

ABSTRACT
Because the pathogenesis of enterovirus 71 (EV71) remains mostly ambiguous, identifying the factors that mediate viral binding and entry to host cells is indispensable to ultimately uncover the mechanisms that underlie virus infection and pathogenesis. Despite the identification of several receptors/attachment molecules for EV71, the binding, entry, and infection mechanisms of EV71 remain unclear. Herein, we employed glycoproteomic approaches to identify human nucleolin as a novel binding receptor for EV71. Glycoproteins purified by lectin chromatography from the membrane extraction of human cells were treated with sialidase, followed by immunoprecipitation with EV71 particles. Among the 16 proteins identified by tandem mass spectrometry analysis, cell surface nucleolin attracted our attention. We found that EV71 interacted directly with nucleolin via the VP1 capsid protein and that an antinucleolin antibody reduced the binding of EV71 to human cells. In addition, the knockdown of cell surface nucleolin decreased EV71 binding, infection, and production in human cells. Furthermore, the expression of human nucleolin on the cell surface of a mouse cell line increased EV71 binding and conferred EV71 infection and production in the cells. These results strongly indicate that human nucleolin can mediate EV71 binding to and infection of cells. Our findings also demonstrate that the use of glycoproteomic approaches is a reliable methodology to discover novel receptors for pathogens.
IMPORTANCE Outbreaks of EV71 have been reported in Asia-Pacific countries and have caused thousands of deaths in young children during the last 2 decades. The discovery of new EV71-interacting molecules to understand the infection mechanism has become an emergent issue. Hence, this study uses glycoproteomic approaches to comprehensively investigate the EV71-interacting glycoproteins. Several EV71-interacting glycoproteins are identified, and the role of cell surface nucleolin in mediating the attachment and entry of EV71 is characterized and validated. Our findings not only indicate a novel target for uncovering the EV71 infection mechanism and anti-EV71 drug discovery but also provide a new strategy for virus receptor identification.
INTRODUCTION
Enterovirus 71 (EV71), a member of the enterovirus A species of the Picornaviridae family, is known to cause hand-foot-and-mouth disease (1). Severe EV71 infections in children result in several neurological complications, such as encephalitis, aseptic meningitis, pulmonary edema, and acute flaccid paralysis (2). The infection of EV71 is initiated in the intestine and then spreads to the central nervous system, which results in a high mortality rate in infected children (3). Because virus receptors determine the host range, tissue tropism, and pathogenesis (4–6), the identification of factors that mediate the recognition and/or entry of EV71 to host cells is essential to decipher infection mechanisms. Several receptors or attachment molecules for EV71 have been identified, including scavenger receptor B2 (SCARB2), P-selectin glycoprotein ligand-1 (PSGL-1), sialylated glycoprotein, dendritic cell-specific ICAM 3-grabbing nonintegrin, annexin II, vimentin, and polysaccharide (heparin sulfate) (7–13). EV71 strains can be divided into different subgenotypes based on sequence homology (14). Human SCARB2 (hSCARB2) mediates the entry of EV71 strains or genotypes tested (15). SCARB2 not only facilitates the infection of EV71 but is also involved in virus internalization and the viral RNA uncoating of EV71 (16). Unlike SCARB2, PSGL-1 mediates the infection of some, but not all, EV71 strains or genotypes (7, 17). In addition, PSGL-1 participates in EV71 virus binding but not virus entry and viral RNA release (16). Although EV71 uses multiple receptors (18), none of the antireceptor or antiattachment molecule antibodies can completely abolish the infection of host cells by EV71 (7–12, 18). Undiscovered receptors or cofactors that are involved in the binding and infection of EV71 urgently need to be identified.
Glycoproteomics coupled with mass spectrometric analyses have been used to identify the functions of glycoproteins, such as tumor markers (19–26). Although cell surface glycoproteins are known to participate in the recognition, binding, and infection of pathogens (27), this advanced methodology has never been applied to the discovery of virus receptors prior to this study.
We previously demonstrated that a cell surface monosaccharide, sialic acid, can mediate EV71 binding and infection (9). Blocking EV71-sialic acid interactions reduces EV71 binding and subsequent viral replication. In addition, EV71 can interact with sialylated, desialylated, or deglycosylated SCARB2 (9, 15). These findings suggest that sialylation may be a common modification for EV71 receptors, and the removal of sialic acids does not affect the binding of EV71 to receptors. In the present study, targeted glycoproteomic approaches were applied to further investigate the undiscovered binding receptors from sialylated membrane proteins. Sialylated glycoproteins were purified from the cell membrane extract by lectin chromatography and treated with sialidase, followed by pulldown with EV71 particles. Sixteen proteins were identified by mass spectrometry, and nucleolin (NCL), a multifunctional binding protein (28–30), attracted our attention. The interaction between NCL with EV71 and the roles of NCL in the binding and infection of EV71 to cells were then carefully evaluated and characterized.
MATERIALS AND METHODS
Cells and viruses.
The RD (rhabdomyosarcoma) and NIH 3T3 cell lines were maintained at 37°C in medium according to the instructions of the American Type Culture Collection. The EV71 strains, including the clinical isolates 89-N0363 (subgenogroup B4), 87-N6356 (subgenogroup C2), 94-N2873 (subgenogroup C4), and 97-M448 (subgenogroup B5) and a mouse-adapted strain (MP4), were propagated and titrated in RD cells (31).
Plaque assay and CCID50.
For plaque assays, RD cells were seeded in plates overnight, infected with virus for 1 h (h), incubated in medium with 1% methylcellulose at 35°C for 72 h, and stained with crystal violet. The 50% cell culture infective doses (CCID50) were determined as previously described (31). Briefly, RD cells in suspension were infected with virus, seeded in 96-well plates, and then incubated at 35°C for 1 week to observe the cytopathic effect (CPE). The CCID50 was calculated following the method of Reed and Muench (32).
Isolation and identification of EV71-interacting glycoproteins.
RD cells were harvested and homogenized, and the membrane proteins were obtained as previously described (9). Cells were harvested and homogenized in ice-cold homogenization buffer (20 mM Tris-HCl, pH 7.5, 2.0 mM EDTA, 1.0 mM dithiothreitol [DTT], and protein inhibitor cocktail) using a sonicator (Chrom Tech), and the cell lysate was centrifuged at 90 × g for 10 min to remove the cell debris. The supernatants were collected and subjected to ultracentrifugation at 25,000 × g (4°C) for 20 min. The membrane protein pellets were washed with homogenization buffer (containing 1% NP-40) three times and resuspended in homogenization buffer. The washed membrane proteins were subjected to a lectin affinity chromatography column packaged with Maackia amurensis agglutinin (MAA) and Sambucus nigra agglutinin (SNA) lectin agarose beads (EY Laboratories) to bind the α2-3 and α2-6 sialic acid lectins, respectively. The sialylated glycoproteins were eluted with ethylenediamine, and all of the fractions were collected and subjected to an immunoprecipitation assay. The purified sialylated glycoproteins were incubated with sialidase at 4°C for 16 h, followed by mixing with EV71 particles, anti-EV71 antibody, and protein G-agarose beads and incubation at 4°C overnight. The EV71-interacting proteins were centrifuged, washed with buffer, eluted with glycine-HCl, neutralized with NaOH, concentrated, analyzed with silver staining and Western blotting, and subjected to protein identification.
Protein identification.
