

ABSTRACT
In naked viruses, membrane breaching is a key step that must be performed for genome transfer into the target cells. Despite its importance, the mechanisms behind this process remain poorly understood. The small protein VP4, encoded by the genomes of most viruses of the order Picornavirales, has been shown to be involved in membrane alterations. Here we analyzed the permeabilization activity of the natively nonmyristoylated VP4 protein from triatoma virus (TrV), a virus belonging to the Dicistroviridae family within the Picornavirales order. The VP4 protein was produced as a C-terminal maltose binding protein (MBP) fusion to achieve its successful expression. This recombinant VP4 protein is able to produce membrane permeabilization in model membranes in a membrane composition-dependent manner. The induced permeability was also influenced by the pH, being greater at higher pH values. We demonstrate that the permeabilization activity elicited by the protein occurs through discrete pores that are inserted on the membrane. Sizing experiments using fluorescent dextrans, cryo-electron microscopy imaging, and other, additional techniques showed that recombinant VP4 forms heterogeneous proteolipidic pores rather than common proteinaceous channels. These results suggest that the VP4 protein may be involved in the membrane alterations required for genome transfer or cell entry steps during dicistrovirus infection.
IMPORTANCE During viral infection, viruses need to overcome the membrane barrier in order to enter the cell and replicate their genome. In nonenveloped viruses membrane fusion is not possible, and hence, other mechanisms are implemented. Among other proteins, like the capsid-forming proteins and the proteins required for viral replication, several viruses of the order Picornaviridae contain a small protein called VP4 that has been shown to be involved in membrane alterations. Here we show that the triatoma virus VP4 protein is able to produce membrane permeabilization in model membranes by the formation of heterogeneous dynamic pores. These pores formed by VP4 may be involved in the genome transfer or cell entry steps during viral infection.
INTRODUCTION
The positive single-stranded RNA (ssRNA) virus triatoma virus (TrV) belongs to the Dicistroviridae family in the Picornavirales order. It is a lethal pathogen of the bloodsucking insect Triatoma infestans and of other insect species belonging to the Triatominae subfamily (1, 2). These insects are the main vectors for the transmission of the protozoon Trypanosoma cruzi, the causative agent of Chagas disease (American trypanosomiasis), a neglected tropical disease endemic in Latin America (3). Thus, TrV has been proposed to be a biological control agent that could be used to prevent the spread of this pathogen (4–6).
Dicistroviruses were initially referred to as picorna-like viruses due to their similarities with members of the Picornaviridae family: the nonenveloped icosahedral capsid structure, the positive ssRNA genome, and the capsid protein composition. However, dicistroviruses are characterized by the possession of two different and separated open reading frames (ORFs), which was the main reason for the establishment of this new family (7). The first ORF codes for the nonstructural proteins (helicase, protease, and RNA-dependent RNA polymerase). On the other hand, the second ORF codes for the four structural proteins which build the capsid in the order N terminus-VP2-VP4-VP3-VP1-C terminus as a unique protein precursor called P1. Once the P1 precursor has been cleaved into VP0 (VP2 plus VP4), VP3, and VP1 by the protease encoded by the viral genome and after particle assembly, VP0 undergoes autoproteolytic cleavage into its components by an unknown mechanism that takes place only in RNA-encapsulating TrV particles (8, 9).
Contrary to rhinoviruses and enteroviruses from the related Picornaviridae family, in which the structural proteins are ordered as N terminus-VP4-VP2-VP3-VP1-nonstructural protein-C terminus, the order in dicistroviruses, in which the VP4 N terminus is bound to VP2, prevents the attachment of the highly hydrophobic saturated fatty acid myristoyl to the α amino group of the N terminus of VP4. Furthermore, the N-myristoylation process occurs at the N-terminal glycine residue, and this reaction is unlikely in TrV VP4 because its first residue is an alanine. This observation was experimentally verified by mass spectrometry (8) and illustrates a significant difference between the picornavirus and dicistrovirus VP4 proteins.
The knowledge regarding VP4 in dicistroviruses is still very limited, and therefore, its characteristics and functionality have been assumed to be similar to those of picornavirus VP4 proteins, which are the most closely related proteins that have been studied. However, the structural studies that have been carried out with TrV show significant differences between the dicistrovirus and picornavirus VP4 proteins. In addition to not being myristoylated, the solution of the crystallographic structure of TrV uncovered another intriguing difference. The TrV capsid structure was solved at high resolution (10), and it was shown to be composed of 60 copies of the three major structural proteins (VP1, VP2, and VP3), all of which are folded in a jelly roll-like core. Surprisingly the small protein VP4 (5.5 kDa) was not observed either in the X-ray crystallographic structure or in cryo-electron microscopy (cryo-EM) three-dimensional reconstructions (10, 11), even though it is known to be present in infectious TrV particles (8). These data showed that the TrV VP4 protein is disordered within the capsid, contrary to picornavirus VP4, which is largely ordered inside the particle in a manner that matches the icosahedral capsid symmetry (12, 13).
In picornaviruses, the small hydrophobic protein VP4 (5 to 7 kDa) was proposed to be involved in the membrane alterations required for genome transfer to the cytoplasm of cells during infection (14, 15). Moreover, a recent study (16) has shown that human rhinovirus VP4 interacts with model membranes, inducing permeability by the formation of multimeric proteinaceous pores. The permeabilization activity was enhanced by the myristoylation of VP4 and was higher at acidic pH values, in concordance with the genome delivery mechanisms in acidic endosomes proposed for some picornaviruses like poliovirus and rhinoviruses (17–21).
In contrast, in TrV the low endosomal pH was excluded as a genome release trigger because it has been shown that TrV is not acid labile (22). Moreover, empty capsids (after genome delivery) do not display any conformational changes similar to those in picornaviruses (11). Therefore, a different mechanism of uncoating was proposed by characterizing the capsid disassembly and genome release using atomic force microscopy (AFM) and native mass spectrometry techniques (22). An increase of the pH to 8 to 8.5 destabilizes the genome-capsid interactions, ultimately resulting in a capsid unable to resist the outward forces of the genome that trigger the disassembly of the capsid into pentons and the release of the genome along with VP4. Even though this alkali-mediated disassembly seems unlikely to be the natural trigger for uncoating, this model was supported by the fact that some areas of the intestinal tract of the insect have pH values of 8.9 (21).
Considering all of these differences with previously studied VP4 proteins and with the aim of addressing the function of the triatoma virus VP4 protein, in this study we have cloned, expressed, and purified VP4 as a maltose binding protein (MBP) fusion to characterize its ability to promote membrane permeabilization. Here, we show for the first time that VP4 induces membrane permeability in a pH- and membrane composition-dependent manner through the formation of structurally heterogeneous proteolipidic pores.
MATERIALS AND METHODS
Materials.
