

ABSTRACT
Japanese encephalitis virus (JEV) strains can be separated into 5 genotypes (g1 to g5) based on sequence similarity. JEV g5 strains have been rarely isolated and are poorly characterized. We report here the full characterization of a g5 virus generated using a cDNA-based technology and its comparison with a widely studied g3 strain. We did not observe any major differences between those viruses when their infectious cycles were studied in various cell lines in vitro. Interestingly, the JEV g5 strain was highly pathogenic when inoculated to BALB/c mice, which are known to be largely resistant to JEV g3 infection. The study of chimeric viruses between JEV g3 and g5 showed that there was a poor viral clearance of viruses that express JEV g5 structural proteins in BALB/c mice blood, which correlated with viral invasion of the central nervous system and encephalitis. In addition, using an in vitro model of the blood-brain barrier, we were able to show that JEV g5 does not have an enhanced capacity for entering the central nervous system, compared to JEV g3. Overall, in addition to providing a first characterization of the understudied JEV g5, our work highlights the importance of sustaining an early viremia in the development of JEV encephalitis.
IMPORTANCE Genotype 5 viruses are genetically and serologically distinct from other JEV genotypes and can been associated with human encephalitis, which warrants the need for their characterization. In this study, we characterized the in vitro and in vivo properties of a JEV g5 strain and showed that it was more neuropathogenic in a mouse model than a well-characterized JEV g3 strain. The enhanced virulence of JEV g5 was associated with poor viral clearance but not with enhanced crossing of the blood-brain barrier, thus providing new insights into JEV pathogenesis.
INTRODUCTION
Japanese encephalitis virus (JEV) is a member of the Flavivirus genus in the Flaviviridae family. JEV has a positive-sense RNA genome encoding a single polyprotein. This polyprotein is processed by host- and JEV-encoded proteases into 10 proteins: three structural proteins (core [C], premembrane [prM], and envelope [E]) and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5).
JEV is a significant human pathogen and the causative agent for Japanese encephalitis, one of the most important viral encephalitides of medical interest in Asia, with an incidence of approximately 67,900 cases per year, among which are about 20,000 fatal cases (1). About 20 to 30% of the symptomatic human cases are fatal, while 30 to 50% of survivors can develop long-term neurological sequelae (2). JEV is maintained in an enzootic cycle between Culex tritaeniorhynchus mosquitoes and amplifying vertebrate hosts, such as water birds and domestic swine (3). Humans and several other animals can also be infected, but since they do not develop a sufficient level of viremia to infect mosquitoes, they are thought to be dead-end hosts (4).
Phylogenetic studies based on the viral envelope protein sequences allow the division of JEV strains into five genotypes (g1 to g5). From the isolation of the prototype strain of JEV in 1935 until recently, most of the circulating strains of JEV belonged to g3 and were at the origin of major epidemics in Southeast Asian countries (5). Recently, a shift in prevalence from JEV g3 to g1 has been observed in several Asian countries (6–8), while some strains of JEV g5 have been occasionally isolated in China in 2009 (9) and in South Korea in 2010 (10). The JEV g5 prototype strain, strain Muar, was originally isolated in 1952 from an encephalitis patient in Malaysia (11) and was found to be genetically and serologically distinct from other genotypes (12–14). No other JEV g5 strain had been identified until 2009-2010, and little characterization of this virus has been made. The recent finding that JEV g5 is still circulating in Asia (9, 10) has highlighted the need for an in-depth characterization of g5 viruses, especially given that they share little sequence identity with JEV strains belonging to the well-studied genotype 1 or 3.
A recent report described the construction of molecular virology tools that will help characterize the prototype strain Muar (15). In the present study, we used a cDNA-based technology to produce a g5 virus derived from the recently isolated strain XZ0934 (9). The pathogenicity of this molecular clone of JEV g5 was analyzed and compared to a well-characterized JEV g3 strain. We showed that while BALB/c mice were largely resistant to JEV g3, they were sensitive to JEV g5 infection and developed viral encephalitis. The marked neuropathogenicity of JEV g5 for BALB/c mice was mostly dependent on the virus capacity to sustain an early viremia in mice. The study of chimeric JEV between g3 and g5 demonstrated the implication of the structural protein region in the neuroinvasive properties of JEV g5 in BALB/c mice.
MATERIALS AND METHODS
Cells.
Mosquito Aedes albopictus C6/36 cells were maintained at 28°C in Leibovitz medium (L15) supplemented with 10% heat-inactivated fetal bovine serum (FBS). Baby hamster kidney-derived BHK-21, chicken fibroblast-derived DF-1, human neuroblastoma-derived SK-N-SH, and human kidney-derived HEK293T cells were maintained at 37°C in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS. JEV replicon cells were cultured in DMEM supplemented with 10% Tet System Approved FBS (catalog no. 631106; Clontech).
JEV-RP-9 production (genotype 3).
JEV g3 strain RP-9 is a plaque-purified variant of the NT109 strain, isolated from Culex tritaeniorhynchus mosquitoes in Taiwan in 1985 (16). A molecular cDNA clone of JEV-RP-9 was kindly provided by Yi-Lin Ling (17). This plasmid was modified to ensure correct propagation in bacteria, through site-directed mutagenesis of a bacterial cryptic promoter, located between positions 1787 and 1873, that had not yet been identified in the genome of JEV RP-9 (18). We first used the primer pairs 5′-CAAGCTCAGTGAAGTTGACATCAGGCCACCTG-3′/5′-CAGGTGGCCTGATGTCAACTTCACTGAGCTTG-3′ and 5′-GGCCACCTGAAATGCAGGCTGAAAATGG-3′/5′-CCATTTTCAGCCTGCATTTCAGGTGGCC-3′ to introduce silent mutations predicted to disrupt the bacterial promoter (mutations are underlined); then, we reintroduced a missing nucleotide at position A1915 using the primers 5′-AGAAAAATTCTCGTTCGCAAAAAATCCGGCGGACAC-3′ and 5′-GTGTCCGCCGGATTTTTTGCGAACGAGAATTTTTCT-3′. A nonsilent mutation at position 3216, which changed the isoleucine at position 247 in the NS1 protein to a valine, was also reverted to the wild-type sequence using primers 5′-CATCATTCCGCATACCATAGCCGGACCAAAAAGCAA-3′ and 5′-TTGCTTTTTGGTCCGGCTATGGTATGCGGAATGATG-3′. The resulting plasmid, pBR322(CMV)-JEV-RP9, could then be stably propagated at 30°C in Stbl2 cells (catalog no. 10268-019; Life Technologies). To produce infectious virus, the molecular clone was transfected into HEK293T cells using Lipofectamine 2000 (catalog no. 11668-019; Life Technologies). At 3 days posttransfection, viral supernatants were collected and used to infect C6/36 cells in order to grow final virus stocks for experiments.
JEV-XZ0934 production (genotype 5).
JEV g5 strain XZ0934 was isolated from Culex tritaeniorhynchus mosquitoes in China in 2009 (9). Four different plasmids containing partial cDNA of JEV-XZ0934 (GenBank accession number JF915894) were synthesized by GeneCust. The first plasmid was designed so that a cytomegalovirus (CMV) promoter sequence was fused to a sequence encompassing nucleotides 1 to 2226 of JEV-XZ0934. The second and third plasmids contained cDNA sequences encompassing nucleotides 2183 to 5595 and 5564 to 8184 of JEV-XZ0934. The last plasmid contained a cDNA sequence encompassing nucleotides 8149 to 10983, immediately adjacent to the hepatitis delta virus (HDV) ribozyme, and was followed by a simian virus 40 (SV40) poly(A) sequence. The sequence of interest in each plasmid was amplified by PCR using Phusion High-Fidelity DNA polymerase (catalog no. F-530S; Thermo Scientific) and the primer pairs 5′-AGGGCATCGGTCGACTAGTA-3′/5′-GTCTCTGAGCACCCTTAAGAGTGGTCGTGAAAGCCTTTCCCAGT-3′; 5′-ACTGGGAAAGGCTTTCACGACCACTCTTAAGGGTGCTCAGAGAC-3′/5′-TGGAGTCAGGGAAGGGGTCAGTCGTTCCTGGT-3′; 5′-ACCAGGAACGACTGACCCCTTCCCTGACTCCA-3′/5′-AGGCCATTTCCAGAACACGCAGAGTGCGTTGCTCCT-3′ and 5′-AGGAGCAACGCACTCTGCGTGTTCTGGAAATGGCCT-3′/5′-ATCGATTTAAGATACATTGA-3′. Each fragment shared an overlap of 44, 32, and 36 nucleotides, respectively, with the next fragment, which permitted assembly of the viral full-length genome after transfection into HEK293T cells using Lipofectamine 2000 (catalog no. 11668-019; Life Technologies). At 3 days posttransfection, viral supernatants were collected and used to infect C6/36 cells in order to grow final virus stocks for experiments. Viral RNA was extracted from the supernatants using NucleoSpin RNA (catalog no. 740955; Macherey-Nagel) according to the manufacturer's instructions. Most of the JEV genome was amplified by reverse transcription (RT)-PCR using the SuperScript III One-Step RT-PCR system with Platinum Taq DNA polymerase (catalog no. 12574-018; Life Technologies) and the primer pairs 5′-AGAAGTTTATCTGTGTGAAC-3′/5′-GTCTCTGAGCACCCTTAAGAGTGGTCGTGAAAGCCTTTCCCAGT-3′; 5′-AATTGGAACCACCATTTGGA-3′/5′-GAGTGATCTCATTCTCGTCCCA-3′; 5′-AGATGGTGTTGAGGAAAGTGA-3′/5′-AACCCAATCGCAGACAAAAC-3′; 5′-CTCAGTTCCACTGGGTGGTT-3′/5′-AACCATTTCAAAGCCTGGTG-3′; 5′-GACCTCGGACGAATGCTATC-3′/5′-TTTGGGTCCTCTCCAAACTG-3′ and 5′-TCCATACATGCCAAAGGTGA-3′/5′-AGATCCTGTGTTCTTCCTCA-3′. The PCR fragments were then sequenced by Eurofins Genomics.
JEV S-g5/NS-g3 chimeric virus production.