Bands of interest were cut from the SDS-PAGE gel and rinsed in solution (50% methanol and 5% acetic acid in water). The gel species were dehydrated in acetonitrile, reduced with 10 mM DTT, and carboxyamidomethylated using 100 mM indole acetic acid (IAA) in the dark. The samples were dehydrated again with acetonitrile and then digested with sequencing-grade trypsin at 37°C overnight. Tryptic peptides were sequentially extracted with extraction buffer (50% acetonitrile and 5% formic acid in water) and desalted with Zip-Tip prior to mass analysis. The peptide digests were analyzed by the Mass Solution Technology Co. using a Thermo LTQ (linear trap quadrupole) Velos mass spectrometer for protein identification. The protein identification analyses were repeated in at least three independent experiments. Mascot software (Matrix Science, Boston, MA) was used to analyze the data. Mascot searches were performed with the following parameters: treatment with enzyme trypsin, fixed modifications due to carbamidomethyl (C), variable modifications due to deamidation (NQ) and oxidation (M), peptide tolerance of ±0.5 Da, and fragments mass tolerance of ±0.5 Da.
Silver staining.
The EV71-precipitated proteins were subjected to 10% polyacrylamide gel electrophoresis under nonreducing conditions. The gels were stained with a ProteoSilver silver stain kit (Sigma-Aldrich).
Virus overlay protein binding assay (VOPBA).
The EV71-precipitated proteins were subjected to 10% polyacrylamide gel electrophoresis under nonreducing conditions and transferred to nitrocellulose membranes, which were blocked with binding buffer (1% bovine serum albumin [BSA], 154 mM NaCl, 0.05% Tween 20, and 1 mM CaCl2) at 4°C for 16 h. The membranes were incubated with EV71 particles in binding buffer at 4°C for 16 h with gentle rocking. After three washes with binding buffer, the membranes were incubated with anti-EV71 antibody (clone 422-8D-4C-4D) at room temperature for 2 h, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature for 1 h. After three washes, the images were captured with a Fujifilm LAS-3000 imaging system.
ELISA.
Microtiter plates were coated with serially diluted BSA, EV71 particles, and recombinant VP1 and VP2 proteins in coating buffer (0.1 M Na2HPO4) at 4°C for 16 h. Recombinant human nucleolin (hNCL) protein (10 ng/ml; Abnova) was added to the wells and incubated at 37°C for 2 h. After the unbound proteins were washed away with 0.5% Tween 20 in phosphate-buffered saline (PBS), anti-NCL antibody (Abcam) was added to the wells and incubated at 37°C for 2 h. After a washing step, HRP-conjugated secondary antibody was added to the wells and incubated at 37°C for 1 h, followed by the addition of HRP substrates (Millipore). The binding signals were detected using an enzyme-linked immunosorbent assay (ELISA) reader (PerkinElmer).
Immunofluorescence assay.
The RD cells were seeded on chamber slides and infected with isolate 87-N6356 (10× CCID50) at 4°C for 1 h. The unbound viral particles were removed with PBS, and the cells were fixed with 4% formaldehyde for 5 min. The cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI) or primary antibodies against EV71, NCL, or SCARB2 (Abcam) at 37°C for 2 h, followed by incubation with fluorescence-conjugated secondary antibodies at 37°C for 1 h. The slides were then covered with glass coverslips and mounting medium, and the images were analyzed and captured using a confocal microscope (Olympus) at a ×400 magnification.
Detection of cell surface antigens.
The cells (wild type, vector only, RNA interference [RNAi] knockdown, or overexpression) were washed twice with fluorescence-activated cell sorting (FACS) buffer (0.1% NaN3 and 4% fetal calf serum [FCS] in 1× PBS), fixed with 4% paraformaldehyde at room temperature for 5 min, and incubated with primary antibody at 4°C for 1 h. After cells were washed with FACS buffer, Alexa 488-conjugated anti-mouse or anti-rabbit IgG antibody was added and incubated at 4°C for 0.5 h. The binding was detected using a FACSCalibur instrument (BD Biosciences).
Antibody blocking assay.
RD cells in suspension were treated with control antibody, anti-SCARB2 antibody, or anti-NCL antibody (50 μg/ml) at 37°C for 1 h. After the unbound antibody was washed away, the cells were incubated with 87-N6356 (10× CCID50) at 4°C for 3 h, washed, fixed with 4% paraformaldehyde, and incubated with anti-EV71 antibody (clone 422-8D-4C-4D) at room temperature for 2 h. The cells were incubated with Alexa 488-conjugated secondary antibody at 4°C for 1 h and analyzed by flow cytometry.
Knockdown of SCARB2 or NCL.
The short hairpin RNA (shRNA) sequences were purchased from the National RNAi Core Facility, Academia Sinica, Taiwan, and the sequences were as follows: sh-SCARB2, CCGGGCCCGATATCTCTCCCTATTTCTCGAGAAATAGGGAGAGATATCGGGCTTTTT; sh-NCL-1, CCGGAGTAAAGGGATTGCTTATATTCTCGAGAATATAAGCAATCCCTTTACTTTTTTG; and sh-NCL-2, CCGGGCGATCTATTTCCCTGTACTACTCGAGTAGTACAGGGAAATAGATCGCTTTTTG. The plasmid that contained the shRNA was transfected into RD cells using TurboFect transfection reagent (Thermo). Cells were subjected to Western blotting and flow cytometry to determine the total and cell surface SCARB2 and NCL, respectively, at 24 h posttransfection. The cells were subjected to virus binding and infection assays at 48 h posttransfection.
Overexpression of hNCL in NIH 3T3 and L929 cells.
The full-length human nucleolin gene (hncl) was amplified from the cDNA library of RD cells using the primer pair 5′-CATGAATTCATGGTGAAGCTCGCG-3′ and 5′-GACTCTAGAACAACCCCACGAACG-3′, cloned into an expression vector, p3×FLAG-Myc-CMV-26 (which carries three copies of a FLAG tag; Sigma-Aldrich), and transfected into NIH 3T3 or L929 cells using TurboFect transfection reagent (Thermo) according to the manufacturer's instructions. The stably expressing cell clones were selected in medium containing Geneticin (Sigma-Aldrich). The overexpression levels of cell surface hNCL and total NCL were analyzed by flow cytometry and Western blotting.
Virus binding assay.
A virus binding assay was performed as previously described (9). Briefly, cells were seeded in six-well plates overnight and infected with 87-N6356 (10× CCID50) for 1 h at 4°C. The unbound virus was washed away with a buffer, and the cell lysate was collected at different times to analyze the viral proteins, RNA, or viral particles by Western blotting, quantitative reverse transcription-PCR (qRT-PCR), or flow cytometry, respectively.
Whole-mount immunoelectron microscopy of EV71-infected RD cells.
EV71-infected RD cells were placed on nickel grids in the chamber at 37°C overnight and then fixed with 1% paraformaldehyde for 30 min. After 2 h of blocking with 3% bovine serum albumin in PBS, the specimens were incubated with gold-labeled SCARB2 antibody (3-nm beads) and gold-labeled NCL antibody (13-nm beads) for 60 min and rinsed in PBS. After labeling, specimens were fixed with 0.1% glutaraldehyde for 5 min at room temperature, stained with ammonium molybdate, and air dried. Specimens were viewed in a JEM-1400 electron microscope at 120 kV.
Statistical analysis.
The data were analyzed with Student's t test (two-tailed) using GraphPad Prism, version 5, software. P values of less than 0.05 were considered statistically significant.
RESULTS
Identification and characterization of EV71-interacting glycoproteins.
