerging bunyavirus which can cause a severe hemorrhagic fever-like disease with high case fatality rates in humans, posing a serious health threat. However, there are no specific antivirals available, and the pathogenesis and virus-host interactions are largely unclear. Here, we demonstrated that SFTSV can inhibit type I IFN antiviral signaling by the NSs-mediated hijacking of STAT2 and STAT1 into viral IBs, highlighting the interesting role of viral IBs in virus-host interactions as the virus-built jail. Sequestering signaling molecules into IBs represents a novel and, perhaps, also a general mechanism of viral suppression of IFN signaling, the understanding of which may benefit the study of viral pathogenesis and the development of antiviral therapies.
INTRODUCTION
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging infectious disease with a high case fatality rate of up to 30% (1–3). The causative agent is a novel phlebovirus of the Bunyaviridae family, namely, SFTS virus (SFTSV), which was identified first in China and subsequently was found in South Korea (4, 5) and Japan (6). Recently, another phlebovirus genetically closely related to SFTSV was isolated in the United States (7). Emerging bunyavirus infection has become a substantial threat to public health; however, the pathogenesis is largely unknown, and there are no vaccines or specific antivirals available. The SFTSV genome consists of three single-stranded RNA segments. The large (L) and medium (M) segments are of negative polarity and encode the RNA-dependent RNA polymerase and the glycoprotein precursor, respectively, while the small segment (S) encodes the nucleoprotein (NP) and the nonstructural protein (NSs) by an ambisense strategy. Although little is known on SFTSV-host interactions, studies have suggested that the NSs protein is implicated in viral suppression of host antiviral innate immunity; thus, it likely is contributing to viral pathogenesis (8–10).
Antiviral innate immune response is initiated through the recognition of virus infection by cellular pattern recognition receptors (PRRs), such as transmembrane toll-like receptor 3 (TLR3) and cytosolic RIG-I-like receptors RIG-I and MDA5 (11). Upon recognition, PRRs trigger the signaling cascades that lead to the induction of type I interferons (IFNs) through the activation of transcription factors, such as interferon regulatory factors (IRFs) 3 and 7 and NF-κB. The newly synthesized and secreted type I IFNs bind to their receptors on the cell surface and result in the phosphorylation of signal transducer and activator of transcription 2 (STAT2) and 1 (STAT1), transcription factors that are the key components of type I IFN signaling pathway, by Janus kinases (JAKs). The phosphorylated STATs then heterodimerize and assemble with a third protein, IRF9, to form the heterotrimeric interferon-stimulated gene factor 3 (ISGF3). As the activated transcription factor complex, ISGF3 rapidly translocates to the cell nucleus and binds to the IFN-stimulated response element (ISRE), a crucial regulatory element in the IFN-stimulated gene (ISG) promoters, leading to the induction of more than 300 ISGs and establishment of the host antiviral state (12–15). The induction of some ISGs (such as IRF7) in turn can contribute to the expression of more type I IFNs, leading to the amplification of IFN response (16).
Evidence for the critical role of type I IFN response in preventing infection lies in the fact that many viruses have evolved various strategies to subvert the host defense line by counteracting the early IFN induction or subsequent IFN signaling (17–19). In cells infected with bunyaviruses, it is well known that the early IFN induction phase often is targeted by multiple mechanisms (20–22), while little is known on bunyavirus-mediated suppression of IFN signaling. Previous studies by us and others have suggested that SFTSV can target host kinases TBK1/IKKε to inhibit IFN induction (8–10); however, it is unknown whether SFTSV interferes with IFN signaling.
In the present study, we demonstrated that indeed, SFTSV can suppress type I IFN signaling. Moreover, the expression of NSs protein alone by transient transfection can inhibit IFN-induced activation of ISRE and expression of ISGs, indicating the role of NSs as an IFN signaling antagonist. Furthermore, NSs interacts with STAT2 and relocalizes STAT2 and STAT1 into viral inclusion bodies (IBs), blocking STAT2 phosphorylation and nuclear translocation of both STATs and the establishment of the host antiviral state. These findings demonstrate a clear instance for bunyavirus-mediated inhibition of IFN signaling by a novel viral IB-associated mechanism and highlight the versatile roles of NSs and IBs in virus-host interactions.
MATERIALS AND METHODS
Cells and virus.
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; GIBCO) at 37°C in a 5% CO2 atmosphere. HEK293, HepG2, and Vero cells were maintained in DMEM supplemented with 10% newborn calf serum at 37°C in a 5% CO2 atmosphere. SFTSV WCH-2011/HN/China/isolate97 (23) was expanded in Vero or HEK293 cells in a biosafety level 3 laboratory.
Plasmids.
The firefly luciferase reporter plasmid for the ISRE promoter and the Renilla luciferase control plasmid for the constitutively active herpes simplex virus (HSV)-thymidine kinase (TK) promoter (pRL-TK) were kindly supplied by Hong-Bing Shu (Wuhan University, China). Open reading frames (ORFs) encoding NP or NSs were amplified by reverse transcription-PCR (RT-PCR) from SFTSV genomic RNA and cloned into expression vector pCAGGSP7 with or without an S tag or hemagglutinin (HA) tag as described previously (8). Expression plasmids for the full-length or truncated STAT2 proteins C-terminally fused with an HA tag were constructed by standard molecular biology techniques.