A silent mutation that created a unique restriction site (AflII) at positions 2208 to 2213 (residues 705 and 706 of the viral polyprotein) was introduced directly in pBR322(CMV)-JEV-RP9 through PCR mutagenesis using primers 5′-ACTGGGAAAGGCTTTCACGACCACTCTTAAGGGTGCTCAGAGAC-3′ and 5′-GTCTCTGAGCACCCTTAAGAGTGGTCGTGAAAGCCTTTCCCAGT-3′ (the AflII site is underlined). The resulting pBR322(CMV)-JEV-RP9(AflII) plasmid was used as the template to generate the chimeric JEV. The fragment corresponding to nucleotides 114 to 2213 and flanked by the unique sites ApaI and AflII was replaced with the homologous fragments of JEV g5 strain XZ0934 (nucleotides 114 to 2213, obtained from a cDNA synthesized by GeneCust). The resulting plasmid had the JEV-RP-9 5′- and 3′-noncoding regions as well as the coding sequences for all JEV-RP-9 nonstructural proteins, but most of the structural protein genes were derived from JEV-XZ0934. As described for JEV-RP-9 production, the resulting infectious clone was propagated in Stbl2 cells and transfected to cells to produce JEV S-g5/NS-g3, followed by amplification in C6/36 cells. The progeny virus was sequenced as described above.
JEV S-g3/NS-g5 chimeric virus production.
The chimeric JEV S-g3/NS-g5 virus was produced as described for JEV-XZ0934, except that the first PCR fragment containing the CMV promoter sequence was fused to a sequence encompassing nucleotides 1 to 2229 of JEV-RP-9, instead of nucleotides 1 to 2226 of JEV-XZ0934. This fragment was amplified from pBR322(CMV)-JEV-RP9 using the primers 5′-AGGGCATCGGTCGACTAGTA-3′ and 5′-GTCTCTGAGCACCCTTAAGAGTGGTCGTGAAAGCCTTTCCCAGT-3′. As described for JEV-XZ0934 production, the different PCR fragments were cotransfected to cells to produce JEV S-g3/NS-g5, followed by amplification in C6/36 cells. The progeny virus was sequenced as described above. Recombination events over the consensus sequence of nucleotides 2205 to 2215 led to the production of a chimeric JEV that is composed of the region of nucleotides 1 to 2215 of JEV-RP-9 followed by the region of nucleotides 2216 to 10983 of JEV-XZ0934.
Antibodies.
Mouse hybridomas producing the monoclonal antibody 4G2 anti-flavivirus E were purchased from ATCC (catalog no. HB-112), and a highly purified antibody preparation was produced by RD Biotech (Besançon, France). Mouse monoclonal antibody anti-JEV NS5 and rabbit polyclonal antibody anti-JEV C were kindly provided by Yoshiharu Matsuura (19, 20). The antibody against calnexin was purchased from Enzo Life Sciences (catalog no. ADI-SPA-865). Horseradish peroxidase (HRP)-conjugated goat anti-mouse and anti-rabbit IgG antibodies were obtained from Bio-Rad Laboratories (catalog no. 170-6516 and 170-6515, respectively). Alexa Fluor 488-conjugated goat anti-mouse IgG antibody and Cy3-conjugated donkey anti-rabbit IgG antibody were obtained from Jackson ImmunoResearch (catalog no. 115-545-003 and 711-165-152, respectively).
RT-qPCR.
For quantitative RT-PCR (RT-qPCR), RNA was extracted from samples using NucleoSpin RNA (catalog no. 740955; Macherey-Nagel) according to the manufacturer's instructions. The quantitation of a given target RNA was done on 8 μl or 200 ng of RNA using the QuantiTect SYBR green RT-PCR kit (catalog no. 204243; Qiagen) according to the manufacturer's instructions. The MiniOpticon real-time PCR system (Bio-Rad) was used to measure SYBR green fluorescence with the following program: reverse transcription step at 50°C (30 min), followed by an initial PCR activation step at 95°C (15 min), 40 cycles of denaturation at 94°C (15 s), annealing at 58 to 60°C depending on the primer pairs (30 s), and extension at 72°C (30 s). Results were analyzed using the CFX Manager software (Bio-Rad).
For viral genome quantification, we used a primer pair designed to equally amplify JEV-RP-9 and JEV-XZ0934 genomes: 5′-AGAAAAGARATGAGGTGTGGKAGTG-3′ and 5′-ACGGCTTCCCACATTTGA-3′, where R and K represent a mix of G and A and of G and T, respectively. The RNA standard used for quantification of JEV copy numbers was an in vitro transcript synthesized from a ClaI-linearized JEV-RP-9 replicon plasmid, J-R2A (21). In vitro transcripts were synthesized using a MEGAscript SP6 transcription kit (catalog no. AM1330; Life technologies) according to the manufacturer's instructions, and the absence of template DNA after DNase digestion was monitored through absence of qPCR amplification from the in vitro transcript.
The different mouse host genes were amplified using the following primer pairs: for Aif1, 5′-GGATTTGCAGGGAGGAAAAG-3′ and 5′-TGGGATCATCGAGGAATTG-3′; for Il6, 5′-TCCTACCCCAATTTCCAATGC-3′ and 5′-TGAATTGGATGGTCTTGGTCCT-3′; for Trail, 5′-CCCTGCTTGCAGGTTAAGAG-3′ and 5′-GGCCTAAGGTCTTTCCATCC-3′; for Icam1, 5′-ATAACTGGACTATAATCATTCTG-3′ and 5′-AGCCTTCTGTAACTTGTAT-3′; for Ptprc, 5′-GCCCAAACAAATTACACAT-3′ and 5′-TTAGGCGTTTCTGGAATC-3′. Target gene expression was normalized to the expression of the reference gene for hypoxanthine guanine phosphoribosyl transferase (Hprt), measured using the primers 5′-CTGGTGAAAAGGACCTCTCG-3′ and 5′-TGAAGTACTCATTATAGTCAAGGGCA-3′.
FFA.
For focus-forming assays (FFA), BHK-21 cells were seeded in 24-well plates. Tenfold dilutions of virus samples were prepared in duplicate in DMEM, and 200 μl of each dilution was added to the cells. The plates were incubated for 1 h at 37°C. Unadsorbed virus was removed, after which 1 ml of DMEM supplemented with 1.6% carboxymethyl cellulose (CMC), 10 mM HEPES buffer, 72 mM sodium bicarbonate, and 2% FBS was added to each well, followed by incubation at 37°C for 32 h. The CMC overlay was aspirated, and the cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min, followed by permeabilization with 0.1% Triton X-100 for 5 min. After fixation, the cells were washed with phosphate-buffered saline (PBS) and incubated for 1 h at room temperature with anti-E antibody (4G2), followed by incubation with HRP-conjugated anti-mouse IgG antibody. The plates were developed with the Vector VIP peroxidase substrate kit (catalog no. SK-4600; Vector Laboratories) according to the manufacturer's instructions.
Western blotting.
Protein lysates were prepared by cell lysis in radioimmunoprecipitation assay (RIPA) buffer (catalog no. RB4476; Bio Basic) containing protease inhibitors (catalog no. 11873580001; Roche). Equal amounts of proteins or purified reporter viral particles (RVPs) were loaded on a NuPAGE Novex 4 to 12% bis-Tris protein gel (Life Technologies) and transferred to a polyvinylidene difluoride (PVDF) membrane (catalog no. 170-4156; Bio-Rad) using the Trans-Blot Turbo Transfer system (Bio-Rad). After blocking the membrane for 1 h at room temperature in PBS-Tween (PBS-T) plus 5% milk, the blot was incubated overnight at 4°C with appropriate dilutions of the primary antibodies. The membrane was then washed in PBS-T and then incubated for 1 h at room temperature in the presence of HRP-conjugated secondary antibodies. After washes in PBS-T, the membrane was developed with Pierce ECL Western blotting substrate (catalog no. 32106; Thermo Scientific) and exposed to film.
Endo-H assay.
Equal amounts of protein lysates were subjected to digestion with endo-β-N-acetylglucosaminidase H (endo-H; catalog no. P0702S; New England BioLabs) in the presence of the manufacturer's buffer G5 and protease inhibitors (catalog no. 11873580001; Roche) for 30 min at 37°C. Undigested and digested samples were separated on a Novex 8% Tris-glycine protein gel (Life Technologies) and then analyzed by Western blotting with anti-JEV E 4G2 antibody as described above.
Immunofluorescence assay.
Cells were grown on coverslips and fixed with methanol for 15 min at −20°C. After fixation, the cells were washed with PBS, and JEV proteins were detected with appropriate dilutions of the primary antibodies, followed by incubation with fluorophore-conjugated secondary antibodies. The coverslips were mounted with ProLong Gold Antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; catalog no. P36931; Life Technologies). The slides were examined using a fluorescence microscope (Axioplan 2 Imaging; Zeiss).
Mouse experiments.
Three-week-old female BALB/c or C57BL/6 mice were housed under pathogen-free conditions at the Institut Pasteur animal facility. The protocols and subsequent experiments were ethically approved by the Ethic Committee for Control of Experiments on Animals (CETEA) at the Institut Pasteur and declared to the French Ministère de l'Enseignement Supérieur et de la Recherche (no. 000762.1) in accordance with European regulations. Experiments were conducted in accordance with the guidelines of the Office Laboratory of Animal Care at the Institut Pasteur. Groups of mice were intraperitoneally inoculated with various doses of JEV diluted in 100 μl of Dulbecco's PBS (DPBS) supplemented with 0.2% endotoxin-free serum albumin or intracranially inoculated with JEV diluted in 20 μl of DPBS supplemented with 0.2% endotoxin-free serum albumin.
Mice were bled by puncturing at the retro-orbital sinus level. To measure the amount of viral RNA in blood, 50 μl of blood was processed with the RNA lysis buffer from the NucleoSpin RNA kit (catalog no. 740955; Macherey-Nagel) according to the manufacturer's instructions. For enzyme-linked immunosorbent assay (ELISA) and seroneutralization assay, the blood was collected using a Capiject capillary blood collection tube (catalog no. 3T-MG; Terumo), and the serum was separated after centrifugation for 10 min at 4,000 × g. To measure the amount of viral RNA in tissues, each organ was collected and stored at −80°C until RNA extractions were performed. At that time, each sample was ground in DMEM (30%, wt/vol) using a tissue-homogenizing kit (catalog no. KT03961-1-009.2; Precellys). RNA was extracted from 1/10 of the homogenate following instructions from the NucleoSpin RNA kit (catalog no. 740955; Macherey-Nagel).