To identify the EV71-interacting sialylated glycoproteins on the cell surface, the membrane fraction of RD cells was extracted by ultracentrifugation and purified using an MAA/SNA lectin-agarose column. The membrane fraction was confirmed by the presence of E-cadherin, as detected by Western blotting (data not shown), and the bands of the sialylated membrane proteins were validated by Western blotting for MAA and SNA lectins (Fig. 1A). To prevent the binding of EV71 to sialic acids (9, 33), the purified sialylated glycoproteins were treated with sialidase. The EV71-interacting glycoproteins were then isolated by immunoprecipitation with virus particles. The silver staining results of the SDS-PAGE analysis of the EV71-immunoprecipitated proteins revealed several bands, including an obvious band located between 95 and 130 kDa (Fig. 1B). The interactions of EV71 with the immunoprecipitated proteins were also confirmed by EV71 VOPBA (Fig. 1C). All of the bands observed on the SDS-PAGE gel were collected and analyzed by mass spectrometry, and 16 proteins were identified (Table 1). We then focused on a 100-kDa, multifunctional sialylated glycoprotein, NCL, which had been reported as a receptor for HIV and human respiratory syncytial virus (RSV) (28, 29, 34–36). First, we evaluated the binding of EV71 to NCL (in cell lysates) with an immunoprecipitation assay. EV71 can interact with NCL, and the removal of sialic acids reduced the binding of EV71 to NCL by 30% (Fig. 1D). The EV71 capsid consists of VP1, VP2, VP3, and VP4, with VP4 on the inner surface (37). We found that the binding of EV71 or VP1 protein to pure, unglycosylated NCL protein was dependent on the virus/protein concentration (Fig. 1E), suggesting that the VP1-NCL interaction may be responsible for the binding of EV71 to host cells during EV71 infection. However, NCL did not interact with VP2 and VP3 (Fig. 1E and unpublished data). The results also showed that EV71 can interact with desialylated or unglycosylated NCL. Furthermore, pure NCL protein can diminish the binding of EV71 to RD cells (data not shown). We then confirmed that both NCL and SCARB2, a well-studied EV71 receptor, are expressed on the cell surface of RD cells. The colocalization of NCL with EV71 on the RD cell surface was observed by confocal microscopy (Fig. 1F). Both the anti-SCARB2 and anti-NCL antibodies successfully reduced the binding of EV71 to RD cells (Fig. 1G). Taken together, these findings suggest that EV71 can interact with cell surface NCL.
FIG 1.

Identification and characterization of EV71-interacting glycoproteins. (A) Membrane proteins column purified with (+) or without (−) MAA or SNA were separated by SDS-PAGE and immunoblotted with HRP-conjugated MAA and HRP-conjugated SNA lectins. (B) The silver staining of EV71-immunoprecipitated desialylated membrane proteins is shown. (C) The VOPBA of desialylated membrane proteins binding to EV71 particles is shown. Red arrows indicate the bands observed on VOPBA. (D) RD cell lysates were immunoprecipitated with EV71 particles (IP EV) and immunoblotted with antinucleolin (anti-NCL) antibody and HRP-conjugated secondary antibody (left panel). Sialylated membrane proteins, with or without sialidase treatment, were immunoprecipitated with EV71 particles and immunoblotted with anti-NCL antibody and HRP-conjugated secondary antibody (right panel). (E) The binding of serially diluted BSA, EV71 particles, VP1, and VP2 recombinant proteins (from 1 pg/well to 10 ng/well) with pure NCL protein was determined by ELISA. (F) The colocalization (yellow) of EV71 (green) with NCL (red) was observed by confocal microscopy in infected RD cells. Nuclei were stained in blue by DAPI. (G) Anti-SCARB2 or anti-NCL antibody (50 μg/ml) significantly reduced the binding of EV71 to RD cells as determined by flow cytometry. The data are representative of the means ± standard deviations (error bars) of ≥3 samples per group (**, P < 0.01).
TABLE 1.
List of EV71-interacting proteins identified by glycoproteomic approaches
| Protein name | GeneInfo Identifier sequence identification no. | Mol wt | Mascot score | No. of peptide matches |
|---|---|---|---|---|
| Alpha-1 type XV collagen | gi|461397 | 142,412 | 21 | 2 |
| CGI-72 protein | gi|1685049 | 48,129 | 51 | 3 |
| Epidermal cytokeratin 2 | gi|181402 | 66,110 | 63 | 8 |
| Erythrocyte alpha adducin | gi|28382 | 81,377 | 26 | 3 |
| High-risk human papillomavirus E6 oncoproteins, targeted protein E6TP1 alpha | gi|4151328 | 198,570 | 44 | 5 |
| Histone deacetylase 5 | gi|4754909 | 122,714 | 35 | 1 |
| Hr1b domain from Prk1 | gi|159163170 | 9,000 | 22 | 3 |
| Human selenoprotein S (chain A) | gi|149243517 | 10,487 | 32 | 3 |
| Dff40 and Dff45 N-terminal domain complex (chain B) | gi|14278227 | 16,302 | 297 | 13 |
| Nucleolin | gi|189306 | 76,355 | 46 | 8 |
| Phosphatidylinositol 3-kinase regulator, subunit alpha, isoform 1 | gi|32455248 | 83,889 | 58 | 16 |
| Plasminogen | gi|38051823 | 93,263 | 56 | 2 |
| RAF proto-oncogene serine-threonine protein kinase | gi|4506401 | 73,803 | 27 | 4 |
| Type I inner root sheath-specific keratin 25 irs4 | gi|31074643 | 51,191 | 30 | 6 |
| Tyrosine kinase | gi|1161364 | 62,052 | 41 | 14 |
| Zinc metalloendopeptidase | gi|11493589 | 121,646 | 41 | 7 |
Knockdown of cell surface NCL reduces EV71 binding, infection, and production in RD cells.
To determine the influence of NCL deficiency on EV71 infection, the expression of NCL on RD cells was knocked down with shRNA. The knockdown of SCARB2 expression on RD cells served as the control. We first confirmed that NCL shRNA treatment did not affect the cell surface-expressed SCARB2 and vice versa (Fig. 2A). Flow cytometry showed that cell surface NCL expression was also reduced in the NCL-knockdown cells (Fig. 2B). NCL knockdown significantly reduced the binding of both human and mouse-adapted EV71 strains to RD cells by 50 to 70% (Fig. 2C). Decreased levels of bound EV71 virus were also observed using qRT-PCR and Western blotting analyses (Fig. 2D). In addition, double knockdown of SCARB2 and nucleolin in RD cells showed a further reduction in an EV71 binding assay (Fig. 2E). The EV71 RNA levels were decreased by 25% and 45% in the NCL-knockdown cells at 3 and 6 h postinfection (p.i.), respectively (Fig. 2F). To assess the effect of NCL or SCARB2 knockdown on EV71 production, the amounts of virus in the supernatant of infected cells were measured by determining the CCID50 values. Both the NCL- and the SCARB2-knockdown cells showed significantly decreased CCID50 values compared with those of cells transfected with an empty vector at 9 h p.i. (Fig. 2G). In addition, we used five EV71 strains for the binding assay. MP4 is a strain that belongs to genotype C2 and has been adapted four times in mice to increase neurovirulence (38). The strains 87-N6356 in subgenogroup C2 and 97-M448 in subgenogroup B5 were isolated from EV71-infected patients with mild symptoms, and strains 89-N0363 in subgenogroup B4 and 94-N2873 in subgenogroup C4 were isolated from cases with neurological complications. The binding of all five EV71 strains to NCL-knockdown cells was significantly lower than that of cells transfected with an empty vector (Fig. 2H). These results suggest that NCL is involved in the binding and infection of EV71 to host cells.
FIG 2.