Antibodies and reagents.
Rabbit polyclonal antibodies to STAT2 or phosphotyrosine 690-STAT2 (p-STAT2) and monoclonal antibodies to STAT1 or phosphotyrosine 701-STAT1 (p-STAT1) were purchased from Santa Cruz Biotechnology (SCBT). The rabbit polyclonal antibody to S tag was from Abcam. Mouse monoclonal antibodies (MAbs) against HA tag or β-actin were from Beyotime Institute of Biotechnology (Beyotime). Rabbit and mouse anti-NSs antisera were raised against the NSs protein produced in Escherichia coli. Secondary antibodies were goat anti-mouse IgG-fluorescein isothiocyanate (FITC) (Proteintech) and goat anti-rabbit IgG-Cy5 (Abcam). Human recombinant IFN-α2b was purchased from PBL Biomedical Laboratories. In some experiments, recombinant human IFN-β (Peprotech Inc.) also was used for treatments, and results similar to those of IFN-α treatment could be obtained; therefore, they are not shown.
Reporter gene assay.
HEK293 cells cultured in 24-well plates were cotransfected with 100 ng ISRE reporter plasmid, 20 ng pRL-TK plasmid, and the indicated amounts of expression plasmids for NSs or NP per well using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The total amount of DNA was kept constant through the addition of empty plasmids (vectors). Thirty-six hours posttransfection, cells were treated with human IFN-α or were left untreated for 18 h. Luciferase activities then were measured with a dual-luciferase reporter (DLR) assay kit (Promega). For the reporter gene assays with SFTSV infection, cells cotransfected with the ISRE reporter plasmid and pRL-TK were mock infected or infected with SFTSV at 12 h posttransfection. One day postinfection, cells were treated with IFN-α or were left untreated for 18 h before measurements of luciferase activities. TK promoter-driven Renilla luciferase expression was not affected by SFTSV infection, viral protein expression, or IFN treatments; thus, it is a valid control in our DLR experiments. For data presentation, firefly luciferase activities were normalized to Renilla luciferase activities to show the relative luciferase activities, or fold activation over the untreated control was further calculated.
Quantitative real-time PCR.
Total RNA was isolated from HEK293 cells using a total RNA purification kit (GeneMark, Taiwan). RT of RNA was performed using a cDNA reverse transcription kit (Promega). Quantitative real-time PCR was performed with an SYBR green real-time PCR kit (Toyobo), and the PCR primers used in this study are listed in Table 1. Relative mRNA levels were calculated by the 2−ΔΔCT method with GAPDH mRNA as an internal control and were shown as relative fold change by normalizing to the untreated-control samples.
TABLE 1.
List of primers for real-time quantitative PCR
| Primer namea | Primer sequence (5′ to 3′) |
|---|---|
| OAS1-F | CATCCGCCTAGTCAAGCACTG |
| OAS1-R | CACCACCCAAGTTTCCTGTAG |
| MxA-F | CTACACACCGTGACGGATATG |
| MxA-R | CGAGCTGGATTGGAAAGCCC |
| ISG15-F | CACCGTGTTCATGAATCTGC |
| ISG15-R | CTTTATTTCCGGCCCTTGAT |
| ISG56-F | CCTCCTTGGGTTCGTCTACA |
| ISG56-R | GGCTGATATCTGGGTGCCTA |
| GAPDH-F | ACCACAGTCCATGCCATCAC |
| GAPDH-R | TCCACCACCCTGTTGCTGTA |
F, forward primer; R, reverse primer.
Protein-protein interaction analysis.
For the identification of transient-expression protein interactions, S-tag pulldown assays were used as previously described (8). Briefly, transfected cells were lysed in the lysis buffer (25 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) supplemented with a cocktail protease inhibitor (Roche). Supernatants of the cell lysates then were mixed with the S-protein agarose slurry (Merck Novagen) by rotating at 4°C for 4 h. After washing extensively with the lysis buffer and phosphate-buffered saline (PBS), the beads were treated in 1× SDS sample buffer and boiled for 5 min, followed by SDS-PAGE and Western blot analyses.
For coimmunoprecipitation (Co-IP) assays, mock- or SFTSV-infected HEK293 cells (∼5 × 107) were lysed in the lysis buffer as described above. The cell lysate supernatants first were pretreated with preimmune serum and protein A/G-Plus agarose (SCBT). After centrifugation, the pretreated supernatants then were incubated with anti-NSs antiserum at 4°C for 1 h and mixed with protein A/G Plus-agarose at 4°C overnight. After extensive washes, immunoprecipitates were subjected to SDS-PAGE and Western blot analyses.
Immunofluorescence and confocal microscopy.
After fixation with 4% paraformaldehyde-PBS, transfected or infected cells were incubated in 0.5% Triton X-100-PBS for permeabilization and blocked with 2.5% bovine serum albumin (BSA) (Biosharp) and 2.5% normal goat serum (Jackson ImmunoResearch) in PBS. Cells then were treated with primary antibodies for 1 h at room temperature or overnight at 4°C and stained with secondary antibodies for 1 h at room temperature. For visualization of nuclei, cells were incubated with Hoechst 33258 (Beyotime) for 3 min at room temperature. Image acquisition was performed with a Nikon Ti confocal microscope and Volocity software (PerkinElmer).