Detection of antibodies by ELISA.
Soluble recombinant proteins SNAP-JEV.EDIII, which are composed of JEV g3 or g5 E protein domain III fused in frame to the C terminus of SNAP-tag, were produced in stable S2 cells and purified on chelating column chromatography and then a Superdex column (22). The total protein concentration of purified recombinant JEV proteins tagged with SNAP was determined using a bicinchoninic acid (BCA) protein assay kit (catalog no. 23225; Thermo Scientific), and 100 ng of highly purified SNAP-JEV.EDIII g3 or g5 was diluted in DPBS and used to coat 96-well plates at 4°C overnight. Recombinant SNAP protein served as a negative antigen control. Plates were incubated at 40°C with mouse serum sample diluted at 1:50 in PBS-T plus 3% milk. The plates were then washed in PBS-T and incubated in the presence of HRP-conjugated anti-mouse IgG antibody. After washes in PBS-T, plates were finally incubated with TMB substrate (catalog no. 50-76-00; KPL), and absorbance was measured at 450 nm. Sera obtained from DPBS-inoculated mice served as negative controls, and experimental samples values were calculated as a fold increase over the DPBS values.
JEV replicon cell line.
The JEV-RP-9 replicon plasmid J-R2A was kindly provided by Yi-Lin Ling (21). First, the J-R2A plasmid was modified so that the hepatitis delta virus ribozyme was placed immediately adjacent to the JEV-RP-9 3′-end and was followed by an SV40 poly(A) sequence. To do so, the corresponding sequence in the pBR322(CMV)-JEV-RP-9 plasmid was excised through digestion with NsiI and ClaI and cloned into the similarly treated J-R2A. Next, the plasmid was modified to replace the SP6 promoter with an inducible PTRE3G promoter (Clontech). The PTRE3G promoter was amplified from the pTRE3G vector (catalog no. 631173; Clontech) using the primers 5′-CTCGAGTTTACTCCCTATCAGTGA-3′ (XhoI site underlined) and 5′-TCACACAGATAAACTTCTCGGTTCACTAAACGAGCT-3′ (JEV-RP-9 nucleotides 1 to 18 underlined). Nucleotides 1 to 249 of the JEV-RP-9 genome were amplified using the primers 5′-AGCTCGTTTAGTGAACCGAGAAGTTTATCTGTGTGA-3′ (PTRE3G promoter nucleotides 291 to 308 underlined) and 5′-TGATAAGAGCCAGCACGAATCG-3′. The primers were designed so that both fragments shared a sequence homology of 36 nucleotides. A second round of PCR using these first two fragments allowed the amplification of a fragment composed of the PTRE3G promoter fused to the nucleotides 1 to 249 of JEV-RP-9. This fragment was digested with XhoI and ApaI and cloned into the J-R2A plasmid treated with SalI and ApaI. The resulting pTRE3G-JEV-RP9.replicon plasmid was amplified in Stbl2 cells (catalog no. 10268-019; Life Technologies). HEK293T cells were cotransfected with the pTRE3G-JEV-RP9.replicon and the pTK-Hyg selection vector (catalog no. 631750; Clontech), and stable cells were selected with 50 μg/ml of hygromycin.
RVP production.
The fragment encompassing the structural genes of JEV-RP-9 was amplified using the primers 5′-GAAGATCTATGACTAAAAAACCAGGAGGGCCCGGT-3′ (BglII site underlined) and 5′-TTCTGCAGTCAAGCATGCACATTGGTCGCTAAGA-3′ (PstI site underlined). The fragment was digested with BglII and PstI and cloned into the similarly treated pTRE3G vector (catalog no. 631173; Clontech). The resulting pTRE3G-JEV-RP9.CprME plasmid was amplified in Stbl2 cells (catalog no. 10268-019; Life Technologies). The pTRE3G-JEV-XZ0934.CprME plasmid containing JEV-XZ0934 structural genes was designed in the same way as the pTRE3G-JEV-RP9.CprME plasmid and was synthesized by GeneCust.
To produce JEV g3 or JEV g5 RVPs, HEK293T-JEV-RP9.replicon cells were plated in a 10-cm dish and then transfected with pTRE3G-JEV-RP9.CprME or pTRE3G-JEV-XZ0934.CprME, respectively, using Lipofectamine 2000 (catalog no. 11668-019; Life Technologies) according to the manufacturer's instructions. The expression of the JEV replicon and structural genes was induced using the Tet-Express system (catalog no. 631177; Clontech) according to the manufacturer's instructions. The supernatants containing RVPs were collected at 48 h postinduction and clarified by centrifugation for 5 min at 1,000 × g, and aliquots were stored at −80°C.
For RVP purification, the clarified supernatant was loaded over a sucrose cushion consisting of 15% sucrose in TNE (10 mM Tris-HCl [pH 7.5], 2.5 mM EDTA, 50 mM NaCl) and centrifuged at 100,000 × g for 2.5 h at 4°C. The supernatants were discarded, and the purified RVPs were suspended in TNE buffer.
For the infectivity assays, BHK-21 cells were seeded in 24-well or 96-well tissue culture plates in DMEM supplemented with 2% FBS. Then, purified RVPs or portions of supernatants containing RVPs were added to the cells, and the plates were incubated for 1 h at 37°C. Unadsorbed RVPs were removed, after which DMEM supplemented with 2% FBS was added to the cells, followed by incubation at 37°C. At 24 h postinfection, the samples were processed according to the instructions in the Renilla luciferase assay system (24-well format; catalog no. E2820; Promega) or the Renilla-Glo luciferase assay system (96-well format; catalog no. E2720; Promega). The Renilla luciferase signal was read using a Centro XS3 LB960 (Berthold Technologies) plate reader.
Seroneutralization assay.
The serum samples were decomplemented by heating at 56°C for 30 min and were 2-fold serially diluted in DMEM supplemented with 2% FBS, with a starting dilution of 1:20. Each dilution was incubated for 1 h at 37°C with an equal volume of purified g3 or g5 RVP. Remaining RVP infectivity was assayed on BHK cells seeded in a 96-well plate, as described above. Sera obtained from DPBS-inoculated mice served as negative controls. The infectivity values obtained with the negative-control samples were set as 100% infectivity, and the rest of the experimental values were calculated reciprocally to those samples.
Brain microvascular endothelial cells and pericytes.
Brain microvascular endothelial cells and pericytes were isolated from 6-week-old female BALB/c mice and cultured according to previously reported methods (23, 24). Briefly, the brains from 5 adult mice were harvested and diced in 10 ml of DMEM-HEPES medium. After centrifugation at 290 × g for 5 min, the pellets were digested with 5 ml of papain solution (catalog no. LK003178; Worthington Biochemical) and 250 μl of DNase solution (catalog no. LK003172; Worthington Biochemical), both diluted in DMEM-HEPES, for 70 min at 37°C. The digested solution was then triturated to break the microvessels through 10 passages with a 19-gauge needle, followed by 10 passages with a 21-gauge needle. The brain homogenate was then mixed with a solution of 22% (vol/vol) bovine serum albumin (BSA) and centrifuged at 1,360 × g for 10 min. The blood vessel cell pellets were subsequently suspended with endothelial cell growth medium and washed by spinning down at 290 × g for 5 min. The obtained cells were finally plated in endothelial cell growth medium on collagen-coated (catalog no. 3440-100-01; Trevigen) 6-well plates and cultured at 37°C until reaching confluence. The endothelial cell growth medium is composed of endothelial basal medium EBM-2 (catalog no. 190860; Lonza), 5% FBS, 1.4 μM hydroxycortisone (catalog no. H0135-1MG; Sigma), 5 μg/ml ascorbic acid (catalog no. H4544-25G; Sigma), 1% chemically defined lipid concentrate (catalog no. 11905-031; Life Technologies), 10 mM HEPES, and 1 mg/ml basic fibroblast growth factor (catalog no. F0291-25UG; Sigma). To grow endothelial cells, one set of cells was cultured in endothelial cell growth medium and 4 μg/ml of puromycin for 10 days. To grow pericytes, another set of cells was grown for 7 days in endothelial cell growth medium in the absence of puromycin and then switched to pericyte growth medium, composed of pericyte basal medium (catalog no. 1201-b; ScienCell), 2% FBS, and pericyte growth supplement (catalog no. 1252; ScienCell).
To determine the purity of the cultures, each cell preparation was examined in an immunofluorescence assay. We found that the majority of endothelial cells were expressing the CD31 cell marker (catalog no. PA5-16301; Thermo Scientific) while the majority of pericytes were expressing the NG2 cell marker (catalog no. PA5-17199; Thermo Scientific). The endothelial cells were seeded onto collagen-coated Transwell inserts (catalog no. 3450; Corning) at a density of 1 × 105 cells per well,3 days prior to experiments. The pericytes were seeded onto collagen-coated 12-well plates at a density of 2 × 104 cells per well 3 days prior to experiments. On the day of the experiment, a coculture was established by putting the culture inserts containing the endothelial cells into the plates containing the pericytes.
Statistical analysis.
An unpaired t test was used to compare quantitative data, and a log rank (Mantel-Cox) test was used to compare survival data. GraphPad Prism was used for all statistical analysis.
RESULTS
Characterization of a JEV g5 strain derived from JEV-XZ0934 cDNA.
We decided to characterize a representative JEV g5 strain, JEV-XZ0934, recently isolated from Culex tritaeniorhynchus mosquitoes in China in 2009 (9). The genome of JEV-XZ0934 was assembled in cells by cotransfection in HEK293T cells of four overlapping fragments corresponding to the viral genome, flanked by a CMV promoter and an SV40 polyadenylation signal. To allow for correct termination of the recombined full-length genome, a hepatitis delta virus (HDV) ribozyme sequence was included at the 3′-end of the genome. The virus recovered in the supernatants of the transfected cells was amplified in C6/36 cells, and the viral RNA was extracted and sequenced to ensure that no mutation had arisen during viral production. As a representative strain of JEV g3, we selected the well-characterized JEV-RP-9, isolated from Culex tritaeniorhynchus mosquitoes in Taiwan in 1985 (16, 17). JEV-RP-9 was produced in parallel with JEV-XZ0934 by transfection of a molecular clone in HEK293T cells, followed by amplification in C6/36 cells. For simplification, and since those viruses were chosen as representative strains for genotypes 3 and 5, JEV-RP-9 and JEV-XZ0934 are here referred to as JEV g3 and JEV g5, respectively.