The knockdown of cell surface NCL reduces EV71 binding, infection, and production in RD cells. RD cells were transfected without vector (RD) or with an empty vector (Vector) or the vector containing sh-SCARB2, sh-NCL-1, or sh-NCL-2. The expression levels of SCARB2 and NCL were determined by Western blotting (A) and flow cytometry (B). (C) The binding of an EV71 clinical isolate (87-N6356) or mouse-adapted strain (MP4) to RD cells was determined by flow cytometry. (D) The binding of EV71 (87-N6356) to vector-, sh-NCL-1-, sh-NCL-2-, and sh-SCARB2-transfected RD cells was determined by qRT-PCR (top) and Western blotting (bottom). (E) The binding of EV71 to vector, sh-NCL-1-, sh-SCARB2- and sh-NCL-1–sh-SCARB2-transfected RD cells was determined by qRT-PCR. (F) The relative RNA levels of EV71 (87-N6356) in RD cells at 3 and 6 h p.i. were determined by qRT-PCR. The RNA level of vector cells at 3 h was defined as 100%. (G) The 50% cell culture infective dose (CCID50) values of RD cells infected with EV71 (87-N6356) were evaluated. (H) The binding of different genotypes of EV71 (MP4, C2, B4, C4, and B5 strains) to RD cells was measured by flow cytometry. The mean fluorescence intensity value of MP4-infected vector cells was defined as 100%. The data are representative of the means ± standard deviations (error bars) of ≥3 samples per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The expression of hNCL on the surface of a mouse cell line increases EV71 binding, infection, and production in the cells.
The mouse embryonic fibroblast cell line NIH 3T3 is relatively resistant to EV71 infection. To determine whether the expression of hNCL on the cell surface can increase EV71 binding, infection, and production, NIH 3T3 cells were transfected with a vector containing the hncl gene tagged with FLAG. Two cell clones, NCL1 and NCL2, were established. The results of flow cytometric analysis using anti-FLAG antibody detected increased FLAG levels on the surface of both NCL1 and NCL2 cells compared with levels on NIH 3T3 cells transfected with an empty vector (Fig. 3A), suggesting that hNCL was expressed on the surface of both NCL1 and NCL2 cells. Notably, a high FLAG level was detected on NCL2 cells compared with levels on NCL1 cells, suggesting that NCL2 cells express more hNCL on the cell surface than NCL1 cells. Western blotting detected enhanced NCL levels in both NCL1 and NCL2 cells compared with levels in NIH 3T3 cells transfected with an empty vector using anti-NCL antibody, which recognizes both human and mouse NCL proteins, which have similar sizes (Fig. 3B, top panel). Western blotting using an anti-FLAG antibody detected FLAG in both NCL1 and NCL2 cells but not in NIH 3T3 cells transfected with an empty vector (Fig. 3B, middle panel).
FIG 3.

The expression of hNCL increases EV71 binding to a mouse cell line. NIH 3T3 cells were transfected without vector (No vector), an empty vector (Vector), or the vector containing the hncl gene tagged with FLAG (NCL1 and NCL2). (A) The expression of FLAG on the cell surface was determined by flow cytometry. A histogram (top panel) and the quantitative results of the mean fluorescence intensity (MFI) of FLAG-positive cells are shown. (B) The expression levels of NCL, FLAG, and β-actin in cells were detected by Western blotting. (C) The binding of different genotypes of EV71 (MP4, C2, B4, C4, and B5 strains) to RD cells, NIH 3T3 cells transfected with an empty vector (Vector), and NIH 3T3 (NCL2) cells was determined by flow cytometry. The expression of NCL and β-actin in cells detected by Western blotting (D) and the binding of different genotypes of EV71 (MP4, B4, C4, and B5 strains) to RD cells, L929 cells transfected with an empty vector (Vector), and L929 (NCL) cells determined by flow cytometry (E) are shown. The value of MP4-infected RD cells was defined as 100%. The data are representative of the means ± standard deviations (error bars) of ≥3 samples per group. *, P < 0.05; ***, P < 0.001.
Because NCL2 cells express more hNCL on the cell surface than NCL1 cells, NCL2 cells were utilized to evaluate EV71 binding to cells. The results of a binding assay showed that the relative virus binding to NIH 3T3 cells transfected with an empty vector was not very efficient compared with that to RD cells (Fig. 3C). The expression of hNCL significantly increased virus binding to NCL2 cells by more than 2.6-fold compared with that to NIH 3T3 cells transfected with an empty vector (P < 0.05). Remarkably, the levels of virus binding to NCL2 cells were high compared with levels to RD cells. The binding intensities of virus to RD cells, NIH 3T3 cells transfected with an empty vector, and NCL2 cells were comparable irrespective of the genotypes or virus strains. Enhanced EV71 binding by the expression of hNCL was also observed in the mouse L929 cell line (Fig. 3D and E).
We further monitored virus-induced CPE in RD cells, NIH 3T3 cells transfected with an empty vector, and NCL2 cells at 0, 3, and 9 h p.i. A clear CPE was observed in both RD and NCL2 cells but was not observed in NIH 3T3 cells transfected with an empty vector at 9 h p.i. (Fig. 4A). The quantitative data also showed that the virus-induced CPE in NCL2 cells significantly increased compared with that in NIH 3T3 cells transfected with an empty vector at 3 and 9 h p.i. (Fig. 4B).
FIG 4.

The expression of hNCL increases the number of mouse cells infected by EV71. CPE induced by EV71 in RD, NIH 3T3 (Vector), and NIH 3T3 (NCL2) cells at 0, 3, and 9 h p.i. with the 87-N6356 strain (A) and the percentages of cells with CPE (B) are shown. The data are representative of the means ± standard deviations (error bars) of ≥3 samples per group (*, P < 0.05).
In addition, we examined viral growth in RD, NIH 3T3 (empty vector), and NCL2 cells at 0, 12, 24, and 48 h p.i. (multiplicity of infection [MOI] of 10; isolate 87-N6356). Virus growth increased rapidly (>10,000-fold) in RD cells at 24 h p.i. and was maintained at a high level until 48 h p.i. (Fig. 5). Virus growth in NCL2 cells also increased continuously from 0 to 48 h p.i. Furthermore, the virus titer in NCL2 cells was 100-fold higher than that in NIH 3T3 cells transfected with an empty vector at 48 h p.i. The increased virus production suggests that the expression of hNCL enhances the attachment and infection of EV71 to mouse cells.
FIG 5.
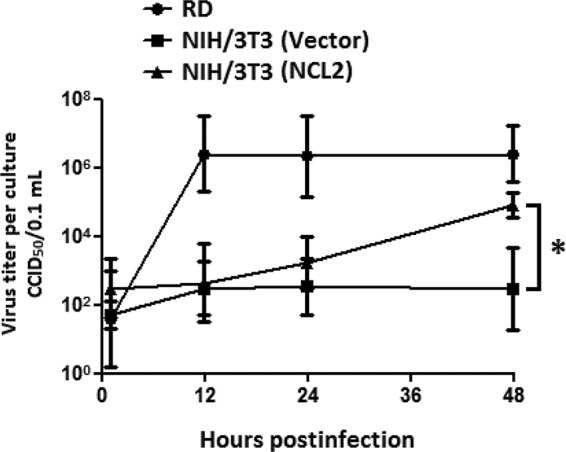
The expression of hNCL increases EV71 production in mouse cells. The data are representative of the means ± standard deviations (error bars) of ≥3 samples per group (*, P < 0.05).
NCL and SCARB2 are associated in uninfected cells, and EV71 infection increases the level of NCL associated with SCARB2 in cells.