Western blot analysis.
Protein samples were subjected to SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). After blocking with 5% BSA in Tris-buffered saline-Tween 20 (TBS-T), the membrane was probed with primary antibodies and then horseradish peroxidase-conjugated secondary antibodies (Proteintech) in 1% BSA–TBS-T. Protein bands were detected by an enhanced chemiluminescence (ECL) kit (Thermo Fisher Scientific).
Statistical analysis.
A two-tailed Student's t test was used to evaluate the data by IBM SPSS for Windows, and a P value of <0.05 was considered statistically significant.
RESULTS
SFTSV infection suppresses type I IFN signaling.
Type I IFN signaling results in the activation of the ISRE promoter and mounts the transcription of ISGs. To investigate whether SFTSV can interfere with the IFN signaling, we examined the effect of SFTSV infection on IFN-triggered ISRE activation by dual-luciferase reporter (DLR) gene assays. HEK293 cells were cotransfected with a reporter plasmid for ISRE promoter-driven expression of firefly luciferase and a control plasmid for TK promoter-driven expression of Renilla luciferase. Twelve hours after transfection, cells were mock infected or infected with SFTSV at a multiplicity of infection (MOI) of 5, treated with IFN-α or left untreated for 18 h at 24 h postinfection (p.i.), and lysed for measuring luciferase activities. As shown in Fig. 1A, IFN-α treatment induced a strong (∼89-fold) ISRE activation in mock-infected cells; however, the activation was largely blocked in SFTSV-infected cells (Fig. 1A), suggesting that SFTSV can interfere with IFN signaling. Meanwhile, TK promoter-driven Renilla luciferase expression was not noticeably affected by SFTSV infection (Fig. 1B), indicating the specific inhibition of IFN-α-triggered ISRE activation by SFTSV.
FIG 1.

SFTSV infection suppresses type I IFN signaling. (A) SFTSV infection inhibits IFN-α-triggered activation of the ISRE promoter. HEK293 cells were cotransfected with the ISRE reporter plasmid and the Renilla luciferase control plasmid (pRL-TK). Twelve hours posttransfection, cells were mock infected or infected with SFTSV at an MOI of 5 for 24 h and treated with IFN-α (1,000 U/ml) or left untreated for 18 h before the measurement of luciferase activities. Relative luciferase activity (Rel. Luc. Act.) is shown on the left, and fold activation (over untreated groups) of ISRE by IFN-α is shown on the right. (B) TK promoter-driven Renilla luciferase activities measured in panel A are shown. (C) SFTSV infection suppresses IFN-induced gene expression. HEK293 cells were mock infected or infected with SFTSV for 24 h and treated with IFN-α (200 U/ml) or left untreated for 10 h. Expression of ISGs, including OAS1, MxA, ISG15, and ISG56, was measured by real-time quantitative PCR. Data are presented as means ± standard deviations (SD) (n = 3).
Furthermore, we assessed IFN-α-induced expression of several ISGs, including oligoadenylate synthetase 1 (OAS1), myxovirus-resistance A (MxA), ISG15, and ISG56, by real-time quantitative PCR analyses. As expected, SFTSV infection greatly suppresses the induction of all examined ISGs (Fig. 1C), further confirming the inhibition of IFN signaling by SFTSV.
The NSs protein of SFTSV is an antagonist of type I IFN signaling.
Bunyavirus NSs proteins are considered to be multifunctional; however, as nonstructural proteins, their expression usually can occur only after several hours with viral infection (24). Here, we analyzed the kinetics of SFTSV NSs expression with time in HEK293 cells. In immunofluorescence assays (IFA), although a weak expression of NSs occasionally could be seen at 4 h p.i., significant expression was not able to be observed until 6 h p.i. in most cells (Fig. 2A). Accordingly, only after 6 h with SFTSV infection could NSs expression be detected by Western blotting (Fig. 2B). Thus, there likely is a lack of NSs functioning at the very early phase of SFTSV infection, although NSs has the capacity to interfere with IFN induction by targeting TBK1/IKKε (8–10).
FIG 2.

NSs functions as an antagonist of type I IFN signaling. (A and B) The kinetics of NSs expression during SFTSV infection. HEK293 cells were infected with SFTSV at an MOI of 10. At the indicated times p.i., they were either fixed for immunofluorescence assays (IFA) with the rabbit anti-NSs antiserum (A) or harvested for Western blot (WB) analysis using antibodies against the indicated proteins. β-Actin was detected as the sample loading control. (C) NSs expression inhibits IFN-α-induced activation of the ISRE promoter. HEK293 cells were cotransfected with the reporter plasmids, along with plasmids expressing the indicated viral proteins, or the control plasmid (vector). At 36 h posttransfection, cells were treated with IFN-α (200 U/ml) or left untreated for 18 h and then were harvested for measuring luciferase activities or monitoring protein expression through WB analysis. (D) NSs expression suppresses IFN-α-induced expression of ISGs. HEK293 cells cultured in 24-well plates were transfected with the control plasmid (vector) or the NSs expression plasmid (800 ng per well) and treated with IFN-α (200 u/ml) or left untreated for 10 h. Expression of ISGs was measured by real-time PCR. Graphs show means ± SD (n = 3). IFN-elicited transcriptional induction of all four ISGs was inhibited in cells transfected with the NSs expression plasmid compared with those transfected with the control plasmid (*, P < 0.05; Student's t test).