The fluorescent probes 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) and p-xylene-bis-pyridinium bromide (DPX) were purchased from Molecular Probes, while fluoroisothiocyanate-dextran 4 (FD4) and fluoroisothiocyanate-dextran 40 (FD40) were from Sigma-Aldrich. Sodium hydrosulfite (dithionite) was purchased from Merck. All the lipids [l-α-phosphatidic acid (PA), l-α-phosphatidylcholine (PC), l-α-phosphatidylserine (PS), l-α-phosphatidylinositol (PI), l-α-lysophosphatidylcholine (Lyso-PC), sphingomyelin (SM), 1-palmitoyl-2-oleoyl-sn-glycerol (DAG), l-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (PE-Rh), and l-α-phosphatidylethanolamine-N-(4-nitrobenzo-2-oxa-1,3-diazole) (PE-NBD)] were purchased from Avanti Polar Lipids.
Sequence alignment and transmembrane region prediction.
A comparative sequence analysis was performed by aligning the amino acid sequences of the following viruses: TrV, cricket paralysis virus (CrPV), Drosophila C virus (DCV), poliovirus, and human rhinovirus 16 (HRV16). The alignments were performed using the software package T-Coffee (23). The presence of possible transmembrane helices in the TrV VP4 protein was predicted using the hydrophobicity-based software TopPred, which is available online (24, 25).
Cloning and expression of VP4.
The cDNA fragment corresponding to the VP4 protein of TrV was obtained by reverse transcription-PCR using the RNA extracted from TrV and the following primers: sense primer 5′-GGAATTCCATATGGCAGGTAAAGAACAGTTAGGTCC-3′ containing an NdeI restriction site (underlined) and antisense primer 5′-TCCGGGATCCCTAAAAACCTAAAGCTGACAATAACCC-3′ including a BamHI restriction site (underlined). The resulting PCR product was cloned into plasmid pET28a. After confirming the correctness of the cloned sequence by DNA sequencing, the pET28a-VP4 recombinant plasmid was transformed into Escherichia coli strain Rosetta 2(DE3)pLysS. A 0.5-ml overnight growth culture was used to inoculate 10 ml of LB medium supplemented with 50 μM kanamycin. When this culture, incubated at 37°C, reached an A600 of 0.6 to 0.8, protein expression was induced by adding 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Culture growth at 37°C was monitored by measuring the turbidity (A600) at different times. The culture growth controls consisted of (i) a noninduced culture and (ii) an induced culture expressing the TrV VP1 protein.
Cloning and purification of recombinant MBP-VP4.
The cDNA fragment corresponding to the VP4 protein of TrV was obtained as described above. The resulting PCR product was cloned after the tobacco etch virus (TEV) protease site into the pET28-HMT plasmid (a modified pET28a plasmid that carries a His-tagged maltose binding protein with a C-terminal TEV site). After confirming the correctness of the cloned sequence by DNA sequencing, the pET28-HMT-VP4 recombinant plasmid was transformed into E. coli strain BL21(DE3). A 50-ml overnight growth culture was used to inoculate 1 liter of LB medium supplemented with 50 μM kanamycin. When this culture, incubated at 37°C, reached an A600 of 0.6 to 0.8, protein expression was induced by adding 0.5 mM IPTG for 3 h at 37°C. Cells were harvested by centrifugation at 5,000 × g for 15 min at 4°C.
The cell pellet was resuspended in binding buffer (50 mM Tris, pH 7.5, 0.5 M NaCl, 30 mM imidazole) supplemented with Complete protease inhibitor cocktail (Roche) and disrupted by a high-pressure homogenizer at 4°C. The lysate was incubated for 20 min with 25 U/ml Benzonase nuclease (Invitrogen), and cell debris and protein precipitates were removed by centrifugation at 30,000 × g for 30 min at 4°C. The supernatant was applied to a His-Trap (GE) affinity column, and MBP-VP4 was eluted by a 30 to 500 mM linear gradient of imidazole. Fractions containing the protein were checked by SDS-PAGE and Western blotting, and pure protein fractions were dialyzed overnight against the assay buffer (50 mM Tris, pH 8.0, 200 mM NaCl).
Preparation of liposomes.
Large unilamellar vesicles (LUVs) were prepared by the extrusion method (26) with some modifications. Briefly, lipids dissolved in a 2:1 (mol/mol) chloroform-methanol mixture were dried using a nitrogen stream, and the residual solvent was eliminated under vacuum. Lipid dry film was hydrated by adding the assay buffer, followed by 10 freeze-thaw cycles at 50°C. The multilamellar vesicles (MLVs) obtained were turned onto LUVs of 400 nm in diameter by extruding the suspension through polycarbonate filters of the respective size. The size was confirmed by dynamic light scattering using a Zetasizer instrument (Malvern). The lipid concentration in the liposome solution was calculated by determination of the phosphorus concentration (27).
Membrane binding assay.
LUVs made of PA-PC (1:1, in a molar basis) and 0.5% (mol/mol) PE-Rh in 50 mM Tris, pH 8.0, 200 mM NaCl, as described above, were incubated either with MBP-VP4 or with MBP in the same buffer at a molar lipid/protein ratio of 500:1 (lipid concentration, 1.5 mM; protein concentration, 3 μM) for 30 min at 25°C. To assess protein-membrane binding, a liposome flotation assay was performed as previously described (28). Briefly, the protein-liposome mixture was adjusted to a sucrose concentration of 1.4 M and loaded on the bottom of a sucrose gradient containing two more fractions (0.8 and 0.5 M sucrose from bottom to top, respectively). The gradients were centrifuged at 400,000 × g for 3 h at 4°C and were then fractionated in four different fractions. The top, first fraction corresponds to the liposome-containing fraction, whereas the bottom fraction contains nonbonded protein. The protein location in the gradient was analyzed by SDS-PAGE.
Leakage assays.
LUVs were prepared at 100 μM as described above but in the presence of 25 mM ANTS and 90 mM of its quencher, DPX, in the assay buffer. Nonencapsulated dyes were removed by passing the sample through a Sephadex G-25 PD-10 column (GE).
Leakage of the vesicle contents was measured in 50 mM Tris, pH 8.0, 200 mM NaCl by a modified ANTS-DPX assay (29). In this case, ANTS fluorescence was recorded continuously (excitation at 355 nm, emission at 520 nm) after MBP-VP4 addition in a Fluoromax spectrometer at 25°C with continuous stirring. When the fluorescence intensity reached equilibrium, Triton X-100 was added to induce 100% release. The absence of leakage (0%) was assigned to the fluorescence of the vesicles before protein addition, and the maximum leakage (100%) corresponded to the fluorescence obtained after vesicle lysis by 0.5% (mol/mol) Triton X-100. The leakage was calculated using equation 1:
| (1) |
where F is the fluorescence after protein addition, F0 is the fluorescence of the intact vesicles, and F100 is the fluorescence after addition of Triton X-100.