The titers of both C6/36-grown viral stocks in BHK-21 cells were determined, and the apparition of infection foci was detected using 4G2, a pan-flavivirus antibody directed against the E protein. We noted that the JEV g5 foci were of smaller size than JEV g3 foci (Fig. 1A). The viral RNA contents extracted from the viral stocks were quantified, and we observed a strict correlation between JEV RNA copies and JEV titers (Fig. 1B), which showed that the two JEV genotypes' viral stocks had a comparable specific infectivity (Fig. 1C) and were both suitable for further comparative analysis.
FIG 1.
Production and characterization of JEV g3 and g5 viral stocks. JEV g3 and g5 were produced after transfection of infectious DNA into mammalian cells, followed by amplification of viral stocks in C6/36 cells. (A) Example of infectious foci developed for JEV g3 and g5 after FFA in BHK-21 cells. (B) Qualification of viral stocks grown from C6/36 cells: infectious virus contents of the supernatants were analyzed by titration on BHK-21 cells (dashed bars), while viral RNA was quantified by RT-qPCR (gray bars). A representative experiment (n > 3) is shown. The error bars represent the standard deviations of the means (the assays were done in duplicate). (C) The specific infectivity was calculated as the number of viral RNA molecules per infectious unit of each virus.
Comparison of JEV g3 and g5 growth in cultured cell lines.
Next, we studied the kinetics of JEV g3 and g5 viral production in representative cell lines, such as mosquito C6/36 cells, chicken fibroblasts DF-1 cells, or human neuroblastoma SK-N-SH cells. We infected the cells at a multiplicity of infection (MOI) of 1 and analyzed the progeny virus production at 24, 48, and 72 h postinfection using a standard focus-forming assay (FFA). Although we noted that JEV g5 viral production was less efficient in SK-N-SH cells than was JEV g3 production, the two viruses had comparable viral growths in the different cell lines tested (Fig. 2A).
FIG 2.

Analysis of JEV g3 and g5 infectious cycle in vitro. (A) Aedes albopictus-derived C6/36 cells, chicken fibroblast-derived DF-1 cells, or human neuroblastoma-derived SK-N-SH cells were infected with JEV g3 or g5 at an MOI of 1. The infectious virus released to the supernatants at 24, 48, and 72 h postinfection was quantified by FFA. The error bars represent the standard deviations of the means (titrations were done in duplicate). Asterisks indicate that the differences between experimental samples at each time point are statistically significant, using the unpaired t test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; not significant, P > 0.05). (B) The cell lysates of infected SK-N-SH cells were analyzed for the presence of JEV E and NS5 proteins, as well as calnexin (CNX) by Western blotting. (C) Trafficking of E proteins through the secretory pathway was analyzed in SK-N-SH cell lysates 48 h postinfection. Cell lysates were treated with endo-H and then analyzed by Western blotting with anti-JEV E antibody. (D) SK-N-SH cells were analyzed by immunofluorescence staining for the presence of the JEV C (red), E (green), and NS5 (green) proteins. The images were taken at a magnification of ×200. For each panel, results from a representative experiment (n > 2 repeats) are shown.
Next, we analyzed the steady-state accumulation of the structural E protein and the nonstructural NS5 protein in infected SK-N-SH cells. While JEV g5 accumulated higher levels of NS5 protein than did JEV g3, both viruses accumulated comparable levels of intracellular E protein (Fig. 2B). We observed that the E protein of JEV g5 migrated faster than JEV g3 E protein (Fig. 2B). Since it is not predicted that these E proteins have significantly different molecular weights (g3, 53.44 kDa; g5, 53.71 kDa), we asked whether this faster migration of JEV g5 E protein could be linked to a different state of posttranslational modification, such as N-glycosylation. To verify that JEV g5 E protein was N-glycosylated on position 154, as in other JEV strains (25), we treated cell lysates with endo-H, an enzyme that removes immature high-mannose glycans from endoplasmic reticulum (ER)-associated proteins (26). As shown in Fig. 2C, both JEV g3- and g5-infected cells contained a pool of E proteins that were sensitive to endo-H treatment, thus indicating that both E proteins were glycosylated.
Last, the subcellular distribution of JEV C, E, and NS5 proteins within infected SK-N-SH cells was examined by immunofluorescence at 48 h postinfection (Fig. 2D). We noted that the JEV g5 E protein accumulation pattern was more discrete within infected cells while JEV g5 NS5 presented a slightly stronger staining, which was concordant with the higher levels of intracellular accumulation as observed by Western blotting (Fig. 2B).
Taken together, these results showed that the use of cDNA-based technology allows the production of a JEV g5 that is as competent as JEV g3 at infecting representative cell lines.
Study of JEV g5 pathogenicity in a mouse model.
The pathogenic properties of JEV g3 and g5 were evaluated in two different inbred mice that differ in their susceptibility to JEV. Similar to previous findings (27), we observed no dose dependence in the survival curve of sensitive mice following peripheral injection with JEV (data not shown), and consequently we chose to inoculate 103 FFU of JEV in the following in vivo experiments.
First, 3-week-old C57BL/6 mice were injected intraperitoneally with 103 FFU of JEV. These mice are highly susceptible to JEV, and we observed the development of symptoms linked to an invasion of the central nervous system (CNS), such as limb paralysis and encephalitis. The survival rate ranged between 0 and 17% after infection with JEV g3 or g5 (Fig. 3A), with a comparable mean survival time of 10 days. This confirmed that both viruses are neuroinvasive and pathogenic in a susceptible inbred mouse strain.
FIG 3.

Analysis of JEV g3 and g5 infection in vivo. Groups of 3-week-old C57BL/6 (A) or BALB/c (B) mice were monitored for survival after intraperitoneal (IP) injection with 103 FFU of JEV g3 or g5 (n = 12 per group) or after intracranial (IC) injection with 10 FFU of either virus (n = 6 per group). Results from a representative experiment (n > 2 repeats) are shown. Asterisks indicate that the differences between survival curves are statistically significant using the log rank (Mantel-Cox) test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; ns, not significant, P > 0.05). (C) Sera were collected from mice at 20 days postinoculation and anti-JEV IgGs were quantified by ELISA using recombinant proteins corresponding to the domain III (DIII) of either JEV g3 or g5 E protein. The ELISA absorbance values were measured at 450 nm and were represented as a fold increase relative to the absorbance values from sera of mice inoculated with DPBS. Each symbol represents an individual mouse, and values from 3 independent experiments are presented.
Unlike C57BL/6, the BALB/c mouse strain is known to present inherent resistance to JEV infection (28, 29). To evaluate the susceptibility of BALB/c mice to JEV, we first inoculated 3-week-old mice intracranially with 10 FFU of virus. All mice died within 8 days postinoculation, thus indicating that JEV g3 and g5 were both neurovirulent in the BALB/c mouse model (Fig. 3B). Next, 3-week-old BALB/c mice were injected intraperitoneally with 103 FFU of JEV. While the majority of JEV g3-infected mice remained asymptomatic and survived the inoculation, a small portion (less than 10%) developed typical symptoms of encephalitis and succumbed to JEV infection (Fig. 3B). All surviving mice seroconverted and had high JEV-specific antibody titers (20 days postinoculation; Fig. 3C). Interestingly, when BALB/c mice were injected with JEV g5, the survival rate was below 25%, with a mean survival of 11 ± 2 days (Fig. 3B). The mice presented limb paralysis and encephalitis, and the onset of symptoms spanned 5 to 14 days. These results show that JEV g5 has neuroinvasive properties in BALB/c mice, an inbred mouse strain known to be usually resistant to other JEV strains.
Humoral response to JEV in BALB/c mice.
Since the humoral response plays a critical role in protecting mice from infection with neuroinvasive flaviviruses (27, 30), we asked whether the susceptibility of BALB/c mice to JEV g5 could be associated to a failure at eliciting neutralizing antibodies. First, we monitored the apparition of antibodies directed against JEV E domain III (DIII), which are known to exhibit potent neutralizing activity (31, 32). The apparition and titers of JEV E DIII-specific IgGs were similar for JEV g3 and g5 (Fig. 3C). It is worth noting that the mice surviving JEV g5 inoculation did not produce higher IgG titers than those of mice succumbing to the infection, suggesting that the total production of anti-JEV IgG was not directly related to protection against JEV g5.
Next, the JEV neutralization potency of mice sera was evaluated using single-cycle reporter viral particles (RVPs). RVPs were produced in cells stably transformed with a JEV-RP-9 (g3) subgenomic replicon expressing the viral nonstructural proteins and a Renilla luciferase reporter. Those cells were transfected with a plasmid that expresses either JEV g3 or JEV g5 structural proteins (C, prM, and E), leading to successful release of RVPs (Fig. 4A and B). Successful entry of the recombinant RVPs into new target cells leads to genome release and subsequent expression of a luciferase reporter gene (Fig. 4B). Such a system has been shown to be sensitive and potent for use in seroneutralization assays (33). To detect JEV-neutralizing antibodies, each RVP preparation was incubated with corresponding immune sera collected from BALB/c mice at 5, 7, 9, or 20 days post-JEV inoculation, and the remaining RVP infectivity was assayed by infection on BHK-21 cells. Interestingly, anti-JEV g3 neutralizing antibodies were detected as early as 5 to 7 days postinoculation, whereas g5-neutralizing antibodies were not consistently observed within the first 9 days of JEV infection (Fig. 4C). Nonetheless, by 20 days postinoculation, the mice surviving JEV g5 inoculation had raised potent neutralizing antibodies against g5 RVPs (Fig. 4C).
FIG 4.