For a more profound impact on host immune responses, we hypothesized that SFTSV NSs also are involved in the inhibition of IFN signaling. To test the hypothesis, we examined the effects of NSs expression upon IFN-induced activation of the ISRE promoter. As shown in the DLR assay, the activation of the ISRE promoter by IFN-α was remarkably reduced in cells transfected with the NSs expression plasmid, even at a very low transfection dosage, indicating that NSs is a robust antagonist of IFN signaling (Fig. 2C, left). In contrast, NP expression did not display such an inhibitory activity (Fig. 2C, right).
IFN-induced expression of ISGs also was compared in cells transfected with the NSs expression plasmid versus the empty plasmid (vector) by real-time quantitative PCR. In the absence of IFN-α treatment, NSs expression did not influence the expression of ISGs analyzed (Fig. 2D). Addition of IFN-α resulted in the notable induction of ISGs in the absence of NSs expression; however, the induction was significantly weaker in cells transfected with the NSs expression plasmid (Fig. 2D). These results further suggest that NSs contributes to the viral antagonism of IFN signaling. Additionally, it should be pointed out that the inhibitory capacity of NSs likely was underestimated by examination with transient expression experiments, because a portion of the cells were left untransfected.
NSs interacts with STAT2.
For viral suppression of IFN signaling, STAT1 and STAT2 often are targeted (17, 25). To unravel the mechanism by which NSs interferes with the IFN signaling, we investigated whether NSs targets signaling proteins such as STATs in the type I IFN signaling pathway. SFTSV-infected HEK293 cells were lysed for coimmunoprecipitation assays using the rabbit anti-NSs antiserum. Interestingly, endogenous STAT2, but not other signaling molecules, including STAT1, was detected abundantly in the NSs coimmunoprecipitates (Fig. 3A). The interaction of NSs with STAT2 also was examined by S-tag pulldown assay with transient expression proteins in HEK293 cells (Fig. 3B). Both endogenous and overexpressed STAT2 could be detected in the coprecipitates of NSs fused with an S tag (NSs-S) (Fig. 3B), further confirming the interaction of viral NSs with cellular STAT2 and also indicating that NSs targeting of STAT2 is independent of SFTSV infection and other viral protein expression.
FIG 3.

NSs interacts with STAT2. (A) Identification of the NSs-STAT2 interaction in the context of SFTSV infection by coimmunoprecipitation (Co-IP) assays. Lysate supernatants of mock- or SFTSV-infected HEK293 cells first were pretreated with preimmune serum as described in Materials and Methods and then used for Co-IP assay with the rabbit anti-NSs antiserum. Immunoprecipitates (IP) and cell lysates (lysate input) were subjected to WB analysis using the indicated antibodies. (B) Identification of the interaction between transient-expression NSs and overexpression or endogenous STAT2 by S-tag pulldown (S-pulldown) assay. HEK293 cells were cotransfected with the plasmids expressing the indicated proteins, NSs fused with S tag (NSs-S) or STAT2, or control plasmids (vector). At 48 h posttransfection, cells were lysed for S-pulldown assays. S-pulldown products and cell lysates were analyzed by WB with the indicated antibodies.
Mapping of the domain(s) of STAT2 required for the NSs-STAT2 interaction.
To identify the domain(s) of STAT2 required for the interaction with NSs, a series of plasmids encoding N-terminal-truncated STAT2 proteins fused with an HA tag (Fig. 4A) were generated. Full-length or truncated STAT2 expression constructs, along with the NSs-S plasmid or the corresponding control plasmids, were cotransfected into HEK293T cells, and protein interactions were examined by S-tag pulldown assays. As indicated in Fig. 4B, deletions of the N-terminal domain (NTD) and coiled-coil domain (CCD) appear to result in no or only slight influence on coprecipitation of the truncated proteins with NSs-S, whereas further deletion of the DNA-binding domain (DBD) leads to the greatest impairment, suggesting that the N-terminal DBD is required for the efficient interaction of STAT2 with NSs. Furthermore, we investigated whether an interaction between DBD and NSs can occur. As shown in the pulldown assay, DBD indeed could be detected in NSs-S coprecipitates (Fig. 4C), indicating that DBD is a NSs-binding domain of STAT2.
FIG 4.

DNA-binding domain of STAT2 is required for the efficient STAT2-NSs interaction. (A) Linear representation of the organization of full-length or N-terminal-truncated STAT2 proteins C-terminally fused with HA tag. NTD, N-terminal domain; CCD, coiled-coil domain; DBD, DNA-binding domain; LD, linker domain; SH2, Src-homology domain-2; TAD, transactivation domain; pY, tyrosine (690) phosphorylation site. The N-terminal-truncated STAT2 proteins were named T1, T2, T3, T4, and T5, as indicated. (B) Analysis of the interactions between NSs and full-length or N-terminal-truncated STAT2. HEK293T cells were cotransfected with the NSs-S expression plasmid, along with the plasmids encoding full-length or N-terminal-truncated STAT2, or the control plasmids (vector). At 48 h posttransfection, protein interactions were examined by S-pulldown assays. Cell lysates (input) and S-pulldown products were subjected to WB analysis using the indicated antibodies. (C) Identification of the NSs-DBD interaction. HEK293T cells were cotransfected with the NSs-S expression plasmid, together with a plasmid encoding the DBD of STAT2 or the corresponding control plasmid (−). Protein interactions then were analyzed as described for panel B.