For the pore-sizing experiments, leakage assays were complementarily performed using two probes of different sizes: FD4 and FD40 (30). Both LUV suspensions were prepared as described above in the presence of 100 mg/ml of each dye, and the fluorescence emission was measured at 530 nm upon excitation at 465 nm.
Entry assays.
The entry of solutes into vesicles was measured by selective reduction of the 4-nitrobenzo-2-oxa-1,3-diazole (NBD)-labeled lipids in the inner leaflet of the vesicle membranes by an extravesicular solute as previously described (31). LUVs (100 μM) containing 0.6% (mol/mol) NBD-PE in 50 mM Tris, pH 8.0, 200 mM NaCl were incubated with the protein in the same buffer for 2 h at 25°C. Freshly prepared 0.6 M sodium hydrosulfite (dithionite) in 1 M Tris, pH 10, was then added to the mixture to achieve a final dithionite concentration of 12 mM. The reduction of the NBD group by dithionite can be followed as a fluorescence emission decrease (excitation at 460 nm, emission at 540 nm). The degree of permeabilization was calculated from the percentage of inner NBD reduced by equation 2:
| (2) |
where F is the measured fluorescence for the sample, F0 is the fluorescence after the addition of dithionite to intact vesicles (in which the outer leaflet NBD was reduced), and F100 is the fluorescence after addition of 0.5% (mol/mol) Triton X-100.
Cryo-EM.
For cryo-EM two-dimensional (2D) image analysis of membrane disruption due to the effect of the VP4 protein, vitrified grids of LUVs that had been incubated with the protein in 50 mM Tris, pH 8.0, 200 mM NaCl were prepared at different protein/lipid ratios. As a control, vitrified grids of LUVs without the presence of the VP4 protein were also prepared following standard procedures. Three microliters of sample was applied to glow-discharged 200-mesh Quantifoil holey EM grids and vitrified in liquid ethane that had been cooled with liquid nitrogen using a Vitrobot system (FEI).
Different vitrified grids were observed on a JEM-2200FS/CR (JEOL, Ltd.) field emission gun (FEG) transmission electron microscope operated at 200 kV at the liquid nitrogen temperature. An in-column omega energy filter helped to record images with an improved signal-to-noise ratio by zero-loss filtering, and the energy slit width was set at 15 eV. Zero-tilted two-dimensional digital micrographs were recorded on a 4K×4K Ultrascan4000T charge-coupled-device camera (Gatan Inc.) under low-dose conditions (on the order of 10 to 20 electrons/Å2 per exposure) with an underfocus range of from 1.5 to 4.0 μm at nominal magnifications of from ×40,000 to ×80,000, producing a final pixel size of from 2.6 Å/pixel to 1.2 Å/pixel. Digital images were collected using DigitalMicrograph software (Gatan Inc.), and 2D image analysis was performed with ImageJ software.
Statistical analyses for membrane discontinuities were performed by counting ∼60 different unilamellar liposomes at each protein concentration.
Hemolysis assay.
The hemolysis assay was conducted as previously described (32). Briefly, different protein solutions (MBP-VP4, MBP, or a hemolytic control [adenylate cyclase]) were mixed with a human erythrocyte cell suspension in 50 mM Tris, pH 8.0, 200 mM NaCl (prepared in such a way that an 80-fold dilution gave an A412 of 0.6). Cells and protein mixtures were incubated at room temperature for 3 h and centrifuged at 4,000 × g for 10 min at 4°C. The absorbance of the supernatants was read at 412 nm. The blank (zero hemolysis) consisted of a mixture of protein buffer and erythrocytes.
RESULTS AND DISCUSSION
Sequence analysis of VP4.
As described in the introduction, VP4 proteins from viruses of the order Picornavirales are small hydrophobic peptides encoded by the structural gene. Specifically, TrV VP4 is a small 5.5-kDa peptide composed of 57 amino acids. Hydrophobicity-based analysis (Fig. 1A) predicts that this protein may contain two transmembrane regions of 20 residues structured as helices separated by a loop formed by 12 amino acids. These transmembrane helices could be involved in the membrane-related activity of VP4 (described in the following subsections).
FIG 1.

VP4 sequence analysis. (A) Transmembrane helix (TMH) prediction performed using TopPred software as described in Materials and Methods. Two helices are predicted; the first one is from residues 5 to 25, and the second one is from residues 37 to 57. (B) Sequence alignment of the VP4 proteins from TrV and other dicistroviruses: cricket paralysis virus (CrPV) and Drosophila C virus (DCV). (C) Sequence alignment of the VP4 proteins from TrV and the picornaviruses human rhinovirus 16 (HRV16) and poliovirus (Polio). (B, C) Conserved residues are in a white font on a black background, and similar residues are framed.
The sequence alignment of the TrV VP4 protein with dicistroviral and picornaviral proteins is shown in Fig. 1B and C, respectively. As expected, the similarity between the VP4 proteins from TrV and dicistroviruses was higher (T-Coffee scores, 837 for TrV VP4 and CrPV VP4 and 799 for TrV VP4 and DCV VP4) than the similarity between TrV VP4 and the VP4 proteins from picornaviruses (T-Coffee scores, 544 for TrV VP4 and HRV16 VP4 and 759 for TrV VP4 and poliovirus VP4). Moreover, the few conserved amino acids between TrV and picornaviruses did not seem to be significant, since they were mostly not conserved in dicistroviruses. The high similarity between the dicistrovirus VP4 proteins, in addition to being related to the function of VP4, may also be related to a common mechanism of action. On the contrary, the diminished similarity found when TrV VP4 was compared to picornavirus VP4 proteins, especially to the VP4 protein of rhinovirus, may imply a different functionality.
VP4 can be successfully expressed as an MBP fusion.
As described in the introduction, VP4 proteins from viruses of the order Picornavirales are small hydrophobic peptides encoded by the structural gene. To characterize the TrV VP4 protein, it must be expressed as a soluble and stable protein. This effort was initiated by direct cloning of VP4 into different plasmids and expression of VP4 in E. coli strain Rosetta 2(DE3)pLysS. These attempts resulted in an interesting phenomenon: at 20 min after the induction of VP4 expression, the cloudy culture turned into a clear solution, suggesting that a cell death process was occurring.
In order to monitor cell death, culture turbidity (absorbance at 600 nm) was measured as a function of time (Fig. 2). As observed in Fig. 2, both the lack of induction and the induction of another viral control protein (VP1) allowed normal growth of the culture. However, when the induced protein was VP4, the culture turbidity dropped by approximately 80%. This drop in turbidity can be interpreted not only as a cell death process but also as a drastic reduction of the number of structured microorganisms. The presence of VP4 was checked by SDS-PAGE and by antihistidine Western blotting, but these attempts failed. The small amount of synthesized protein prior to cell death and the leakage of the internal contents of the cell into the external medium may explain the absence of detectable amounts of protein. The presence of VP4 in the external medium was also checked through ammonium sulfate precipitation, but this attempt did not yield results. The lack of significant amounts of protein prevented further biochemical and biophysical analyses of the VP4 protein.