Production of JEV reporter viral particles for seroneutralization experiments. (A and B) In order to produce RVPs, a stable HEK293T cell line expressing a Tet-Express-inducible JEV-RP-9 (g3) replicon RNA, which encodes a Renilla luciferase reporter in place of the JEV structural proteins, was transfected with a plasmid encoding either JEV g3 or g5 structural genes under the control of a Tet-Express-inducible promoter. The expression of the JEV replicon and structural genes was induced, and cell lysates and RVPs containing supernatants were collected at 48 h postinduction. The supernatants from cells that had not been transfected with the JEV structural genes served as a control. Results from a representative experiment (n > 3 repeats) are shown. (A) The cell lysates were analyzed by Western blotting for JEV E and calnexin (CNX) as a loading control. The RVPs released in the supernatants were purified and analyzed by Western blotting using JEV E antibody (extracellular). (B) The successful production of RVPs was detected using an infectivity assay, in which BHK-21 cells were infected with 200 μl of supernatants. Productive delivery of the JEV replicon was quantified by measuring Renilla expression at 24 h postinfection (dashed bars). The content of RVPs in the culture supernatants was also analyzed by RT-qPCR, and the replicon RNA level quantification (gray bars) was plotted along the values obtained from the corresponding infectivity assay. RLU, Renilla light units. (C and D) For seroneutralization assays, JEV g3 or g5 RVPs were incubated with dilutions of sera collected from mice inoculated with 103 FFU of either JEV g3 or JEV g5. Sera from 6 individual mice were used in each experiment, and sera collected from DPBS injected mice served as a control. After incubation, the RVPs were used to infect BHK-21 cells. Intracellular Renilla luciferase activity was quantified at 24 h postinfection as a measure of successful RVP entry. Infectivity was measured as a function of the Renilla luciferase activity obtained with the control sera. Results from representative experiments (n = 2 repeats) are shown. Asterisks indicate that the differences between experimental samples are statistically significant at each time point, or at each dilution, using the unpaired t test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; not significant, P > 0.05). (C) To measure the seroneutralization activity in sera collected at 5, 7, 9, and 20 days postinfection, g3 RVPs were incubated with a single dilution (1:20) of g3-inoculated mouse sera, while g5 RVPs were incubated with the same dilution of g5-inoculated mouse sera. (D) JEV g3 (gray) or g5 (white) RVPs were incubated with serial dilutions (1:20 to 1:320) of sera collected at 20 days from mice inoculated with either JEV g3 (left) or JEV g5 (right).
Additionally, we investigated whether the anti-JEV antibodies raised in mice could have cross-protective capacity against JEV g3 or JEV g5. We observed that immune sera collected at 20 days from BALB/c mice surviving JEV challenge had a similar neutralization potency against their homologous RVP (Fig. 4C and D). Interestingly, while sera obtained from JEV g3-inoculated mice equally neutralized both JEV g3 and g5 RVPs (Fig. 4D, left), immune sera from JEV g5-inoculated mice exerted a lower neutralizing activity against JEV g3 RVPs than against JEV g5 RVPs (Fig. 4D, right).
Characterization of JEV g5 infection in BALB/c mice.
Among the multiple factors governing JEV pathogenesis, viremia in blood and in peripheral organs was shown to be important for successful invasion of the CNS by JEV and by related flaviviruses (27, 30, 34). RNA was extracted from blood samples at 2, 3, and 4 days after peripheral inoculation of BALB/c mice. At 2 days postinoculation, viral RNA could be detected by RT-qPCR in blood samples and we noted that the levels of JEV g5 RNA were significantly higher than those of JEV g3 (Fig. 5A). A day 3 postinoculation, sustained viremia could still be detected in JEV g5-infected animals but not in JEV g3-inoculated mice (Fig. 5A). At 4 days postinoculation, viral RNA could no longer be detected in the blood samples regardless of the virus tested (Fig. 5A). The levels of JEV RNA were also measured in the spleens, livers, kidneys, and brains of BALB/c mice at 5, 7, and 9 days postinoculation. We observed little to no infection of these peripheral organs by either JEV strain (Fig. 5B to D). In brains, high levels of JEV g5 but no JEV g3 could be detected from 7 to 9 days postinoculation (Fig. 6A). The mice that had high levels of JEV in brain presented symptoms resulting from CNS invasion at the time of collection.
FIG 5.

Measure of viral burden in BALB/c mice infected with JEV g3 or g5. (A) Total blood was collected from mice inoculated with 103 FFU of JEV g3 or g5 at 2, 3, and 4 days postinfection, and total RNA was extracted. JEV RNA was quantified by RT-qPCR, and the limit of detection of the assay, corresponding to values obtained from mice injected with DPBS, is shown as a dotted line. Each sign represents an individual mouse, and results from a representative experiment (n = 2 repeats) are shown. Asterisks indicate that the differences between experimental samples are statistically significant at each time point using the unpaired t test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; not significant, P > 0.05). The spleen (B), liver (C), and kidney (D) were collected from mice inoculated with 103 FFU of JEV g3 or g5 at 5, 7, and 9 days postinfection. Total RNA was extracted from each organ, and JEV RNA was quantified by RT-qPCR. The results were normalized to HPRT mRNA and are expressed as the relative fold increase over RNA from DPBS-infected controls. The limit of detection of the assay, corresponding to values obtained from mice injected with DPBS, is shown as a dashed line. Each sign represents an individual mouse, and results from a representative experiment (n = 2 repeats) is shown.
FIG 6.

Relative RNA levels of markers specific for inflammatory responses in the brain of infected mice. (A to F) Brains were collected from mice inoculated with 103 FFU of JEV g3 or g5 at 5, 7, and 9 days postinfection. Total RNA was extracted, and JEV RNA (A) and mRNA for AIF-1 (B), IL-6 (C), TRAIL (D), ICAM-1 (E), and CD45 (F) were quantified by RT-qPCR. The results were normalized to hypoxanthine phosphoribosyltransferase (HPRT) mRNA and are expressed as the relative fold increase over RNA from DPBS-infected controls. The limit of detection of the assay, corresponding to values obtained from mice injected with DPBS, is shown as a dotted line. Each symbol represents an individual mouse, and results from a representative experiment (n = 2 repeats) are shown.
Next, we analyzed the expression pattern of genes commonly involved in different aspects of neuropathogenesis, such as microglia activation (AIF-1 mRNA) (Fig. 6B) and synthesis of inflammatory cytokines (interleukin-6 [IL-6], IL-1β, and tumor necrosis factor alpha [TNF-α] mRNA) (Fig. 6 and data not shown) or activation of cell death pathways (TRAIL mRNA) (Fig. 6D). We observed a concomitant activation of those genes in the brains of BALB/c mice that had been inoculated with JEV g5 and that presented an infection of the CNS (Fig. 6B to D). JEV g5 infection of brains also resulted in the activation of the intercellular adhesion molecule ICAM-1 (Fig. 6E), which promotes the transmigration of immune cells across the vascular endothelium. Accordingly, we observed infiltration of immune cells in JEV g5-infected brains (CD45 mRNA [Fig. 6F]). Importantly, all of those genes were induced only once JEV g5 RNA could be detected in mice brains, and their expression was not induced in the brains of mice challenged with JEV g3.
Viral determinants of JEV g5 pathogenicity.
In an effort to understand the molecular basis of the marked neuropathogenicity of JEV g5, we generated two mutant viruses in which the structural protein region of one virus was replaced with that of the other virus. The first chimeric virus had the JEV-RP-9 (g3) 5′- and 3′-untranslated region (UTR) sequences, as well as the coding sequences for all JEV-RP-9 nonstructural proteins, but had most of its structural proteins derived from JEV-XZ0934 (g5) (Fig. 7A). For simplification, the resulting virus is here referred to as JEV S-g5/NS-g3. A reciprocal chimeric virus that had the structural proteins of JEV-RP-9 and the nonstructural proteins of JEV-XZ0934 was also constructed and is referred to as JEV S-g3/NS-g5 (Fig. 7A). For each chimeric JEV, there was no change in viral growth in cultured cell lines compared to the parental viruses (data not shown). Interestingly, the chimeric virus that had the nonstructural proteins of JEV g3, i.e., JEV S-g5/NS-g3, developed infectious foci similar in size to those of JEV g3 (Fig. 7B). Reciprocally, JEV S-g3/NS-g5, like JEV g5, developed small foci on BHK-21 cells (Fig. 7B), thus showing that the observed difference in focus size is likely linked to a property of JEV nonstructural proteins.
FIG 7.

The use of chimeric viruses links JEV g5 structural genes to pathogenicity in BALB/c mice. (A) Schematic representation of the genomic RNA of JEV g3 (light gray) and g5 (dark gray) and of the chimeric viruses JEV S-g5/NS-g3 and S-g3/NS-g5, which express most of the structural proteins (C, prM, and E [amino acids 1 to 472]) of one genotype, fused to part of the E protein (amino acids 473 to 500) and the nonstructural proteins (NS1 to NS5) of the other. (B) Example of infectious foci developed for JEV g3, g5, S-g5/NS-g3, and S-g3/NS-g5 after FFA in BHK-21 cells. (C) Groups of 3-week-old BALB/c mice (n = 12 per group) were monitored for survival after intraperitoneal (IP) injection with 103 FFU of JEV g3, g5, S-g5/NS-g3, or S-g3/NS-g5. Results from a representative experiment (n > 2 repeats) are shown. Asterisks indicate that the differences between survival curves are statistically significant using the log rank (Mantel-Cox) test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; ns, not significant, P > 0.05). (D) Total blood was collected from mice inoculated with 103 FFU of JEV g3 or S-g5/NS-g3 at 2, 3, and 4 days postinfection, and total RNA was extracted. JEV RNA was quantified by RT-qPCR, and the limit of detection of the assay, corresponding to values obtained from mice injected with DPBS, is shown as a dotted line. Each sign represents an individual mouse, and results from a representative experiment (n = 2 repeats) is shown. Asterisks indicate that the differences between experimental samples are statistically significant at each time point using the unpaired t test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; not significant, P > 0.05). (E) Brains were collected from mice inoculated with 103 FFU of JEV S-g5/NS-g3 at 9 days postinfection. Total RNA was extracted, and JEV RNA (plotted on the left y axis) and mRNA for AIF-1, IL-6, TRAIL, ICAM-1, and CD45 (plotted on the right y axis) were quantified by RT-qPCR. The results were normalized to HPRT mRNA and are expressed as the relative fold increase over RNA from DPBS-infected controls. Each symbol represents an individual mouse, and results from a representative experiment (n = 3 repeats) are shown.