NSs efficiently captures STAT2 and STAT1 into viral IBs.
NSs is localized to cytoplasmic inclusion bodies (IBs) induced by the expression of NSs itself (8). Since NSs strongly interacts with STAT2, we considered that NSs expression influences STAT2 subcellular localization. To validate the consideration, HepG2 cells transfected with the plasmid encoding NSs were fixed to visualize the localization of NSs and endogenous STATs. STAT2 appeared to locate diffusely in cytoplasm in the absence of NSs expression, while it was highly efficiently relocalized into NSs IBs in cells expressing NSs (Fig. 5A, upper), suggesting that NSs can powerfully capture STAT2 into IBs by the interaction with STAT2. Interestingly, a similar relocation of STAT1 into IBs also was seen in NSs-expressing cells (Fig. 5A, lower), although there likely is no direct interaction between NSs and STAT1 (Fig. 3A). Furthermore, similar results could be obtained in the context of SFTSV infection as well (Fig. 5B).
FIG 5.

NSs captures STAT2 and STAT1 into viral inclusion bodies (IBs). HepG2 cells were transfected with the plasmid expressing C-terminally HA-tagged NSs (A) or infected with SFTSV at an MOI of 1 (B). Twenty-four h later, cells were fixed to visualize the subcellular localization of NSs (green) and endogenous STAT2 or STAT1 (red) by IFA. NSs was stained with the anti-HA antibody for transfected cells (A) or the anti-NSs antiserum for SFTSV-infected cells (B), respectively, while STATs were visualized with the corresponding anti-STAT antibodies. Nuclei stained with Hoechst are shown in blue.
NSs-mediated hijacking of STAT2 and STAT1 in IBs blocks type I IFN-triggered nuclear translocation of the transcription factors.
We next investigated whether STATs captured into IBs still can translocate into nuclei in response to type I IFN stimulation. HepG2 cells transfected with plasmids expressing NSs or NP (as a control) were treated with IFN-α or were left untreated for 30 min and fixed to analyze the endogenous STAT localization. In the absence of NSs expression, IFN-α induced STAT2 accumulation into nuclei; however, the nuclear accumulation was remarkably blocked in NSs-expressing cells, and STAT2 was still sequestered in NSs IBs (Fig. 6A, upper). In contrast, NP expression exhibited no appreciable influence on the nuclear translocation of the transcription factor (Fig. 6A, middle), consistent with the observation that NP did not impair IFN signaling (Fig. 2C, left). Similarly, STAT1 nuclear accumulation also was seen to be inhibited in the presence of NSs (Fig. 6A, lower). Furthermore, blockage of STAT nuclear translocation was observed in SFTSV-infected cells as well (Fig. 6B), and similar results could be obtained with IFN-β treatments (data not shown). Collectively, these results suggest that NSs hijacking of STATs is irreversible upon type I IFN stimulation; hence, it suppresses type I IFN-induced nuclear translocation of the transcription factors.
FIG 6.

NSs inhibits the nuclear translocation of STAT2 and STAT1. (A) HepG2 cells were transfected with the plasmids expressing C-terminally HA-tagged NSs or NP, treated with IFN-α (2,000 U/ml) for 30 min, and then fixed to visualize the subcellular localization of viral proteins (green) and endogenous STATs (red) by IFA with the anti-HA or corresponding anti-STAT antibodies, respectively. (B) HepG2 cells infected with SFTSV were treated with IFN-α as described for panel A, and IFA was performed to visualize NSs (green) and STATs (red) with the anti-NSs antiserum and corresponding anti-STAT antibodies, respectively. Nuclei were stained with Hoechst and are shown in blue.
NSs suppresses type I IFN-induced STAT2 phosphorylation.
Tyrosine phosphorylation of STATs is a key activation event before their nuclear translocation during type I IFN signaling. To examined whether NSs also impairs the transcription factor activation, we next analyzed type I IFN-induced phosphorylation of STATs in cells expressing NSs. HEK293 cells transfected with the control plasmid (vector) or the plasmid encoding NSs were treated with IFN-α or left untreated for 30 min, and then cell lysates were subjected to Western blot analyses. Levels of total STAT1 and STAT2 proteins were stable irrespective of NSs expression, while only following the addition of IFN-α were phosphorylated STAT2 and STAT1 able to be detected (Fig. 7A). Upon IFN-α stimulation, no apparent inhibition of STAT1 phosphorylation was observed in cells transfected with the NSs expression plasmid compared with cells transfected with the vector plasmid, whereas STAT2 phosphorylation was significantly diminished in the presence of NSs expression (Fig. 7A), suggesting that NSs specifically inhibits STAT2 phosphorylation. Importantly, specific suppression of type I IFN-induced STAT2 phosphorylation also was observed in cells infected with SFTSV (Fig. 7B). Additionally, similar results could be obtained when cells were treated with IFN-β (data not shown). Taken together, these observations indicate that NSs can block the upstream activation event of STAT2 as well.