FIG 2.
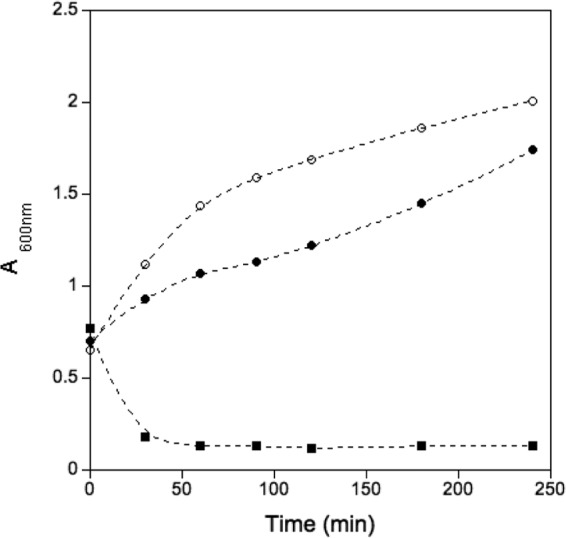
Influence of protein expression on culture turbidity. The turbidity of cultures expressing VP4 (squares) and a control protein (filled circles) was measured after induction by monitoring the absorbance at 600 nm. Results for a bacterial growth control without heterologous protein expression (empty circles) were also measured.
In order to solve this problem, different VP4 expression approaches were assayed. From these approaches, VP4 expression as an MBP fusion protein was selected due to the high yield of soluble protein, because this approach has been used in other cases, and because it has been shown that the MBP fusion protein does not interfere with membrane-related assays (33, 34). The MBP-VP4 (His tag, MBP, the TEV protease site, and VP4 from the N to the C termini) expressed and purified protein was used to test the viability of VP4 cleavage after TEV protease digestion under a variety of conditions (data not shown). However, the VP4 yield and solubility after cleavage were drastically reduced, preventing the use of this strategy. Thus, the whole MBP-VP4 fusion protein was selected to characterize the membrane binding and lytic activity of VP4. For this reason, all experiments were accompanied by MBP (His tag, MBP, and the TEV protease site) controls under the same conditions.
VP4 interacts with lipid membranes.
To assess the interaction of VP4 with membranes, preincubated PA-PC liposomes and MBP-VP4 (or MBP) were loaded in a sucrose flotation gradient as described in Materials and Methods. The fractions of the gradient were analyzed by SDS-PAGE for the presence of the protein and by rhodamine fluorescence for the presence of liposomes. As shown in Fig. 3, MBP-VP4 was found in fraction 1 (F1), thus comigrating with the liposomes. In contrast, MBP control was mainly found in fraction 4 (F4), the bottom fraction, thus showing no such lipid association. Therefore, we can conclude that the interaction of MBP-VP4 with model membranes is driven by VP4.
FIG 3.
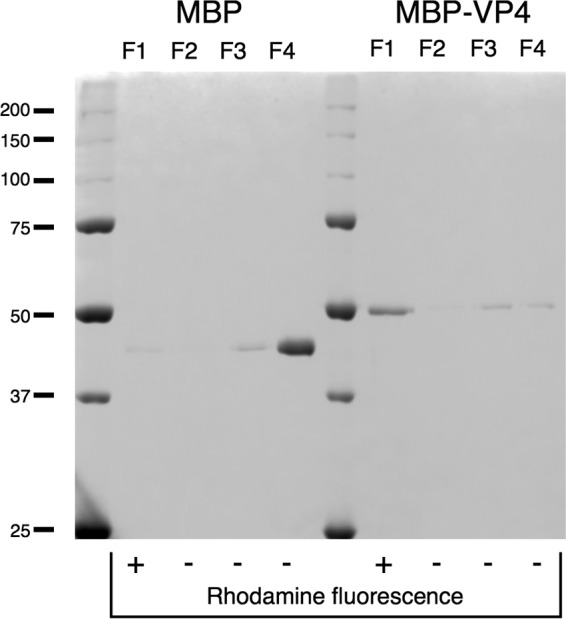
Membrane binding assay. Proteins (MBP-VP4 and control MBP) were incubated with liposome preparations and loaded on the bottom of a sucrose gradient. After ultracentrifugation, gradients were fractionated in four different fractions (F1 to F4, from top to bottom) and analyzed by SDS-PAGE. Liposomes banded at F1. Molecular mass markers (in kDa) are indicated on the left.
MBP-VP4 induces the release of liposome contents in a lipid-dependent manner.
Having proved the interaction between liposomes and MBP-VP4, we examined whether this interaction was able to lead to liposome permeabilization. Vesicles encapsulating ANTS and DPX can be used to measure permeability because, upon leakage, ANTS fluorescence is no longer quenched by DPX due to the high dilution of both molecules (29). Therefore, if permeabilization occurs, fluorescence emission should increase. Leakage curves for 100 μM PA-PC (1:1 molar ratio) liposomes after addition of different amounts of MBP-VP4 are plotted in Fig. 4A. These curves show that the protein is able to permeabilize vesicles. To further characterize the leakage activity, titration curves (Fig. 4B), where saturated leakage values (obtained as shown in Fig. 4A) are plotted as a function of protein concentration, were prepared. These curves show that leakage is protein concentration dependent and that it reaches a plateau (approximately 100% leakage) at high MBP-VP4 concentrations (2 μM). It can also be seen that leakage remains significant even at very low protein doses (e.g., MBP-VP4 at 10 nM with a protein/lipid ratio of 1:10,000 induces a leakage of 24%). This result indicates that vesicle lysis is not a simple effect of membrane destabilization due to huge amounts of membrane-adsorbed protein (like that which happens with the control, MBP) but is mediated by a specific mechanism.
FIG 4.

Membrane permeabilization induced by MBP-VP4. (A) Time course of leakage after addition of MBP-VP4 or MBP at the indicated concentrations. The proteins were added at 0 s, and after 1,000 s, liposomes were disrupted by addition of Triton X-100. (B) Dose-dependent response of MBP-VP4 leakage activity and control MBP at the same concentrations. Data points correspond to the mean values of three independent measurements, and error bars represent standard deviations.
Control experiments using MBP showed that there was no significant leakage for protein at concentrations below 200 nM. Therefore, unless otherwise indicated, a 100 nM MBP-VP4 concentration was selected to perform all experiments to avoid MBP interference.