Groups of 3-week-old BALB/c mice were peripherally injected with the same dose of chimeric or parental JEV. Up to 70% of the mice inoculated with JEV g5 or JEV S-g5/NS-g3 developed an infection and succumbed from a neuropathogenic disease, while no mortality was observed for mice inoculated with JEV g3 or JEV S-g3/NS-g5 virus (Fig. 7C). The mean survival of mice inoculated with JEV S-g5/NS-g3 or JEV g5 was similar (13 ± 1 days). All surviving mice had raised titers of anti-JEV antibodies by 21 days postinoculation (data not shown). Next, we analyzed the development of JEV S-g5/NS-g3 infection in BALB/c mice. Similarly to what had been observed with JEV g5 (Fig. 5A), JEV S-g5/NS-g3 viremia was sustained in blood for a longer time than JEV g3 (Fig. 7D). We also monitored the viral burden in brains and noted that, concomitantly to JEV invasion of the CNS, neuropathogenesis-related genes were induced in the brains of BALB/c mice infected with JEV S-g5/NS-g3 (Fig. 7E). Overall, these results suggest that the JEV structural protein region may play a critical role in the neuropathogenicity of genotype 5.
Susceptibility of mouse brain pericytes to JEV g5 infection.
Although the mechanisms of JEV CNS invasion are not yet clearly defined, it is generally admitted that crossing and/or disruption of the blood-brain barrier (BBB) is a major factor favoring JEV neuropathogenesis (35–38). To assess the capabilities of the two JEV genotypes to cross the BBB, we established an in vitro model for BBB using primary brain microvascular endothelial cells and pericytes. Pericytes are multipotent cells that surround capillaries and were recently shown to be important in regulating the breaching of the endothelial barrier by inducing the production of inflammatory cytokines, such as IL-6, following JEV infection (24). Primary endothelial cells were isolated from BALB/c mice and grown on a Transwell insert above primary pericytes. The endothelial cells were infected at an MOI of 10 with JEV g3 or g5. The crossing of the endothelial barrier and subsequent infection of pericytes were analyzed at 48 and 96 h postinfection. While JEV infection of endothelial cells was not overly productive, we observed that JEV g5 did not infect endothelial cells as efficiently as JEV g3 (Fig. 8A). Both viruses were able to cross the endothelial barrier and to infect productively the pericytes (Fig. 8B). Since JEV g5 levels were not as high as JEV g3 in the pericyte chamber (Fig. 8B), we asked whether this was a consequence of the less efficient infection of endothelial cells (Fig. 8A). Pericytes were infected with JEV g3 or g5 at an MOI of 10, and cells and supernatants were collected at 24 h, 48 h, and 72 h postinfection for analysis. We observed that JEV g3 was more efficient than JEV g5 at infecting BALB/c pericytes (Fig. 8C), leading to a stronger induction of IL-6 mRNA (Fig. 8D). An upregulation of IL-6 protein production is likely to contribute to a greater disruption of the endothelial barrier by JEV g3 but not JEV g5. These results suggest that the marked neuroinvasive properties of JEV g5 were not associated to a greater efficacy of virus to cross the BBB in BALB/c mice.
FIG 8.
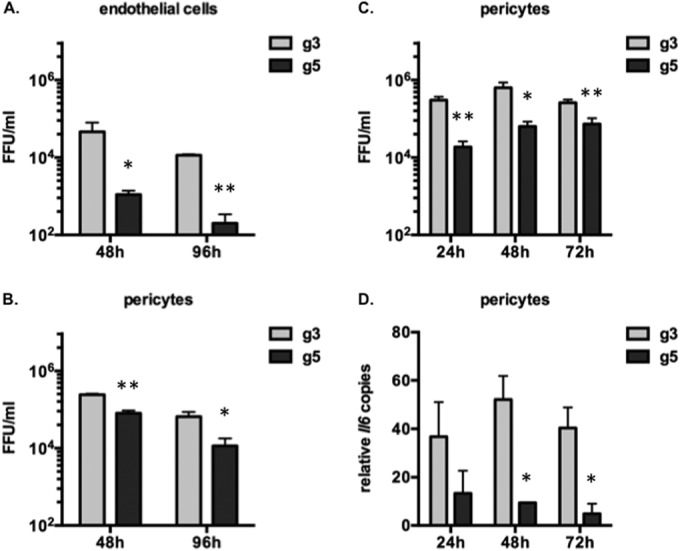
Analysis of JEV g3 and g5 crossing of the blood-brain barrier in vitro. (A and B) Primary endothelial cells and pericytes were isolated from BALB/c mice. Endothelial cells were grown on a Transwell insert, separated from the pericytes that were grown in the lower chamber. The endothelial cells were infected with JEV g3 or g5 at an MOI of 1. The infectious virus released to the supernatants in the upper chamber (A) or the lower chamber (B) at 48 and 96 h postinfection was quantified by FFA. The error bars represent the standard deviations of the means (titrations were done in duplicate), and results from a representative experiment (n > 2 repeats) is shown. Asterisks indicate that the differences between experimental samples are statistically significant at each time point using the unpaired t test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; not significant, P > 0.05). (C and D) Primary BALB/c pericytes were infected with JEV g3 or g5 at an MOI of 1. The infectious virus released to the supernatants at 24, 48, and 72 h postinfection was quantified by FFA (C). Total RNA was also extracted from infected cells, and IL-6 mRNA was quantified by RT-qPCR (D). The results were normalized to HPRT mRNA and are expressed as the relative fold increase over RNA from mock-infected controls. The error bars represent the standard deviations of the means, and results from a representative experiment (n > 2 repeats) are shown. Asterisks indicate that the differences between experimental samples are statistically significant at each time point using the unpaired t test (***, P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; not significant, P > 0.05).
DISCUSSION
The main objective of this study was to improve our knowledge of the poorly characterized genotype 5 JEV strains. To date, the large majority of the studied JEV strains belong to genotypes 1 and 3, which are only 90 to 92% identical to g5 strains at the amino acid level (9, 10). Since the isolation of the prototype strain Muar in 1952 (11), no other g5 strains had been isolated in regions of endemicity for over 50 years. However, during recent campaigns of viral identification in mosquito pools, it was discovered that JEV strains belonging to g5 were still circulating (9, 10). Such a finding raises the interesting question as to what factors might restrict its expansion. JEV g5 was isolated in Culex tritaeniorhynchus (9) and Culex bitaeniorhynchus (10) mosquitoes and was also shown recently to infect experimentally Culex quinquefasciatus and Aedes detritus (39). While it is clear that JEV g5 strains can be efficiently transmitted by mosquitoes, there is further need to evaluate their potential for vector infection and transmission and compare them to those of highly circulating genotypes. Reservoir hosts, such as birds or pigs, develop the high viremia needed for infection of mosquitoes (3) and thus are another important factor in JEV natural transmission. Further investigation of avian and porcine species' capacity for amplifying JEV g5 is needed to address the contribution of reservoir hosts in restricting these viruses' circulation.
Production and characterization of a live JEV g5 using a cDNA-based technology.
In spite of being rarely isolated in areas of endemicity, the fact that a JEV g5 strain had been associated with a case of human encephalitis (11) warrants the need for further characterization of viruses belonging to this genotype. In the present work, we performed assembly of cDNA portions of the JEV-XZ0934 genome within cells and were able to produce and characterize a JEV g5 virus. We systematically compared the infectious properties of this JEV g5 strain with JEV-RP-9, a g3 strain that has been extensively studied in vitro and in vivo (17, 38, 40–42). The biological properties of JEV g5 were evaluated by infecting cell lines from various origins (Fig. 2). Three major phenotypic differences between g3 and g5 viruses could be reported. First, JEV g5 induces the formation of small foci, a feature also observed for the chimeric JEV S-g3/NS-g5 but not the parental JEV g3 (Fig. 7B), thus suggesting that the nonstructural protein region of g5 contributes to this phenotype. While the development of small infection foci in cultured cell lines has been associated to virus attenuation (43, 44), this was not the case with JEV g5, as it was highly pathogenic for mice (Fig. 3A and B). Second, we detected a level of JEV g5 NS5 protein accumulation in infected human neuronal cells that was higher than that observed for JEV g3 (Fig. 2B and D), also observed in other cell lines (data not shown). While such a difference in nonstructural protein accumulation did not appear to have a direct impact on the JEV infectious cycle, it is probable that it has an implication on other aspects of JEV infection that remain to be determined. Third, immunofluorescence analysis showed that JEV g5 E protein displays a weaker signal and a more diffuse pattern inside infected cells than does JEV g3 E protein (Fig. 2D). Intriguingly, JEV g5 E protein exhibited a lower apparent molecular weight on SDS-PAGE than did JEV g3 E protein, which was not associated to a change in its glycosylation profile (Fig. 2B and C). It would be of interest to investigate whether the biochemical characteristics of JEV g5 E protein are linked to the pathogenic properties of viruses expressing this protein.
JEV g5 is highly pathogenic in mice.
One major finding of our study was that JEV g5 displayed a marked neuropathogenicity in BALB/c mice while JEV g3 was essentially attenuated (Fig. 3B). BALB/c mice are known to generally display resistance to peripheral infection with nonneuroadapted JEV g3 strains (28, 29). The viruses that caused mice to develop fatal encephalitis, namely, JEV g5 and chimeric JEV S-g5/NS-g3, presented a sustained viremia in mice inoculated by the peripheral route, while JEV g3 was rapidly cleared in challenged animals (Fig. 5A and 7D). The viral load in blood is known to be an important factor in the development of JEV encephalitis, and the control of early viremia is crucial for disease development in the case of JEV (27, 45) and related encephalitic viruses (30, 34, 46). It was also reported that a better infection of myeloid cells could be linked to an enhanced pathogenesis in a mouse model of JEV infection (47). Hence, we first hypothesized that a greater efficiency at infecting mouse peripheral blood mononuclear cells (PBMCs) could cause JEV g5 sustained early viremia. Nevertheless, we did not observe any differences in the ability of JEV g3 or g5 to infect BALB/c mouse-derived PBMCs in vitro (data not shown). Although systemic infection of peripheral tissues, such as the spleen, could also allow viral amplification and subsequent neuroinvasion (30, 48), we did not observe any significant viral load in peripheral tissues of mice infected with JEV g5 (Fig. 5B to D), thereby excluding systemic infection as a cause for enhanced lethality.