The NCL on the cell surface was previously shown to mediate the binding and subsequent clathrin-dependent endocytosis of lactoferrin (39). In addition, SCARB2 mediates the entry of EV71 via a clathrin-dependent endocytosis mechanism (40). Therefore, we assessed the interaction of NCL and SCARB2 in RD cells in the presence or absence of EV71 infection for 3 h at 4°C. The results of an immunoprecipitation assay performed on cell lysates using anti-SCARB2 or anti-NCL antibody showed that NCL and SCARB2 associated in uninfected cells (Fig. 6A and B, left panels). EV71 antigen, NCL, and SCARB2 were found to associate in infected cells, and the level of NCL that associated with SCARB2 was enhanced, as demonstrated by the increased intensity of NCL bands compared to that in uninfected cells (Fig. 6A and B, right panels). In addition, an immunoprecipitation assay using anti-lactoferrin antibody showed that SCARB2 was not associated with lactoferrin in either uninfected or infected RD cells (unpublished data), suggesting that SCARB2 specifically interacts with only certain proteins, such as NCL. The colocalization of EV71, NCL, and SCARB2 in cells was observed using confocal microscopy (Fig. 6C). In order to look deep inside the nucleolin-SCARB2-EV71 interaction, we stained EV71-infected RD cells with gold-labeled SCARB2 antibody (3-nm beads) and gold-labeled NCL antibody (13-nm beads). The electron microscopy image showed that SCARB2 (Fig. 7, small dots) and nucleolin (large dots) were very close to EV71 particles (white circles), suggesting that both proteins are associated with EV71.
FIG 6.

NCL and SCARB2 associate in uninfected cells, and EV71 infection increases the level of NCL associated with SCARB2 in cells. The lysates of RD cells infected without or with 87-N6356 were harvested at 3 h p.i. and immunoprecipitated (IP) with anti-SCARB2 antibody (A) or anti-NCL antibody (B) prior to immunoblotting with anti-NCL, anti-SCARB2, or anti-EV71 antibody. (C) The colocalization (white) of EV71 antigen (green), NCL (red), and SCARB2 (blue) in RD cells infected with 87-N6356 was examined using a confocal microscope. The data are representative of three independent experiments.
FIG 7.

Detection of NCL, SCARB2, and EV71 in RD cells. We stained EV71-infected RD cells with gold-labeled SCARB2 antibody (3-nm beads) and gold-labeled NCL antibody (13-nm beads). The whole-mount cells were viewed after a negative-staining procedure, followed by conventional transmission electron microscopy. EV71 particles are shown in white circles. Small black dots, SCARB2; large black dots, nucleolin. Insets are higher-magnification images.
DISCUSSION
In this study, we identified NCL as an EV71-interacting protein via glycoproteomic analysis. We also demonstrated that cell surface NCL is involved in the binding and infection of EV71 to host cells. Based on our findings, NCL met all of the criteria that define a novel binding receptor for EV71 (28).
Glycoproteomics, which focuses on the analyses and characterization of glycostructures and the function of glycosylated proteins, has attracted much attention. Proteins that contain the desired glycans were enriched from cell lysates or a protein mixture and quantitatively analyzed by mass spectrometry. Glycoproteomic approaches have been applied to identify new tumor markers (25, 41, 42). In this study, we successfully identified 16 EV71-interacting proteins by glycoproteomic approaches. These candidate proteins include four membrane-associated glycoproteins: erythrocyte alpha adducin, human selenoprotein S, plasminogen, and NCL. We found that treatment with antibody against the four membrane-associated glycoproteins reduced the binding of EV71 to RD cells in a manner similar to treatment with anti-SCARB2 antibody (data not shown). Because NCL is the receptor for HIV and RSV (28, 29, 34–36), we further investigated the roles of NCL in EV71 infection.
NCL is a multifunctional protein that is ubiquitously expressed in growing eukaryotic cells (43). It controls the metabolism of DNA and RNA in the nucleolus (44, 45), shuttles proteins into the nucleus (46), provides posttranscriptional regulation of strategic mRNAs in the cytoplasm (47, 48), and serves as a ligand for proteins, such as midkine and the heparin-binding growth-associated molecule, on the cell surface (49). NCL is also involved in the proliferation of many types of cancer (34, 50–52) and serves as a prognosis marker and therapeutic target for cancer treatment (53–55). Furthermore, NCL also participates in the pathogenic mechanism and mediates the replication of several viruses and/or serves as a cell surface receptor for Escherichia coli O157 (56), Helicobacter pylori (57), RSV (28), adeno-associated virus type-2 (AAV-2) (58), coxsackie B virus (59), and HIV (29). Several nontoxic ligands have been reported to block NCL-pathogen interaction. For instance, pseudopeptide HB-19 treatment prevents the anchorage of HIV to target cells (60). HB-19 treatment also inhibits cancer growth, angiogenesis, and tumorigenesis (61, 62). By binding to cell surface NCL, midkine and pleiotrophin (two heparin-binding growth factors) could inhibit HIV infection of cells (63–65).
According to the investigation of Krust et al., cell surface-expressed NCL is predominantly found on the cell surface of lymphoid organs, including the liver, spleen, thymus, and bone marrow (66). A significant amount of NCL was also found on the cell surface of developing muscle and growing cells (43, 67). Hence, the ubiquitous expression and tissue distribution of NCL not only indicate that NCL is involved in EV71 infections but also suggest that the binding of EV71 to cells of the mononuclear phagocyte system, growing cells, and developing muscle is more efficient than its binding to other cells. Further investigations should be performed to decipher the roles of NCL in the infection of EV71 in young children.
In our previous study, we examined the binding of EV71 with SCARB2, with or without sialidase treatment, by VOPBA and found that desialylation slightly reduced the binding of virus with receptor (9). Yamayoshi et al. also found that the interaction of EV71 with recombinant hSCARB2 was moderately decreased after removing N-glycans from hSCARB2 by enzymatic hydrolysis (15). In this study, we purified sialylated membrane proteins by lectin affinity column (Fig. 1A), with or without sialidase treatment, and subjected them to immunoprecipitation with EV71 particles and immunoblotting with anti-NCL antibody. We found that desialylation reduced the binding of EV71 with NCL by 30% (Fig. 1D, right panel). Although desialylation influenced the EV71-NCL interaction, EV71 can bind with unglycosylated NCL (Fig. 1E). These findings demonstrated that EV71 interacts with not only sialic acid but also sialylated, desialylated, and unglycosylated NCL.
Among the previously identified receptors/attachment molecules, PSGL-1 and SCARB2 have been demonstrated as functional receptors for EV71. SCARB2 is a major receptor for all EV71 strains and some enterovirus A species viruses (8). However, only some EV71 strains or genotypes could attach and infect PSGL-1-expressing cells (7). The functional comparison of SCARB2 and PSGL-1 in EV71 infection has been investigated (16). Although the binding ability of EV71 (PSGL-1-binding strain) to PSGL-1-expressing L929 cells is higher than that to SCARB2-expressing L929 cells, the efficiency of EV71 to infect SCARB2-expressing cells is much higher than that for PSGL-1-expressing cells. The authors suggest that SCARB2 not only mediates the binding of EV71 to host cells but is also involved in the endocytosis and uncoating of EV71. In this study, we showed that the binding of different genotypes of EV71 to hNCL-expressing mouse cells was significantly increased (Fig. 3C and E). The knockdown of NCL could also block the binding of all tested EV71 strains to host cells (Fig. 2H). Combined with the increased CPE and viral yield observed in EV71-resistant cells after the expression of hNCL (Fig. 4 and 5), these findings suggest that NCL could be recognized by all EV71 strains. However, the infection efficiency is not as good as in RD cells. The mechanism of NCL-mediated EV71 infection should be further investigated.
In this study, we found that hNCL directly interacted with viral particles and the capsid protein VP1 of EV71 (Fig. 1C). VP1, an outer surface capsid protein of EV71, is reportedly involved in virus infectivity (68) and immunogenicity (69, 70). VP1 has been shown to interact with the cell surface receptors SCARB2 and PSGL-1 (71, 72) and with the binding molecules lactoferrin and annexin IIA (10, 73). Treatment with peptides derived from VP1 also inhibits the infection of host cells by EV71 (74). EV71-specific interacting domains (in hNCL) and hNCL-specific interacting peptides (in VP1) need to be identified to elucidate the complete infection mechanism of EV71 and identify novel targets for antiviral drug development.