As shown above, MBP-VP4 elicits a huge amount of permeabilization activity compared to the amount elicited by rhinovirus VP4 (16), even though in our case VP4 was not myristoylated. This modification, which consists of the addition of a 14-carbon saturated fatty acid, is found in a variety of proteins and enables them to anchor to membranes (35, 36). Although myristoylation is not needed by a protein to induce membrane permeability, as shown by a huge variety of proteins, e.g., antimicrobial peptides (37, 38) and bacteriophage holins (39, 40), the permeabilization activity of our natively nonmyristoylated VP4 allows us to propose that its membrane insertion mechanism may be different from the myristoyl-driven mechanism of picornaviruses. Moreover, the low sequence identity of TrV VP4 and VP4 from rhinovirus (Fig. 1C) emphasizes the possible existence of a different protein-membrane interaction mode.
To determine if the permeabilization activity is lipid dependent, similar leakage assays were performed using different lipid compositions. As shown in Table 1, MBP-VP4 requires anionic lipids to elicit its leakage activity. Liposomes composed of neutral lipids, such as PC and SM, were not efficiently permeabilized. In contrast, the internal contents of vesicles made of anionic lipids (PI and PS) were spilled to a greater extent by the action of the protein. Among them, PI was the phospholipid that induced the largest amount of leakage. PA liposomes were not used because they were refractory to ANTS-DPX encapsulation. This specificity for anionic lipids may explain the inability of MBP-VP4 to lyse erythrocyte membranes in hemolysis assays (data not shown). The reason for this behavior is because the membrane outer leaflet of these cells is composed of 44.8% PC and 42.1% SM (41), both of which are neutral lipids.
TABLE 1.
Effect of lipid composition on MBP-VP4-induced membrane permeability
| LUV composition | Head group chargea | % leakageb |
|
|---|---|---|---|
| MBP-VP4 | MBP | ||
| PA-PC | − and +/− | 51 | 6 |
| PC | +/− | 13 | 1 |
| SM | +/− | 1 | 0 |
| PS | − | 36 | 3 |
| PI | − | 83 | 7 |
−, anionic groups; +/−, zwitteronic groups.
Leakage activity was calculated as described in Materials and Methods for a protein concentration of 100 nM and a lipid concentration of 100 μM. Mean values of three measurements are indicated.
MBP-VP4 permeabilization activity is pH dependent.
Because the activity of VP4 may be linked to its externalization and as the pH has been shown to be a key factor in VP4 externalization and genome release in both picornaviruses and dicistroviruses, here we investigated the influence of the pH on the function of MBP-VP4. PA-PC (1:1 molar ratio) liposomes encapsulating ANTS-DPX were prepared in different buffers of the same ionic strength but with different pH values: 5.5, 7.5, and 8.0. The possibility that the pH influences the fluorescence properties of ANTS was discarded because all the samples were normalized by liposome solubilization with Triton X-100. As shown in Table 2, the leakage due to MBP-VP4 was significantly more efficient at high pH.
TABLE 2.
Effect of pH on MBP-VP4 leakage in PA-PCa liposomes
| pH | % leakageb |
|
|---|---|---|
| MBP-VP4 | MBP | |
| 8.0 | 50 | 6 |
| 7.5 | 34 | 13 |
| 5.5 | 13 | 15 |
PA and PC were used at a 1:1 molar ratio.
Leakage activity was calculated as described in Materials and Methods for a protein concentration of 100 nM and a lipid concentration of 100 μM. Mean values of three measurements are indicated.
This behavior, which is opposite that observed in picornaviruses (16), is consistent with the disassembly and genome release model proposed for TrV (22). In that model, the alkalization of the solution to approximately pH 8 destabilizes the genome-capsid interactions, triggering the disassembly of the capsid into pentons, accompanied by the release of the genome along with VP4. Although alkaline conditions are not typical during viral infections, this model was supported by the fact that the intestinal tract of the insect (where TrV infects) can reach pH values of 8.9 (42). The high-pH conditions that favor TrV VP4 permeabilization activity are consistent with the above-described model, suggesting that its activity may be optimized for the alkaline conditions encountered during infection.
Liposome contents are leaked via a true pore.
Membrane breaching is a key step in the transfer of the genome of naked viruses. This can be accomplished using different mechanisms, either by the formation of discrete pores or by direct membrane disruption (43). To characterize the mechanism involved in MBP-VP4 vesicle content leakage, previously shown leakage assays were combined with solute entry experiments. These assays were performed using PA-PC (1:1 molar ratio) liposomes containing 0.6% (mol/mol) PE-NBD. Two hours after the addition of the protein, dithionite was added and the entry of the contents was calculated from the percentage of internal NBD reduced by this externally added salt (see Materials and Methods). Since dithionite is membrane impermeant, it can reduce internal fluorophores only if discrete pores allow dithionite to enter through the bilayer. Therefore, this assay allows discrimination of whether the permeabilization phenomenon occurs due to discrete pores or due to membrane integrity disruption (33, 37). As both sides of the membrane are equally labeled with PE-NBD, when dithionite is added the external NBD is reduced (approximately 55%). This value was assigned to 0% solute entry. As shown in Fig. 5A, after reaching equilibrium, the liposomes allowed solutes to enter. A noteworthy finding was that the extent of solute leakage and entry was almost equal. These results suggest that the diffusion of solutes across the membrane occurs through discrete pores.
FIG 5.

Characterization of the leakage activity induced by MBP-VP4 and pore type determination. (A) Solute entry experiment. The extent of solute entry was compared with the extent of solute leakage from 100 μM PA-PC (1:1 molar ratio) liposomes obtained using 25, 50, 100, and 200 nM MBP-VP4. (B) Sizing experiment. The release of compounds of different sizes from 100 μM PA-PC (1:1 molar ratio) liposomes was measured as a function of the protein concentration. Data points correspond to the mean values of three independent measurements, and error bars represent standard deviations.
MBP-VP4 pores are of variable size and structurally heterogeneous.
Previous results allowed us to propose a mechanism of permeabilization through the formation of a discrete pore, but the results did not distinguish between fixed protein pores, such as ion channels, described by the barrel-stave model (44), and dynamic proteolipidic pores, described by the toroidal model (45). To further characterize the nature of these pores, sizing experiments were conducted. Although these assays were initially performed to calculate the cutoff sizes of the solutes diffusing through the MBP-VP4 pore, the data finally obtained (see below) were used to distinguish whether MBP-VP4 forms stable protein pores or dynamic proteolipidic pores. To perform sizing experiments, leakage from vesicles encapsulating ANTS-DPX (Stokes radius, 0.5 nm), FD4 (Stokes radius, 1.8 nm), and FD40 (Stokes radius, 3.0 nm) was measured at different protein concentrations.