FIG 7.
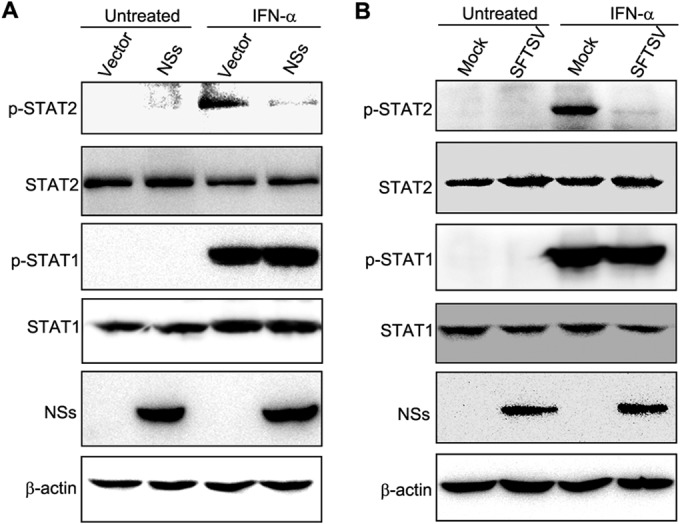
NSs inhibits STAT2 phosphorylation. (A) HEK293 cells transfected with the NSs expression plasmid or the vector were treated with IFN-α (1,000 U/ml) or left untreated for 30 min and were harvested to evaluate the levels of phosphorylated or total STATs by WB using the antibodies against the indicated proteins. (B) HEK293 cells were infected with SFTSV at an MOI of 5. At 24 h p.i., cells were treated with IFN-α (1,000 U/ml) or left untreated for 30 min and harvested for WB as described for panel A.
DISCUSSION
The type I IFN system is the key component of host innate immunity; moreover, it promotes the subsequent development of adaptive immunity (26), limiting viral infection. Thus, many viruses have evolved to be adept in targeting this system, benefiting their replication and spread. Our previous data have suggested that SFTSV can suppress IFN induction, i.e., the early phase of IFN response, through sequestration of host kinases TBK1/IKKε in viral IBs by NSs (8). In the present study, we demonstrated that SFTSV can disrupt IFN signaling through sequestrating STAT2 and STAT1 into IBs also by the NSs protein, giving an overall view for SFTSV-mediated suppression of the type I IFN system (Fig. 8).
FIG 8.

Model for the overall disruption of IFN responses by SFTSV through NSs hijacking of signaling proteins in IBs. Type I IFN response is initiated by the recognition of viral infection by PRRs, followed by the recruitment and activation of crucial kinases, such as TBK1/IKKε. The kinases then activate transcription factors, including IRF3, leading to the IFN induction. Secreted IFNs bind to their receptors (IFNARs) on the cell surface, initiating JAK-STAT signaling and mounting expression of antiviral ISGs. Both TBK1/IKKε in the IFN induction pathway and also STAT2/STAT1 in IFN signaling are sequestrated in viral IBs by SFTSV, resulting in an overall disruption of IFN responses. PM, plasma membrane.
In this study, we identified STAT2 as a target of IFN signaling suppression by SFTSV in human cells. STAT2 is the most highly divergent member of the STAT family in the protein sequences, while other STATs are remarkably conserved among mammalian species, including human and mouse (27). Intriguingly, STAT2 appears to determine the host range across species for some viruses (27–29). For instance, Dengue virus NS5 protein specifically targets human STAT2 but not the counterpart of mouse; hence, mouse STAT2 likely confers restriction on the replication of the virus (29). To date, Homo sapiens is the only species observed to become severely ill with SFTSV natural infection. It will be interesting to examine whether the NSs targeting of STAT2 also is species specific, given that the capacity of repressing IFN response may determine the host range and clinical outcome of viral infection. Additionally, overcoming the restriction through STAT2 humanization of experimental animals may contribute to the development of an immunocompetent animal model of SFTSV infection. In addition, we found that STAT1 also is significantly sequestered into NSs IBs, although we did not detect any notable NSs-STAT1 interaction comparable with that between NSs and STAT2. A similar observation that IRF3 could be arrested into NSs IBs despite no direct interaction of IRF3 with NSs has been reported previously (8, 10). A simple explanation is that the sequestration of these cellular proteins in SFTSV IBs is mediated indirectly by some potential interactions, such as STAT2-STAT1 interaction and TBK1-IRF3 interaction (10).