The restriction of EV71 infection reportedly occurs at the early steps, such as during binding, internalization, and virus uncoating (8). The expression of putative receptors (SCARB2 and PSGL-1) on the cell surface of EV71-resistant cells (L929) transforms these mouse cells to become susceptible to infection by EV71 (7, 8). Although the protein sequence of mouse SCARB2 shows 85.8% identity and 99.9% similarity to human SCARB2, only human SCARB2-transfected L929 cells exhibit EV71 binding and infection (15). In the current study, we found that cell surface murine NCL cannot mediate the binding and infection of EV71 to mouse cells (NIH 3T3) even though the protein sequence of murine NCL exhibits 81.7% identity to hNCL. Nevertheless, the expression of hNCL on NIH 3T3 cells enabled the binding and infection of EV71 to transfected cells. Although the virus titer in NCL2 cells was 10-fold lower than that in RD cells at 48 h p.i. and although the growth rate was not as good as that in RD cells, the viral yield was 100-fold better than that in NIH 3T3 cells transfected with an empty vector (Fig. 5). Although we showed that EV71 binds more efficiently to NCL2 cells (NIH 3T3 cells expressing human NCL) than to RD cells, the viral yield in NCL2 cells is still lower than that of RD cells (Fig. 4 and 5). These results suggest that there should be another block to viral internalization and replication in NIH 3T3 cells. Key molecules that participate in the entry and replication of EV71 in RD and EV71-resistant cells need to be identified to understand the infection mechanism of EV71.
Our findings demonstrate that the use of glycoproteomic technologies is a reliable and powerful tool for the discovery of viral receptors. A novel EV71-binding receptor, namely, cell surface NCL, was successfully identified in EV71-susceptible cell lines. We proved the direct interaction between NCL and EV71 and evaluated the functional role of NCL in EV71 infection. The findings in hNCL-expressing, EV71-resistant cells also strongly suggest that NCL alone can mediate the binding and infection of EV71 to host cells. All of these results confirm that EV71 uses multiple receptors for viral binding and infection (18). Further studies should address the receptor antagonist development for the prevention and treatment of EV71-induced neuron toxicity.
ACKNOWLEDGMENTS
This work was supported by the Ministry of Science and Technology of Taiwan (NSC 102-2321-B-006-006, MOST 103-2320-B-006-041, and 104-2321-B-006-018) and the Headquarters of University Advancement at the National Cheng Kung University, which is sponsored by the Ministry of Education, Taiwan (D103-35B08).
REFERENCES
- 1.Schmidt NJ, Lennette EH, Ho HH. 1974. An apparently new enterovirus isolated from patients with disease of the central nervous system. J Infect Dis 129:304–309. doi: 10.1093/infdis/129.3.304. [DOI] [PubMed] [Google Scholar]
- 2.Ho M. 2000. Enterovirus 71: the virus, its infections and outbreaks. J Microbiol Immunol Infect 33:205–216. [PubMed] [Google Scholar]
- 3.McMinn PC. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev 26:91–107. doi: 10.1111/j.1574-6976.2002.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 4.Lin JY, Chen TC, Weng KF, Chang SC, Chen LL, Shih SR. 2009. Viral and host proteins involved in picornavirus life cycle. J Biomed Sci 16:103. doi: 10.1186/1423-0127-16-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat Rev Microbiol 3:765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]
- 6.Evans DJ, Almond JW. 1998. Cell receptors for picornaviruses as determinants of cell tropism and pathogenesis. Trends Microbiol 6:198–202. doi: 10.1016/S0966-842X(98)01263-3. [DOI] [PubMed] [Google Scholar]
- 7.Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. 2009. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med 15:794–797. doi: 10.1038/nm.1961. [DOI] [PubMed] [Google Scholar]
- 8.Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, Koike S. 2009. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med 15:798–801. doi: 10.1038/nm.1992. [DOI] [PubMed] [Google Scholar]
- 9.Su PY, Liu YT, Chang HY, Huang SW, Wang YF, Yu CK, Wang JR, Chang CF. 2012. Cell surface sialylation affects binding of enterovirus 71 to rhabdomyosarcoma and neuroblastoma cells. BMC Microbiol 12:162. doi: 10.1186/1471-2180-12-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang SL, Chou YT, Wu CN, Ho MS. 2011. Annexin II binds to capsid protein VP1 of enterovirus 71 and enhances viral infectivity. J Virol 85:11809–11820. doi: 10.1128/JVI.00297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan CW, Poh CL, Sam IC, Chan YF. 2013. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J Virol 87:611–620. doi: 10.1128/JVI.02226-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin TY, Hsia SH, Huang YC, Wu CT, Chang LY. 2003. Proinflammatory cytokine reactions in enterovirus 71 infections of the central nervous system. Clin Infect Dis 36:269–274. doi: 10.1086/345905. [DOI] [PubMed] [Google Scholar]
- 13.Du N, Cong H, Tian H, Zhang H, Zhang W, Song L, Tien P. 2014. Cell surface vimentin is an attachment receptor for enterovirus 71. J Virol 88:5816–5833. doi: 10.1128/JVI.03826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang JR, Tuan YC, Tsai HP, Yan JJ, Liu CC, Su IJ. 2002. Change of major genotype of enterovirus 71 in outbreaks of hand-foot-and-mouth disease in Taiwan between 1998 and 2000. J Clin Microbiol 40:10–15. doi: 10.1128/JCM.40.1.10-15.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamayoshi S, Koike S. 2011. Identification of a human SCARB2 region that is important for enterovirus 71 binding and infection. J Virol 85:4937–4946. doi: 10.1128/JVI.02358-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamayoshi S, Ohka S, Fujii K, Koike S. 2013. Functional comparison of SCARB2 and PSGL1 as receptors for enterovirus 71. J Virol 87:3335–3347. doi: 10.1128/JVI.02070-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura Y, Shimizu H. 2012. Cellular receptors for human enterovirus species A. Front Microbiol 3:105. doi: 10.3389/fmicb.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel KP, Bergelson JM. 2009. Receptors identified for hand, foot and mouth virus. Nat Med 15:728–729. doi: 10.1038/nm0709-728. [DOI] [PubMed] [Google Scholar]
- 19.Tian Y, Zhang H. 2010. Glycoproteomics and clinical applications. Proteomics Clin Appl 4:124–132. doi: 10.1002/prca.200900161. [DOI] [PubMed] [Google Scholar]