As shown in Fig. 5B, at the lowest protein concentration, ANTS-DPX and FD4 were released, while FD40 remained inside the vesicles. Moreover, the amount of ANTS-DPX leakage was a little higher than that of FD4. As the protein concentration increased, the leakage of the smaller solutes increased, while the release of the largest compound (FD40) began. Therefore, it can be concluded that the leakage of the solutes of different sizes depends on the protein/lipid ratio. On the other hand, the mechanism of LUV disruption by MBP-VP4 was analyzed by cryo-EM imaging. The pores induced by MBP-VP4 were counted, their size was estimated from the micrographs, and they were clustered in three different groups (Fig. 6A): 6 to 12 nm (Fig. 6B), 13 to 20 nm (Fig. 6C), and larger than 20 nm (Fig. 6D) (pores smaller than 6 nm are very difficult to observe due to technical limitations). This two-dimensional image analysis shows the structural heterogeneity of the pores formed by MBP-VP4. The diameter of the pores was not uniform and increased as the protein concentration increased. Moreover, these pores seemed to be membrane discontinuities. Structure-fixed proteinaceous pores are expected to be of invariable size, structurally homogeneous, and not affected by the protein concentration. In this type of pore, all compounds that fit the pore size are released to the same extent at each protein concentration, and the solutes larger than the pore size are never released, regardless of the protein/lipid ratio. However, in our case, the pores were heterogeneous and their size seemed likely to increase as a function of the protein concentration, arguing against a proteinaceous pore model and being consistent with the dynamic proteolipidic pore model. This pore model has already been proposed for a variety of different peptides and proteins (45–50).
FIG 6.

Cryo-EM imaging. (A) Pore sizes observed by cryo-EM. PA-PC (1:1 molar ratio) liposomes at 500 μM were incubated with different protein concentrations. Membrane interruptions observed in digital micrographs acquired at different nominal magnifications ranging from ×40,000 to ×80,000 were clustered in three different groups: 6 to 12 nm, 13 to 20 nm, and >20 nm. (B to D) Cryo-EM micrographs of membrane disruption. Interruptions in the membrane (black arrows) correspond to the groups with pore sizes of 6 to 12 nm (B), 13 to 20 nm (C) and >20 nm (D).
In the proteolipidic pore model or toroidal pore model, the proteins affect the local curvature of the membrane, bending back the bilayer in such a way that a proteolipidic toroid is formed in the membrane. This membrane deformation depends on the intrinsic curvature of the membrane, which, in turn, is lipid composition dependent (51, 52). Therefore, to provide further evidence for the proteolipidic nature of the pores formed by MBP-VP4, the leakage activity of the protein was measured in the presence of lipids known to change the intrinsic curvature of the membrane. This approach has previously been used to test the validity of the proteolipidic model (51).
Lyso-PC, an inducer of a positive intrinsic curvature, was introduced in PA-PC (1:1 molar ratio) liposomes at different molar percentages by replacing PC with Lyso-PC. As shown in Fig. 7, the incorporation of Lyso-PC into the liposomes induced a linear increase in the leakage activity of MBP-VP4. Otherwise, the incorporation of DAG, an inducer of intrinsic negative curvature, induced a linear decrease in its activity. The membrane curvature on Lyso-PC- or DAG-containing vesicles is linearly dependent on the concentration of these lipids (51, 52), which is consistent with the observed linear correlation between MBP-VP4 activity and Lyso-PC and DAG contents (Fig. 7). Therefore, these results show that the leakage activity of the MBP-VP4 protein is affected by changes in the intrinsic curvature of the membrane induced by Lyso-PC and DAG, arguing in favor of the proteolipidic pore model.
FIG 7.
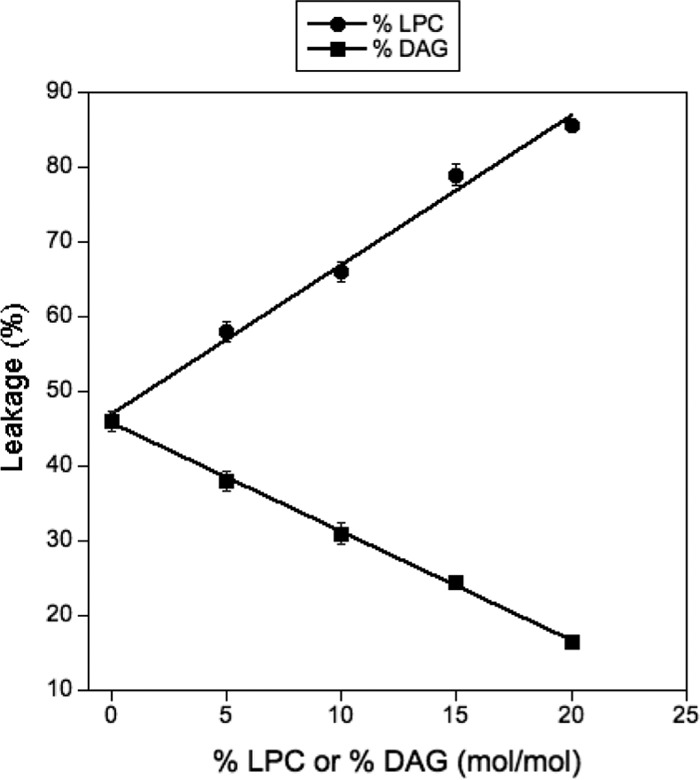
Effect of different concentrations of Lyso-PC (LPC) and DAG on the leakage activity of MBP-VP4. The protein/lipid ratio was 1:1,000 (on a molar basis). Data points correspond to mean values from three independent measurements, and error bars represent standard deviations.
The likely toroidal pores formed by TrV recombinant VP4 protein emphasize the observed differences with the picornaviral VP4 proteins in their sequence, myristoylation, the pH dependence of their membrane permeation, etc. The only fully characterized picornaviral VP4 protein is the one from human rhinovirus 16 (16). This protein elicits its permeabilization activity through the formation of multimeric pores that can be interpreted with the barrel-stave model (44), in opposition to the toroidal model (45) used to explain our case.
ACKNOWLEDGMENTS
R.S.-E. was supported by a predoctoral fellowship from the Basque Government (BG). J.G. was the recipient of a student summer grant from the Fundación Biofísica Bizkaia (FBB), Spain. L.S.-M. holds a JAE-Doc fellowship from CSIC, Spain. This work was partially supported by grants from the BG (MV-2012-2-41, AE-2012-1-44, S-PE12FB001), UPV/EHU (IT849-13, OTRI code 2013.0666), and MICINN (BFU2012-36241), Spain.
We thank Ganeko Bernardo for his interest and for the provision of the pET28-HMT plasmid, as well as Cesar Martin and Helena Ostolaza, who provided the red blood cells for the hemolysis assay. We are also grateful to Aritz Durana for his interest, technical support, and critical comments and to Félix M. Goñi who read and provided critical comments regarding the manuscript.