To counteract the IFN signaling, many viruses have evolved to target some signaling proteins by various strategies, such as suppressing their phosphorylation and activation, blocking their nuclear translocation, or inducing their degradation (17–19). In the present study, we demonstrated that SFTSV disrupts this antiviral signaling by sequestering STATs into viral IBs and suppresses the actions of the transcription factors, highlighting a novel IB-associated mechanism for viral inhibition of IFN signaling. Cytoplasmic IBs have been observed in cells infected with many viruses (30), but their function is largely unknown. In a previous study, we proposed that viral IB functions as a virally built “jail,” imprisoning some host factors and blocking the corresponding cellular processes; thus, it represents the hot spot of virus-host interaction (8). By taking SFTSV as a model, the present study on viral suppression of IFN signaling further supports the proposal. Intriguingly, besides SFTSV NSs, some other virus-encoded proteins localized in IBs often are able to antagonize IFN response through targeting various signaling molecules as well (31–34). For instance, similar to SFTSV, Ebola virus (EBOV) also can inhibit both IFN induction and signaling, and although the inhibitions are mediated by two viral proteins, VP35 and VP24, respectively, both of them are localized in EBOV IBs (33). VP35 can interact with TBK1/IKKε for the targeting of IFN induction (35); VP24 inhibits STAT1 nuclear translocation by interacting with karyopherin α proteins (36, 37). However, Zhang et al. demonstrated that VP24 also can directly interact with STAT1, thereby contributing to IFN signaling inhibition by an additional unidentified mechanism (38). Here, our study suggests the possibility that the sequestration of cellular proteins (TBK1/IKKε, STAT1, etc.) in IBs by VP35 and VP24 accounts for overall EBOV-mediated suppression of the IFN system. Therefore, hijacking signaling molecules into IBs may be a general strategy of viral inhibition of IFN signaling.
In summary, the present study demonstrated the suppression of IFN signaling by SFTSV and unraveled its mechanism therein, presenting the overall view for SFTSV-mediated disruption of the type I IFN system. Hijacking signaling molecules into viral IBs as shown in this study represents a novel IFN signaling antagonism and perhaps also a general mechanism employed by other viruses, including EBOV, the understanding of which may promote the development of therapeutic intervention strategies to combat these deadly pathogen infections.
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation of China (grant numbers 31125003 and 31321001), the National Basic Research Program (973 Program) of China (grant numbers 2010CB530100 and 2013CB911101), and the Science and Technology Basic Work Program (grant number 2013FY113500).
We thank Hong-Bing Shu (Wuhan University, China) for supplying the reporter plasmids and Yan-Yi Wang for critical review of the manuscript.
REFERENCES
- 1.Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL, Zhang L, Zhang QF, Popov VL, Li C, Qu J, Li Q, Zhang YP, Hai R, Wu W, Wang Q, Zhan FX, Wang XJ, Kan B, Wang SW, Wan KL, Jing HQ, Lu JX, Yin WW, Zhou H, Guan XH, Liu JF, Bi ZQ, Liu GH, Ren J, Wang H, Zhao Z, Song JD, He JR, Wan T, Zhang JS, Fu XP, Sun LN, Dong XP, Feng ZJ, Yang WZ, Hong T, Zhang Y, Walker DH, Wang Y, Li DX. 2011. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med 364:1523–1532. doi: 10.1056/NEJMoa1010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu B, Liu L, Huang X, Ma H, Zhang Y, Du Y, Wang P, Tang X, Wang H, Kang K, Zhang S, Zhao G, Wu W, Yang Y, Chen H, Mu F, Chen W. 2011. Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: discovery of a new bunyavirus. PLoS Pathog 7:e1002369. doi: 10.1371/journal.ppat.1002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang YZ, Zhou DJ, Xiong Y, Chen XP, He YW, Sun Q, Yu B, Li J, Dai YA, Tian JH, Qin XC, Jin D, Cui Z, Luo XL, Li W, Lu S, Wang W, Peng JS, Guo WP, Li MH, Li ZJ, Zhang S, Chen C, Wang Y, de Jong MD, Xu J. 2011. Hemorrhagic fever caused by a novel tick-borne bunyavirus in Huaiyangshan, China. Zhonghua Liu Xing Bing Xue Za Zhi 32:209–220. [PubMed] [Google Scholar]
- 4.Denic S, Janbeih J, Nair S, Conca W, Tariq WU, Al-Salam S. 2011. Acute thrombocytopenia, leucopenia, and multiorgan dysfunction: the first case of SFTS bunyavirus outside China? Case Rep Infect Dis 2011:204056. doi: 10.1155/2011/204056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim KH, Yi J, Kim G, Choi SJ, Jun KI, Kim NH, Choe PG, Kim NJ, Lee JK, Oh MD. 2013. Severe fever with thrombocytopenia syndrome, South Korea, 2012. Emerg Infect Dis 19:1892–1894. doi: 10.3201/eid1911.130792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi T, Maeda K, Suzuki T, Ishido A, Shigeoka T, Tominaga T, Kamei T, Honda M, Ninomiya D, Sakai T, Senba T, Kaneyuki S, Sakaguchi S, Satoh A, Hosokawa T, Kawabe Y, Kurihara S, Izumikawa K, Kohno S, Azuma T, Suemori K, Yasukawa M, Mizutani T, Omatsu T, Katayama Y, Miyahara M, Ijuin M, Doi K, Okuda M, Umeki K, Saito T, Fukushima K, Nakajima K, Yoshikawa T, Tani H, Fukushi S, Fukuma A, Ogata M, Shimojima M, Nakajima N, Nagata N, Katano H, Fukumoto H, Sato Y, Hasegawa H, Yamagishi T, Oishi K, Kurane I, Morikawa S, Saijo M. 2014. The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J Infect Dis 209:816–827. doi: 10.1093/infdis/jit603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMullan LK, Folk SM, Kelly AJ, MacNeil A, Goldsmith CS, Metcalfe MG, Batten BC, Albarino CG, Zaki SR, Rollin PE, Nicholson WL, Nichol ST. 2012. A new phlebovirus associated with severe febrile illness in Missouri. N Engl J Med 367:834–841. doi: 10.1056/NEJMoa1203378. [DOI] [PubMed] [Google Scholar]