- 20.Matsuda A, Kuno A, Matsuzaki H, Kawamoto T, Shikanai T, Nakanuma Y, Yamamoto M, Ohkohchi N, Ikehara Y, Shoda J, Hirabayashi J, Narimatsu H. 2013. Glycoproteomics-based cancer marker discovery adopting dual enrichment with Wisteria floribunda agglutinin for high specific glyco-diagnosis of cholangiocarcinoma. J Proteomics 85:1–11. doi: 10.1016/j.jprot.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Fry SA, Sinclair J, Timms JF, Leathem AJ, Dwek MV. 2013. A targeted glycoproteomic approach identifies cadherin-5 as a novel biomarker of metastatic breast cancer. Cancer Lett 328:335–344. doi: 10.1016/j.canlet.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Chen YT, Chong YM, Cheng CW, Ho CL, Tsai HW, Kasten FH, Chen YL, Chang CF. 2013. Identification of novel tumor markers for oral squamous cell carcinoma using glycoproteomic analysis. Clin Chim Acta 420:45–53. doi: 10.1016/j.cca.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 23.Ueda K. 2013. Glycoproteomic strategies: from discovery to clinical application of cancer carbohydrate biomarkers. Proteomics Clin Appl 7:607–617. doi: 10.1002/prca.201200123. [DOI] [PubMed] [Google Scholar]
- 24.Butterfield DA, Owen JB. 2011. Lectin-affinity chromatography brain glycoproteomics and Alzheimer disease: insights into protein alterations consistent with the pathology and progression of this dementing disorder. Proteomics Clin Appl 5:50–56. doi: 10.1002/prca.201000070. [DOI] [PubMed] [Google Scholar]
- 25.Abbott KL, Lim JM, Wells L, Benigno BB, McDonald JF, Pierce M. 2010. Identification of candidate biomarkers with cancer-specific glycosylation in the tissue and serum of endometrioid ovarian cancer patients by glycoproteomic analysis. Proteomics 10:470–481. doi: 10.1002/pmic.200900537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Powlesland AS, Hitchen PG, Parry S, Graham SA, Barrio MM, Elola MT, Mordoh J, Dell A, Drickamer K, Taylor ME. 2009. Targeted glycoproteomic identification of cancer cell glycosylation. Glycobiology 19:899–909. doi: 10.1093/glycob/cwp065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nizet V, Esko JD. 2009. Bacterial and viral infections, p 537–552. In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (ed), Essentials of glycobiology, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 28.Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. 2011. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med 17:1132–1135. doi: 10.1038/nm.2444. [DOI] [PubMed] [Google Scholar]
- 29.Alvarez Losada S, Canto-Nogues C, Munoz-Fernandez MA. 2002. A new possible mechanism of human immunodeficiency virus type 1 infection of neural cells. Neurobiol Dis 11:469–478. doi: 10.1006/nbdi.2002.0566. [DOI] [PubMed] [Google Scholar]
- 30.Nisole S, Krust B, Callebaut C, Guichard G, Muller S, Briand JP, Hovanessian AG. 1999. The anti-HIV pseudopeptide HB-19 forms a complex with the cell-surface-expressed nucleolin independent of heparan sulfate proteoglycans. J Biol Chem 274:27875–27884. doi: 10.1074/jbc.274.39.27875. [DOI] [PubMed] [Google Scholar]
- 31.Hsiung GD. 1994. Hsiung's diagnostic virology. Yale University Press, New Haven, CT. [Google Scholar]
- 32.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg (Lond) 27:493–497. [Google Scholar]
- 33.Yang B, Chuang H, Yang KD. 2009. Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol J 6:141. doi: 10.1186/1743-422X-6-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajrishi MM, Tuteja R, Tuteja N. 2011. Nucleolin: The most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol 4:267–275. doi: 10.4161/cib.4.3.14884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginisty H, Sicard H, Roger B, Bouvet P. 1999. Structure and functions of nucleolin. J Cell Sci 112:761–772. [DOI] [PubMed] [Google Scholar]
- 36.Carpentier M, Morelle W, Coddeville B, Pons A, Masson M, Mazurier J, Legrand D. 2005. Nucleolin undergoes partial N- and O-glycosylations in the extranuclear cell compartment. Biochemistry 44:5804–5815. doi: 10.1021/bi047831s. [DOI] [PubMed] [Google Scholar]
- 37.Plevka P, Perera R, Cardosa J, Kuhn RJ, Rossmann MG. 2012. Crystal structure of human enterovirus 71. Science 336:1274. doi: 10.1126/science.1218713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang YF, Chou CT, Lei HY, Liu CC, Wang SM, Yan JJ, Su IJ, Wang JR, Yeh TM, Chen SH, Yu CK. 2004. A mouse-adapted enterovirus 71 strain causes neurological disease in mice after oral infection. J Virol 78:7916–7924. doi: 10.1128/JVI.78.15.7916-7924.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Legrand D, Vigie K, Said EA, Elass E, Masson M, Slomianny MC, Carpentier M, Briand JP, Mazurier J, Hovanessian AG. 2004. Surface nucleolin participates in both the binding and endocytosis of lactoferrin in target cells. Eur J Biochem 271:303–317. doi: 10.1046/j.1432-1033.2003.03929.x. [DOI] [PubMed] [Google Scholar]
- 40.Lin YW, Lin HY, Tsou YL, Chitra E, Hsiao KN, Shao HY, Liu CC, Sia C, Chong P, Chow YH. 2012. Human SCARB2-mediated entry and endocytosis of EV71. PLoS One 7:e30507. doi: 10.1371/journal.pone.0030507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hongsachart P, Huang-Liu R, Sinchaikul S, Pan FM, Phutrakul S, Chuang YM, Yu CJ, Chen ST. 2009. Glycoproteomic analysis of WGA-bound glycoprotein biomarkers in sera from patients with lung adenocarcinoma. Electrophoresis 30:1206–1220. doi: 10.1002/elps.200800405. [DOI] [PubMed] [Google Scholar]
- 42.Ueda K, Fukase Y, Katagiri T, Ishikawa N, Irie S, Sato TA, Ito H, Nakayama H, Miyagi Y, Tsuchiya E, Kohno N, Shiwa M, Nakamura Y, Daigo Y. 2009. Targeted serum glycoproteomics for the discovery of lung cancer-associated glycosylation disorders using lectin-coupled ProteinChip arrays. Proteomics 9:2182–2192. doi: 10.1002/pmic.200800374. [DOI] [PubMed] [Google Scholar]
- 43.Srivastava M, Pollard HB. 1999. Molecular dissection of nucleolin's role in growth and cell proliferation: new insights. FASEB J 13:1911–1922. [PubMed] [Google Scholar]
- 44.Wang Y, Guan J, Wang H, Leeper D, Iliakis G. 2001. Regulation of DNA replication after heat shock by replication protein a-nucleolin interactions. J Biol Chem 276:20579–20588. doi: 10.1074/jbc.M100874200. [DOI] [PubMed] [Google Scholar]
- 45.Abdelmohsen K, Gorospe M. 2012. RNA-binding protein nucleolin in disease. RNA Biol 9:799–808. doi: 10.4161/rna.19718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barel M, Meibom K, Charbit A. 2010. Nucleolin, a shuttle protein promoting infection of human monocytes by Francisella tularensis. PLoS One 5:e14193. doi: 10.1371/journal.pone.0014193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bremer S, Klein K, Sedlmaier A, Abouzied M, Gieselmann V, Franken S. 2013. Hepatoma-derived growth factor and nucleolin exist in the same ribonucleoprotein complex. BMC Biochem 14:2. doi: 10.1186/1471-2091-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh K, Laughlin J, Kosinski PA, Covey LR. 2004. Nucleolin is a second component of the CD154 mRNA stability complex that regulates mRNA turnover in activated T cells. J Immunol 173:976–985. doi: 10.4049/jimmunol.173.2.976. [DOI] [PubMed] [Google Scholar]
- 49.Take M, Tsutsui J, Obama H, Ozawa M, Nakayama T, Maruyama I, Arima T, Muramatsu T. 1994. Identification of nucleolin as a binding protein for midkine (MK) and heparin-binding growth associated molecule (HB-GAM). J Biochem 116:1063–1068. [DOI] [PubMed] [Google Scholar]