A likely explanation for enhanced JEV g5 virulence is that immune cells elicit stronger host innate and adaptive immune responses following infection with JEV g3 than with JEV g5, thus restricting virus spread. In C57BL/6 mice, it was demonstrated that the control of viremia levels and CNS invasion with another JEV g3 strain strongly depended on the presence of B cells and on the ability to produce antibodies, whereas the T cells had a marginal contribution in mounting an early response to JEV infection (27). While we did not evaluate the contribution of T cells in this work, we note that a T-cell epitope present in JEV g3 E protein (CYHASVTDI) and found to elicit the T-cell response in BALB/c mice (49) was conserved in JEV g5. Nevertheless, it would be worth assessing the contribution of the T-cell response and the involvement of the major histocompatibility complex in BALB/c mouse susceptibility to JEV g5.
An unexpected finding was that JEV g5 did not raise neutralizing antibodies at days 5 to 7 postinoculation while JEV g3 did (Fig. 4C). A blunted early humoral response has already been shown to be a turning point in the development of viral encephalitis mediated by neurovirulent flaviviruses (27, 30, 47). Neutralizing anti-JEV antibodies were detected within the first week of JEV g3 infection, which matches the peak of IgM antibody production. Since levels of anti-JEV IgM in serum are known to be an important factor governing the outcome of JEV or West Nile virus (WNV) infections (50, 51), a blunted IgM response against JEV g5 could be responsible for the observed lack of neutralizing activities of anti-JEV antibodies at early infection times. Such a delay in the mounting of an early protective immunity could explain, in part, the high sensitivity of BALB/c mice to JEV g5 infection.
The levels of anti-JEV IgG antibodies were similarly raised in mice infected with JEV g3 and in those infected with JEV g5 (Fig. 3C). Interestingly, IgG antibodies that were raised against JEV g5 had poor neutralizing activity against JEV g3 (Fig. 4D). Such results could reflect a low propensity for JEV g5 to elicit the production of antibodies directed toward the more conserved epitopes among the JEV genotypes. Importantly, the mice that survived JEV g3 infection had raised levels of neutralizing antibodies that were only slightly less potent at inhibiting JEV g5 than JEV g3 (Fig. 4D). This information is of particular meaning since most of the currently available JEV vaccines were raised against JEV g3 viruses. Thus, it is of importance to evaluate the cross-protective capacity of current vaccination strategies against other circulating JEV genotypes, such as g5 JEV strains.
In our model of JEV encephalitis, it appears that the marked neuropathogenicity of JEV g5 in BALB/c mice is associated mainly to its capacity at invading the CNS. While circulation of cytokines and chemokines is known to facilitate BBB breaking and viral neuroinvasion (48, 52–54), we found that proinflammatory cytokines or immune response-related genes were induced only once the virus could be detected in the mouse brain (Fig. 6). In addition, we did not observe any early activation of genes related to viral infection in peripheral organs other than the brain (data not shown). Experimental infection of mice with JEV has generally led to a similar observation, whereby viral infiltration of the CNS leads to infection of microglia and other resident cells, which in turn activates production of cytokines and chemokines that promote neuronal death and BBB disruption (38, 47, 55, 56). While the JEV g3 strain used in this study was not generally found to be neuroinvasive in BALB/c mice (Fig. 3B), we note that it was competent at activating microglia and subsequent inflammatory markers in the brains of susceptible mice, such as C57BL/6 (Fig. 3A; data not shown). Since JEV infection of endothelial cells is believed to be the virus entry site into the brain (35, 37), we decided to evaluate the capacity of JEV g5 to cross the BBB using an in vitro model (Fig. 8). Surprisingly, we found that primary BALB/c endothelial cells were more permissible to JEV g3 infection than JEV g5. JEV g3 also infected primary BALB/c pericytes at a higher rate than JEV g5, and the infection induced a higher production of IL-6 than did g5. Overall, in this ex vivo model of BBB, JEV g3 appeared to have a better capacity to mediate endothelial cell disruption and CNS invasion, despite the fact that it was poorly neuroinvasive in BALB/c mice. This exemplifies the limit of in vitro models in the comprehensive study of JEV-related encephalitis. In the particular case of the mouse model employing BALB/c mice, a sustained blood viremia and a blunted humoral response are the main factors favoring JEV g5 entry in the CNS and the subsequent development of viral encephalitis.
JEV g5 neuropathogenicity associates with the structural protein region.
Although both JEV g3 and g5 strains are able to infect the CNS, they greatly differed in their capacity at invading the CNS of BALB/c mice. The use of chimeric JEV demonstrated the key role of the structural protein region in the neuroinvasiveness of JEV g5 (Fig. 7). As stated earlier, the percentage of identity between g3 and g5 viruses is unusually low among JEV genotypes. In the particular case of JEV strains RP-9 (g3) and XZ0934 (g5), it spans 91.52% at the amino acid level. Among the structural proteins, the sequence identity is of 79% for C (26 divergent amino acids), 90% for prM (17 divergent amino acids), and 92% for E (42 divergent amino acids). Since JEV S-g5/NS-g3 contains a chimeric E protein between g5 (amino acids 1 to 412) and g3 (amino acids 413 to 500), the number of divergent amino acid residues in E potentially involved in JEV g5 virulence can be restricted to 40. One of the critical issues to be addressed in the future relates to the identification of the E residues that contribute to the pathogenic properties of JEV strain XZ0934.
Limitations.
We acknowledge that our study is based essentially on the use of molecular clones of JEV g3 and g5 and not on wild-type isolates. In particular, the available sequence of strain XZ0934, which we used as a representative JEV g5 strain, was obtained from a single plaque pick and consequently may or may not reflect the average sequence of the viral population. Since only two full-length sequences (strains Muar and XZ0934) (9, 14) and one E protein sequence (strain 1827) (10) of g5 viruses are currently available, it is not yet known if XZ0934 is representative of this particular genotype. It remains therefore to be determined whether the marked neuropathogenicity of JEV strain JEV-XZ0934 can be generalized to other JEV g5 isolates. Comparative sequence analysis of the structural protein region between strains Muar and XZ0934 identified 16 nonconserved amino acid residues (9 in C, 1 in prM, and 6 in E), while there were 2 diverging residues between the E proteins of strains XZ0934 and 10-1827, both of which were isolated more recently. Therefore, it is urgent to determine which viral determinants could contribute to the marked neuropathogenicity of XZ0934 in a mouse model. Such study will broaden our knowledge of the molecular basis of the pathogenicity of JEV strains belonging to genotype 5.
ACKNOWLEDGMENTS
The research leading to these results and M.D.W. received funding from the European Union's Seventh Framework Program for research, technological development and demonstration under grant agreement no. 278433-PREDEMICS.
We thank Yi-Lin Ling for providing the JEV-RP-9 cDNA clone and the JEV-RP-9 replicon and Yoshiharu Matsuura for providing the anti-JEV antibodies.