- 8.Ning YJ, Wang M, Deng M, Shen S, Liu W, Cao WC, Deng F, Wang YY, Hu Z, Wang H. 2014. Viral suppression of innate immunity via spatial isolation of TBK1/IKKepsilon from mitochondrial antiviral platform. J Mol Cell Biol 6:324–337. doi: 10.1093/jmcb/mju015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santiago FW, Covaleda LM, Sanchez-Aparicio MT, Silvas JA, Diaz-Vizarreta AC, Patel JR, Popov V, Yu XJ, Garcia-Sastre A, Aguilar PV. 2014. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J Virol 88:4572–4585. doi: 10.1128/JVI.03021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Qi X, Qu B, Zhang Z, Liang M, Li C, Cardona CJ, Li D, Xing Z. 2014. Evasion of antiviral immunity through sequestering of TBK1/IKKepsilon/IRF3 into viral inclusion bodies. J Virol 88:3067–3076. doi: 10.1128/JVI.03510-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 12.Darnell JE Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 13.Stark GR. 2007. How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev 18:419–423. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark GR, Darnell JE Jr. 2012. The JAK-STAT pathway at twenty. Immunity 36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat Rev Immunol 8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Versteeg GA, Garcia-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr Opin Microbiol 13:508–516. doi: 10.1016/j.mib.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor KE, Mossman KL. 2013. Recent advances in understanding viral evasion of type I interferon. Immunology 138:190–197. doi: 10.1111/imm.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devasthanam A. 2014. Mechanisms underlying the inhibition of interferon signaling by viruses. Virulence 5:270–277. doi: 10.4161/viru.27902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber F, Elliott RM. 2002. Antigenic drift, antigenic shift and interferon antagonists: how bunyaviruses counteract the immune system. Virus Res 88:129–136. doi: 10.1016/S0168-1702(02)00125-9. [DOI] [PubMed] [Google Scholar]
- 21.Hollidge BS, Weiss SR, Soldan SS. 2011. The role of interferon antagonist, non-structural proteins in the pathogenesis and emergence of arboviruses. Viruses 3:629–658. doi: 10.3390/v3060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elliott RM, Weber F. 2009. Bunyaviruses and the type I interferon system. Viruses 1:1003–1021. doi: 10.3390/v1031003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lam TT, Liu W, Bowden TA, Cui N, Zhuang L, Liu K, Zhang YY, Cao WC, Pybus OG. 2013. Evolutionary and molecular analysis of the emergent severe fever with thrombocytopenia syndrome virus. Epidemics 5:1–10. doi: 10.1016/j.epidem.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmaljohn CS, Nichol ST. 2007. Bunyaviridae, p 1741–1789. In Knipe DM, Howley PM (ed), Fields virology, 5th ed, vol 2 Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 25.Ramachandran A, Horvath CM. 2009. Paramyxovirus disruption of interferon signal transduction: STATus report. J Interferon Cytokine Res 29:531–537. doi: 10.1089/jir.2009.0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Bon A, Tough DF. 2002. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol 14:432–436. doi: 10.1016/S0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 27.Chowdhury FZ, Farrar JD. 2013. STAT2: a shape-shifting anti-viral super STAT. JAKSTAT 2:e23633. doi: 10.4161/jkst.23633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parisien JP, Lau JF, Horvath CM. 2002. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol 76:6435–6441. doi: 10.1128/JVI.76.13.6435-6441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashour J, Morrison J, Laurent-Rolle M, Belicha-Villanueva A, Plumlee CR, Bernal-Rubio D, Williams KL, Harris E, Fernandez-Sesma A, Schindler C, Garcia-Sastre A. 2010. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe 8:410–421. doi: 10.1016/j.chom.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storch GA. 2007. Diagnostic virology, p 565–604. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 31.Chenik M, Chebli K, Gaudin Y, Blondel D. 1994. In vivo interaction of rabies virus phosphoprotein (P) and nucleoprotein (N): existence of two N-binding sites on P protein. J Gen Virol 75(Part 11):2889–2896. doi: 10.1099/0022-1317-75-11-2889. [DOI] [PubMed] [Google Scholar]
- 32.Vidy A, Chelbi-Alix M, Blondel D. 2005. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol 79:14411–14420. doi: 10.1128/JVI.79.22.14411-14420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nanbo A, Watanabe S, Halfmann P, Kawaoka Y. 2013. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci Rep 3:1206. doi: 10.1038/srep01206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basler CF, Amarasinghe GK. 2009. Evasion of interferon responses by Ebola and Marburg viruses. J Interferon Cytokine Res 29:511–520. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prins KC, Cardenas WB, Basler CF. 2009. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol 83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol 80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. 2007. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol 81:13469–13477. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang AP, Bornholdt ZA, Liu T, Abelson DM, Lee DE, Li S, Woods VL Jr, Saphire EO. 2012. The Ebola virus interferon antagonist VP24 directly binds STAT1 and has a novel, pyramidal fold. PLoS Pathog 8:e1002550. doi: 10.1371/journal.ppat.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]