- 50.Xu Z, Joshi N, Agarwal A, Dahiya S, Bittner P, Smith E, Taylor S, Piwnica-Worms D, Weber J, Leonard JR. 2012. Knocking down nucleolin expression in gliomas inhibits tumor growth and induces cell cycle arrest. J Neurooncol 108:59–67. doi: 10.1007/s11060-012-0827-2. [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Xu Z, Li D, Cheng S, Fan K, Li C, Li A, Zhang J, Feng M. 2014. Cell surface nucleolin is crucial in the activation of the CXCL12/CXCR4 signaling pathway. Tumour Biol 35:333–338. doi: 10.1007/s13277-013-1044-0. [DOI] [PubMed] [Google Scholar]
- 52.Destouches D, El Khoury D, Hamma-Kourbali Y, Krust B, Albanese P, Katsoris P, Guichard G, Briand JP, Courty J, Hovanessian AG. 2008. Suppression of tumor growth and angiogenesis by a specific antagonist of the cell-surface expressed nucleolin. PLoS One 3:e2518. doi: 10.1371/journal.pone.0002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiu W, Zhou F, Zhang Q, Sun X, Shi X, Liang Y, Wang X, Yue L. 2013. Overexpression of nucleolin and different expression sites both related to the prognosis of gastric cancer. APMIS 121:919–925. doi: 10.1111/apm.12131. [DOI] [PubMed] [Google Scholar]
- 54.Soundararajan S, Wang L, Sridharan V, Chen W, Courtenay-Luck N, Jones D, Spicer EK, Fernandes DJ. 2009. Plasma membrane nucleolin is a receptor for the anticancer aptamer AS1411 in MV4-11 leukemia cells. Mol Pharmacol 76:984–991. doi: 10.1124/mol.109.055947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng L, Liang J, Wang H, Song X, Rashid A, Gomez HF, Corley LJ, Abbruzzese JL, Fleming JB, Evans DB. 2010. High levels of nucleolar expression of nucleolin are associated with better prognosis in patients with stage II pancreatic ductal adenocarcinoma. Clin Cancer Res 16:3734–3742. doi: 10.1158/1078-0432.CCR-09-3411. [DOI] [PubMed] [Google Scholar]
- 56.Sinclair JF, O'Brien AD. 2002. Cell surface-localized nucleolin is a eukaryotic receptor for the adhesin intimin-gamma of enterohemorrhagic Escherichia coli O157:H7. J Biol Chem 277:2876–2885. doi: 10.1074/jbc.M110230200. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe T, Tsuge H, Imagawa T, Kise D, Hirano K, Beppu M, Takahashi A, Yamaguchi K, Fujiki H, Suganuma M. 2010. Nucleolin as cell surface receptor for tumor necrosis factor-alpha inducing protein: a carcinogenic factor of Helicobacter pylori. J Cancer Res Clin Oncol 136:911–921. doi: 10.1007/s00432-009-0733-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qiu J, Brown KE. 1999. A 110-kDa nuclear shuttle protein, nucleolin, specifically binds to adeno-associated virus type 2 (AAV-2) capsid. Virology 257:373–382. doi: 10.1006/viro.1999.9664. [DOI] [PubMed] [Google Scholar]
- 59.de Verdugo UR, Selinka HC, Huber M, Kramer B, Kellermann J, Hofschneider PH, Kandolf R. 1995. Characterization of a 100-kilodalton binding protein for the six serotypes of coxsackie B viruses. J Virol 69:6751–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nisole S, Said EA, Mische C, Prevost MC, Krust B, Bouvet P, Bianco A, Briand JP, Hovanessian AG. 2002. The anti-HIV pentameric pseudopeptide HB-19 binds the C-terminal end of nucleolin and prevents anchorage of virus particles in the plasma membrane of target cells. J Biol Chem 277:20877–20886. doi: 10.1074/jbc.M110024200. [DOI] [PubMed] [Google Scholar]
- 61.Birmpas C, Briand JP, Courty J, Katsoris P. 2012. The pseudopeptide HB-19 binds to cell surface nucleolin and inhibits angiogenesis. Vasc Cell 4:21. doi: 10.1186/2045-824X-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krust B, El Khoury D, Soundaramourty C, Nondier I, Hovanessian AG. 2011. Suppression of tumorigenicity of rhabdoid tumor derived G401 cells by the multivalent HB-19 pseudopeptide that targets surface nucleolin. Biochimie 93:426-433. doi: 10.1016/j.biochi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 63.Hovanessian AG. 2006. Midkine, a cytokine that inhibits HIV infection by binding to the cell surface expressed nucleolin. Cell Res 16:174–181. doi: 10.1038/sj.cr.7310024. [DOI] [PubMed] [Google Scholar]
- 64.Said EA, Krust B, Nisole S, Svab J, Briand JP, Hovanessian AG. 2002. The anti-HIV cytokine midkine binds the cell surface-expressed nucleolin as a low affinity receptor. J Biol Chem 277:37492–37502. doi: 10.1074/jbc.M201194200. [DOI] [PubMed] [Google Scholar]
- 65.Said EA, Courty J, Svab J, Delbe J, Krust B, Hovanessian AG. 2005. Pleiotrophin inhibits HIV infection by binding the cell surface-expressed nucleolin. FEBS J 272:4646–4659. doi: 10.1111/j.1742-4658.2005.04870.x. [DOI] [PubMed] [Google Scholar]
- 66.Krust B, Vienet R, Cardona A, Rougeot C, Jacotot E, Callebaut C, Guichard G, Briand JP, Grognet JM, Hovanessian AG, Edelman L. 2001. The anti-HIV pentameric pseudopeptide HB-19 is preferentially taken up in vivo by lymphoid organs where it forms a complex with nucleolin. Proc Natl Acad Sci U S A 98:14090–14095. doi: 10.1073/pnas.221467298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alete DE, Weeks ME, Hovanession AG, Hawadle M, Stoker AW. 2006. Cell surface nucleolin on developing muscle is a potential ligand for the axonal receptor protein tyrosine phosphatase-sigma. FEBS J 273:4668–4681. doi: 10.1111/j.1742-4658.2006.05471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang SW, Wang YF, Yu CK, Su IJ, Wang JR. 2012. Mutations in VP2 and VP1 capsid proteins increase infectivity and mouse lethality of enterovirus 71 by virus binding and RNA accumulation enhancement. Virology 422:132–143. doi: 10.1016/j.virol.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 69.Xu J, Wang S, Gan W, Zhang W, Ju L, Huang Z, Lu S. 2012. Expression and immunogenicity of novel subunit enterovirus 71 VP1 antigens. Biochem Biophys Res Commun 420:755–761. doi: 10.1016/j.bbrc.2012.03.067. [DOI] [PubMed] [Google Scholar]
- 70.Meng T, Kolpe AB, Kiener TK, Chow VT, Kwang J. 2011. Display of VP1 on the surface of baculovirus and its immunogenicity against heterologous human enterovirus 71 strains in mice. PLoS One 6:e21757. doi: 10.1371/journal.pone.0021757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen P, Song Z, Qi Y, Feng X, Xu N, Sun Y, Wu X, Yao X, Mao Q, Li X, Dong W, Wan X, Huang N, Shen X, Liang Z, Li W. 2012. Molecular determinants of enterovirus 71 viral entry: cleft around GLN-172 on VP1 protein interacts with variable region on scavenge receptor B 2. J Biol Chem 287:6406–6420. doi: 10.1074/jbc.M111.301622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishimura Y, Lee H, Hafenstein S, Kataoka C, Wakita T, Bergelson JM, Shimizu H. 2013. Enterovirus 71 binding to PSGL-1 on leukocytes: VP1-145 acts as a molecular switch to control receptor interaction. PLoS Pathog 9:e1003511. doi: 10.1371/journal.ppat.1003511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin TY, Chu C, Chiu CH. 2002. Lactoferrin inhibits enterovirus 71 infection of human embryonal rhabdomyosarcoma cells in vitro. J Infect Dis 186:1161–1164. doi: 10.1086/343809. [DOI] [PubMed] [Google Scholar]
- 74.Tan CW, Chan YF, Sim KM, Tan EL, Poh CL. 2012. Inhibition of enterovirus 71 (EV-71) infections by a novel antiviral peptide derived from EV-71 capsid protein VP1. PLoS One 7:e34589. doi: 10.1371/journal.pone.0034589. [DOI] [PMC free article] [PubMed] [Google Scholar]