REFERENCES
- 1.Muscio OA, La Torre JL, Scodeller EA. 1988. Characterization of triatoma virus, a picorna-like virus isolated from the triatomine bug Triatoma infestans. J Gen Virol 69(Pt 11):2929–2934. doi: 10.1099/0022-1317-69-11-2929. [DOI] [PubMed] [Google Scholar]
- 2.Marti GA, Echeverría MG, Susevich ML, Ceccarelli S, Balsalobre A, Canale D, Stariolo R, Noireau F, García AL, González-Cifuentes NL, Guhl F, Bacigalupo A, Cattan PE, García A, Villacis AG, Grijalva MJ, Solorzano E, Monroy C, Espinoza-Blanco Y, Cordoca-Benzaquen E, Ruelas-Llerena N, Guzmán-Loayza M, Caceres AG, Vences-Blanco MO, Salazar-Schettino PM, Mojoli A, Rojas de Arias A, Felicaingeli MD, Rivera Mendoza P, Rozas-Dennis GS, Sánchez-Eugenia R, Agirre J, Viguera AR, Hernández-Suárez CM, Vilchez S, Osuna A, Gorla DE, Mougabure-Cueto G, Esteban L, Angulo VM, Querido JFB, Silva MS, Marques T, Gómez-Hernández C, Ramírez LE, Diotaiuti L, Rabinovich JE, Guérin DMA. 2013. Exploration for triatoma virus (TrV) infection in laboratory-reared triatomines of Latin America: a collaborative study. Int J Trop Insect Sci 33:294–304. doi: 10.1017/S1742758413000337. [DOI] [Google Scholar]
- 3.Grayson M, Clayton J, Coura JR, Viñas PA, Petherick A. 2010. Chagas disease. Nature 465:S3–S22. doi: 10.1038/465S3a. [DOI] [PubMed] [Google Scholar]
- 4.Bonning BC, Miller WA. 2010. Dicistroviruses. Annu Rev Entomol 55:129–150. doi: 10.1146/annurev-ento-112408-085457. [DOI] [PubMed] [Google Scholar]
- 5.Czibener C, La Torre JL, Muscio OA, Ugalde RA, Scodeller EA. 2000. Nucleotide sequence analysis of triatoma virus shows that it is a member of a novel group of insect RNA viruses. J Gen Virol 81:1149–1154. [DOI] [PubMed] [Google Scholar]
- 6.Muscio OA, La Torre J, Bonder MA, Scodeller EA. 1997. Triatoma virus pathogenicity in laboratory colonies of Triatoma infestans (Hemiptera:Reduviidae). J Med Entomol 34:253–256. doi: 10.1093/jmedent/34.3.253. [DOI] [PubMed] [Google Scholar]
- 7.Mayo MA. 2002. Virus taxonomy—Houston 2002. Arch Virol 147:1071–1076. doi: 10.1007/s007050200036. [DOI] [PubMed] [Google Scholar]
- 8.Agirre J, Aloria K, Arizmendi JM, Iloro I, Elortza F, Sánchez-Eugenia R, Marti GA, Neumann E, Rey FA, Guérin DMA. 2011. Capsid protein identification and analysis of mature triatoma virus (TrV) virions and naturally occurring empty particles. Virology 409:91–101. doi: 10.1016/j.virol.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 9.Sánchez-Eugenia R, Mendez F, Querido JFB, Sousa Silva M, Guérin DMA, Rodriguez JF. 2015. Triatoma virus structural polyprotein expression, processing and assembly into virus-like particles. J Gen Virol 96:64–73. doi: 10.1099/vir.0.071639-0. [DOI] [PubMed] [Google Scholar]
- 10.Squires G, Pous J, Agirre J, Rozas-Dennis GS, Costabel MD, Marti GA, Navaza J, Bressanelli S, Guérin DMA, Rey FA. 2013. Structure of the triatoma virus capsid. Acta Crystallogr D Biol Crystallogr 69:1026–1037. doi: 10.1107/S0907444913004617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agirre J, Goret G, Legoff M, Sánchez-Eugenia R, Marti GA, Navaza J, Guérin DMA, Neumann E. 2013. Cryo-electron microscopy reconstructions of triatoma virus particles: a clue to unravel genome delivery and capsid disassembly. J Gen Virol 94:1058–1068. doi: 10.1099/vir.0.048553-0. [DOI] [PubMed] [Google Scholar]
- 12.Hogle JM, Chow M, Filman DJ. 1985. Three-dimensional structure of poliovirus at 2.9 Å resolution. Science 229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 13.Verdaguer N, Blaas D, Fita I. 2000. Structure of human rhinovirus serotype 2 (HRV2). J Mol Biol 300:1179–1194. doi: 10.1006/jmbi.2000.3943. [DOI] [PubMed] [Google Scholar]
- 14.Danthi P, Tosteson M, Li Q, Chow M. 2003. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J Virol 77:5266–5274. doi: 10.1128/JVI.77.9.5266-5274.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moscufo N, Chow M. 1992. Myristate-protein interactions in poliovirus: interactions of VP4 threonine 28 contribute to the structural conformation of assembly intermediates and the stability of assembled virions. J Virol 66:6849–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panjwani A, Strauss M, Gold S, Wenham H, Jackson T, Chou JJ, Rowlands DJ, Stonehouse NJ, Hogle JM, Tuthill TJ. 2014. Capsid protein VP4 of human rhinovirus induces membrane permeability by the formation of a size-selective multimeric pore. PLoS Pathog 10:e1004294. doi: 10.1371/journal.ppat.1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bilek G, Matscheko NM, Pickl-Herk A, Weiss VU, Subirats X, Kenndler E, Blaas D. 2011. Liposomal nanocontainers as models for viral infection: monitoring viral genomic RNA transfer through lipid membranes. J Virol 85:8368–8375. doi: 10.1128/JVI.00329-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hogle JM. 2002. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol 56:677–702. doi: 10.1146/annurev.micro.56.012302.160757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs R, Blaas D. 2010. Uncoating of human rhinoviruses. Rev Med Virol 20:281–297. doi: 10.1002/rmv.654. [DOI] [PubMed] [Google Scholar]
- 20.Bayer N, Prchla E, Schwab M, Blaas D, Fuchs R. 1999. Human rhinovirus HRV14 uncoats from early endosomes in the presence of bafilomycin. FEBS Lett 463:175–178. doi: 10.1016/S0014-5793(99)01610-5. [DOI] [PubMed] [Google Scholar]
- 21.Prchla E, Kuechler E, Blaas D, Fuchs R. 1994. Uncoating of human rhinovirus serotype 2 from late endosomes. J Virol 68:3713–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snijder J, Uetrecht C, Rose RJ, Sanchez-Eugenia R, Marti GA, Agirre J, Guerin DMA, Wuite GJL, Heck AJR, Roos WH. 2013. Probing the biophysical interplay between a viral genome and its capsid. Nat Chem 5:502–509. doi: 10.1038/nchem.1627. [DOI] [PubMed] [Google Scholar]
- 23.Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 24.von Heijine G. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol 225:487–494. [DOI] [PubMed] [Google Scholar]
- 25.Claros MG, von Heijine G. 1994. TopPred II: an improved software for membrane protein structure predictions. Comput Appl Biosci 10:685–686. [DOI] [PubMed] [Google Scholar]
- 26.Mayer LD, Hope MJ, Cullis PR. 1986. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim Biophys Acta 858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- 27.Fiske C, Yellapragada S. 1925. The colorimetric determination of phosphorus. J Biol Chem 66:375–400. [Google Scholar]
- 28.Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. 2003. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278:48935–48941. doi: 10.1074/jbc.M306289200. [DOI] [PubMed] [Google Scholar]
- 29.Ellens H, Bentz J, Szoka FC. 1985. Proton- and calcium-induced fusion and destabilization of liposomes. Biochemistry 24:3099–3106. doi: 10.1021/bi00334a005. [DOI] [PubMed] [Google Scholar]
- 30.Ostolaza H, Bartolomé B, Ortiz de Zárate I, de la Cruz F, Goñi FM. 1993. Release of lipid vesicle contents by the bacterial protein toxin α-haemolysin. Biochim Biophys Acta 1147:81–88. doi: 10.1016/0005-2736(93)90318-T. [DOI] [PubMed] [Google Scholar]
- 31.McIntyre JC, Sleight RG. 1991. Fluorescence assay for phospholipid membrane asymmetry. Biochemistry 30:11819–11827. doi: 10.1021/bi00115a012. [DOI] [PubMed] [Google Scholar]
- 32.Cortajarena AL, Goñi FM, Ostolaza H. 2001. Glycophorin as a receptor for Escherichia coli alpha-hemolysin in erythrocytes. J Biol Chem 276:12513–12519. doi: 10.1074/jbc.M006792200. [DOI] [PubMed] [Google Scholar]
- 33.Agirre A, Barco A, Carrasco L, Nieva JL. 2002. Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. J Biol Chem 277:40434–40441. doi: 10.1074/jbc.M205393200. [DOI] [PubMed] [Google Scholar]
- 34.Posada IMD, Sánchez-Magraner L, Hervás JH, Alonso A, Monaco HL, Goñi FM. 2014. Membrane binding of human phospholipid scramblase 1 cytoplasmic domain. Biochim Biophys Acta 1838:1785–1792. doi: 10.1016/j.bbamem.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Bhatnagar RS, Gordon JI. 1997. Understanding covalent modifications of proteins by lipids: where cell biology and biophysics mingle. Trends Cell Biol 7:14–20. doi: 10.1016/S0962-8924(97)10044-7. [DOI] [PubMed] [Google Scholar]
- 36.Yalovsky S, Rodríguez-Concepción M, Gruissem W. 1999. Lipid modifications of proteins—slipping in and out of membranes. Trends Plant Sci 4:439–445. doi: 10.1016/S1360-1385(99)01492-2. [DOI] [PubMed] [Google Scholar]
- 37.Krauson AJ, He J, Wimley WC. 2012. Determining the mechanism of membrane permeabilizing peptides: identification of potent, equilibrium pore-formers. Biochim Biophys Acta 1818:1625–1632. doi: 10.1016/j.bbamem.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Almeida PF, Pokorny A. 2009. Mechanisms of antimicrobial, cytolytic, and cell-penetrating peptides: from kinetics to thermodynamics. Biochemistry 48:8083–8093. doi: 10.1021/bi900914g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gründling A, Manson MD, Young R. 2001. Holins kill without warning. Proc Natl Acad Sci U S A 98:9348–9352. doi: 10.1073/pnas.151247598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young R. 2002. Bacteriophage holins: deadly diversity. J Mol Microbiol Biotechnol 4:21–36. [PubMed] [Google Scholar]
- 41.Virtanen JA, Cheng KH, Somerharju P. 1998. Phospholipid composition of the mammalian red cell membrane can be rationalized by a superlattice model. Proc Natl Acad Sci U S A 95:4964–4969. doi: 10.1073/pnas.95.9.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kollien AH, Grospietsch T, Kleffmann T, Zerbst-Boroffka I, Schaub GA. 2001. Ionic composition of the rectal contents and excreta of the reduviid bug Triatoma infestans. J Insect Physiol 47:739–747. doi: 10.1016/S0022-1910(00)00170-0. [DOI] [PubMed] [Google Scholar]
- 43.Banerjee M, Johnson JE. 2008. Activation, exposure and penetration of virally encoded, membrane-active polypeptides during non-enveloped virus entry. Curr Protein Pept Sci 9:16–27. doi: 10.2174/138920308783565732. [DOI] [PubMed] [Google Scholar]
- 44.Rapaport D, Shais Y. 1991. Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J Biol Chem 266:23769–23775. [PubMed] [Google Scholar]
- 45.Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW. 1996. Membrane pores induced by magainin. Biochemistry 35:13723–13728. doi: 10.1021/bi9620621. [DOI] [PubMed] [Google Scholar]
- 46.Kristan KC, Viero G, Dalla Serra M, Macek P, Anderluh G. 2009. Molecular mechanism of pore formation by actinoporins. Toxicon 54:1125–1134. doi: 10.1016/j.toxicon.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 47.Matsuzaki K, Yoneyama S, Miyajima K. 1997. Pore formation and translocation of melittin. Biophys J 73:831–838. doi: 10.1016/S0006-3495(97)78115-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsuzaki K. 1998. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim Biophys Acta 1376:391–400. doi: 10.1016/S0304-4157(98)00014-8. [DOI] [PubMed] [Google Scholar]
- 49.Lee M-T, Chen F-Y, Huang HW. 2004. Energetics of pore formation induced by membrane active peptides. Biochemistry 43:3590–3599. doi: 10.1021/bi036153r. [DOI] [PubMed] [Google Scholar]
- 50.Sobko AA, Kotova EA, Antonenko YN, Zakharov SD, Cramer WA. 2006. Lipid dependence of the channel properties of a colicin E1-lipid toroidal pore. J Biol Chem 281:14408–14416. doi: 10.1074/jbc.M513634200. [DOI] [PubMed] [Google Scholar]
- 51.Landeta O, Landajuela A, Gil D, Taneva S, Di Primo C, Sot B, Valle M, Frolov VA, Basañez G. 2011. Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. J Biol Chem 286:8213–8230. doi: 10.1074/jbc.M110.165852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zimmerberg J, Kozlov MM. 2006. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol 7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]