REFERENCES
- 1.Campbell GL, Hills SL, Fischer M, Jacobson JA, Hoke CH, Hombach JM, Marfin AA, Solomon T, Tsai TF, Tsu VD, Ginsburg AS. 2011. Estimated global incidence of Japanese encephalitis: a systematic review. Bull W H O 89:766–774. doi: 10.2471/BLT.10.085233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischer M, Hills S, Staples E, Johnson B, Yaich M, Solomon T, Scheld WM, Hammer SM, Hughes JM. 2008. Japanese encephalitis prevention and control: advances, challenges, and new initiatives, p 93–124. In Schel WM, Hammer SM, Hughes JM (ed), Emerging infections 8. American Society for Microbiology, Washington, DC. [Google Scholar]
- 3.Le Flohic G, Porphyre V, Barbazan P, Gonzalez J-P. 2013. Review of climate, landscape, and viral genetics as drivers of the Japanese encephalitis virus ecology. PLoS Negl Trop Dis 7:e2208. doi: 10.1371/journal.pntd.0002208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weaver SC, Barrett ADT. 2004. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol 2:789–801. doi: 10.1038/nrmicro1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuh AJ, Ward MJ, Brown AJL, Barrett ADT. 2013. Phylogeography of Japanese encephalitis virus: genotype is associated with climate. PLoS Negl Trop Dis 7:e2411. doi: 10.1371/journal.pntd.0002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan X-L, Liu H, Wang H-Y, Fu S-H, Liu H-Z, Zhang H-L, Li M-H, Gao X-Y, Wang J-L, Sun X-H, Lu X-J, Zhai Y-G, Meng W-S, He Y, Wang H-Q, Han N, Wei B, Wu Y-G, Feng Y, Yang D-J, Wang L-H, Tang Q, Xia G, Kurane I, Rayner S, Liang G-D. 2011. Emergence of genotype I of Japanese encephalitis virus as the dominant genotype in Asia. J Virol 85:9847–9853. doi: 10.1128/JVI.00825-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao X, Liu H, Wang H, Fu S, Guo Z, Liang G. 2013. Southernmost Asia is the source of Japanese encephalitis virus (genotype 1) diversity from which the viruses disperse and evolve throughout Asia. PLoS Negl Trop Dis 7:e2459. doi: 10.1371/journal.pntd.0002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su C-L, Yang C-F, Teng H-J, Lu L-C, Lin C, Tsai K-H, Chen Y-Y, Chen L-Y, Chang S-F, Shu P-Y. 2014. Molecular epidemiology of Japanese encephalitis virus in mosquitoes in Taiwan during 2005-2012. PLoS Negl Trop Dis 8:e3122. doi: 10.1371/journal.pntd.0003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M-H, Fu S-H, Chen W-X, Wang H-Y, Guo Y-H, Liu Q-Y, Li Y-X, Luo H-M, Da W, Duo Ji DZ, Ye X-M, Liang G-D. 2011. Genotype V Japanese encephalitis virus is emerging. PLoS Negl Trop Dis 5:e1231. doi: 10.1371/journal.pntd.0001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takhampunya R, Kim H-C, Tippayachai B, Kengluecha A, Klein TA, Lee W-J, Grieco J, Evans BP. 2011. Emergence of Japanese encephalitis virus genotype V in the Republic of Korea. Virol J 8:449. doi: 10.1186/1743-422X-8-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hale JH, Lim KA, Chee PH. 1952. Japanese type B encephalitis in Malaya. Ann Trop Med Parasitol 46:220–226. [DOI] [PubMed] [Google Scholar]
- 12.Hasegawa H, Yoshida M, Fujita S, Kobayashi Y. 1994. Comparison of structural proteins among antigenically different Japanese encephalitis virus strains. Vaccine 12:841–844. doi: 10.1016/0264-410X(94)90294-1. [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa H, Yoshida M, Kobayashi Y, Fujita S. 1995. Antigenic analysis of Japanese encephalitis viruses in Asia by using monoclonal antibodies. Vaccine 13:1713–1721. doi: 10.1016/0264-410X(95)00099-M. [DOI] [PubMed] [Google Scholar]
- 14.Mohammed MAF, Galbraith SE, Radford AD, Dove W, Takasaki T, Kurane I, Solomon T. 2011. Molecular phylogenetic and evolutionary analyses of Muar strain of Japanese encephalitis virus reveal it is the missing fifth genotype. Infect Genet Evol 11:855–862. doi: 10.1016/j.meegid.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 15.Ishikawa T, Abe M, Masuda M. 2015. Construction of an infectious molecular clone of Japanese encephalitis virus genotype V and its derivative subgenomic replicon capable of expressing a foreign gene. Virus Res 195:153–161. doi: 10.1016/j.virusres.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Chen LK, Lin YL, Liao CL, Lin CG, Huang YL, Yeh CT, Lai SC, Jan JT, Chin C. 1996. Generation and characterization of organ-tropism mutants of Japanese encephalitis virus in vivo and in vitro. Virology 223:79–88. doi: 10.1006/viro.1996.0457. [DOI] [PubMed] [Google Scholar]
- 17.Liang J-J, Liao C-L, Liao J-T, Lee Y-L, Lin Y-L. 2009. A Japanese encephalitis virus vaccine candidate strain is attenuated by decreasing its interferon antagonistic ability. Vaccine 27:2746–2754. doi: 10.1016/j.vaccine.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Pu S-Y, Wu R-H, Yang C-C, Jao T-M, Tsai M-H, Wang J-C, Lin H-M, Chao Y-S, Yueh A. 2011. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J Virol 85:2927–2941. doi: 10.1128/JVI.01986-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katoh H, Mori Y, Kambara H, Abe T, Fukuhara T, Morita E, Moriishi K, Kamitani W, Matsuura Y. 2011. Heterogeneous nuclear ribonucleoprotein A2 participates in the replication of Japanese encephalitis virus through an interaction with viral proteins and RNA. J Virol 85:10976–10988. doi: 10.1128/JVI.00846-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori Y, Okabayashi T, Yamashita T, Zhao Z, Wakita T, Yasui K, Hasebe F, Tadano M, Konishi E, Moriishi K, Matsuura Y. 2005. Nuclear localization of Japanese encephalitis virus core protein enhances viral replication. J Virol 79:3448–3458. doi: 10.1128/JVI.79.6.3448-3458.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chien H-L, Liao C-L, Lin Y-L. 2011. FUSE binding protein 1 interacts with untranslated regions of Japanese encephalitis virus RNA and negatively regulates viral replication. J Virol 85:4698–4706. doi: 10.1128/JVI.01950-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desprès P, Paulous S, Crublet E. June 2012. Mgmt-based method for obtaining high yield of recombinant protein expression. WO patent 2012076715 A1.
- 23.Boroujerdi A, Tigges U, Welser-Alves JV, Milner R. 2014. Isolation and culture of primary pericytes from mouse brain. Methods Mol Biol 1135:383–392. doi: 10.1007/978-1-4939-0320-7_31. [DOI] [PubMed] [Google Scholar]
- 24.Chen C-J, Ou Y-C, Li J-R, Chang C-Y, Pan H-C, Lai C-Y, Liao S-L, Raung S-L, Chang C-J. 2014. Infection of pericytes in vitro by Japanese encephalitis virus disrupts the integrity of the endothelial barrier. J Virol 88:1150–1161. doi: 10.1128/JVI.02738-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Chen P, Cao R, Gu J. 2011. Mutation of putative N-linked glycosylation sites in Japanese encephalitis virus premembrane and envelope proteins enhances humoral immunity in BALB/C mice after DNA vaccination. Virol J 8:138. doi: 10.1186/1743-422X-8-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freeze HH, Kranz C. 2008. Endoglycosidase and glycoamidase release of N-linked glycans. Curr Protoc Immunol Chapter 8:Unit 8.15. doi: 10.1002/0471142735.im0815s83. [DOI] [PubMed] [Google Scholar]
- 27.Larena M, Regner M, Lee E, Lobigs M. 2011. Pivotal role of antibody and subsidiary contribution of CD8+ T cells to recovery from infection in a murine model of Japanese encephalitis. J Virol 85:5446–5455. doi: 10.1128/JVI.02611-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miura K, Goto N, Suzuki H, Fujisaki Y. 1988. Strain difference of mouse in susceptibility to Japanese encephalitis virus infection. Jikken Dobutsu 37:365–373. [DOI] [PubMed] [Google Scholar]
- 29.Matsuo S, Morita K, Bundo-Morita K, Igarashi A. 1994. Differences in susceptibility to peripheral infection with Japanese encephalitis virus among inbred strains of mouse. Uirusu 44:205–215. (In Japanese.) doi: 10.2222/jsv.44.205. [DOI] [PubMed] [Google Scholar]
- 30.Prow NA, Setoh YX, Biron RM, Sester DP, Kim KS, Hobson-Peters J, Hall RA, Bielefeldt-Ohmann H. 2014. The West Nile virus-like flavivirus Koutango is highly virulent in mice due to delayed viral clearance and the induction of a poor neutralizing antibody response. J Virol 88:9947–9962. doi: 10.1128/JVI.01304-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samuel MA, Diamond MS. 2006. Pathogenesis of West Nile virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol 80:9349–9360. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diamond MS, Shrestha B, Mehlhop E, Sitati E, Engle M. 2003. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol 16:259–278. doi: 10.1089/088282403322396082. [DOI] [PubMed] [Google Scholar]
- 33.Dowd KA, Jost CA, Durbin AP, Whitehead SS, Pierson TC. 2011. A dynamic landscape for antibody binding modulates antibody-mediated neutralization of West Nile virus. PLoS Pathog 7:e1002111. doi: 10.1371/journal.ppat.1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar M, Roe K, Nerurkar PV, Namekar M, Orillo B, Verma S, Nerurkar VR. 2012. Impaired virus clearance, compromised immune response and increased mortality in type 2 diabetic mice infected with West Nile virus. PLoS One 7:e44682. doi: 10.1371/journal.pone.0044682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.German AC, Myint KSA, Mai NTH, Pomeroy I, Phu NH, Tzartos J, Winter P, Collett J, Farrar J, Barrett A, Kipar A, Esiri MM, Solomon T. 2006. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model. Trans R Soc Trop Med Hyg 100:1135–1145. doi: 10.1016/j.trstmh.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 36.Liou ML, Hsu CY. 1998. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res 293:389–394. doi: 10.1007/s004410051130. [DOI] [PubMed] [Google Scholar]
- 37.Liu T-H, Liang L-C, Wang C-C, Liu H-C, Chen W-J. 2008. The blood-brain barrier in the cerebrum is the initial site for the Japanese encephalitis virus entering the central nervous system. J Neurovirol 14:514–521. doi: 10.1080/13550280802339643. [DOI] [PubMed] [Google Scholar]
- 38.Chen S-T, Liu R-S, Wu M-F, Lin Y-L, Chen S-Y, Tan DT-W, Chou T-Y, Tsai I-S, Li L, Hsieh S-L. 2012. CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog 8:e1002655. doi: 10.1371/journal.ppat.1002655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackenzie-Impoinvil L, Impoinvil DE, Galbraith SE, Dillon RJ, Ranson H, Johnson N, Fooks AR, Solomon T, Baylis M. 2015. Evaluation of a temperate climate mosquito, Ochlerotatus detritus (Aedes detritus), as a potential vector of Japanese encephalitis virus. Med Vet Entomol 29:1–9. doi: 10.1111/mve.12083. [DOI] [PubMed] [Google Scholar]
- 40.Yen L-C, Lin Y-L, Sung H-H, Liao J-T, Tsao C-H, Su C-M, Lin C-K, Liao C-L. 2013. Neurovirulent flavivirus can be attenuated in mice by incorporation of neuron-specific microRNA recognition elements into viral genome. Vaccine 31:5915–5922. doi: 10.1016/j.vaccine.2011.09.102. [DOI] [PubMed] [Google Scholar]
- 41.Tu Y-C, Yu C-Y, Liang J-J, Lin E, Liao C-L, Lin Y-L. 2012. Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J Virol 86:10347–10358. doi: 10.1128/JVI.00525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin K-C, Chang H-L, Chang R-Y. 2004. Accumulation of a 3′-terminal genome fragment in Japanese encephalitis virus-infected mammalian and mosquito cells. J Virol 78:5133–5138. doi: 10.1128/JVI.78.10.5133-5138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Y. 2010. Phenotypic and genotypic characteristics of Japanese encephalitis attenuated live vaccine virus SA14-14-2 and their stabilities. Vaccine 28:3635–3641. doi: 10.1016/j.vaccine.2010.02.105. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Z, Date T, Li Y, Kato T, Miyamoto M, Yasui K, Wakita T. 2005. Characterization of the E-138 (Glu/Lys) mutation in Japanese encephalitis virus by using a stable, full-length, infectious cDNA clone. J Gen Virol 86:2209–2220. doi: 10.1099/vir.0.80638-0. [DOI] [PubMed] [Google Scholar]
- 45.Lee E, Lobigs M. 2002. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray Valley encephalitis virus. J Virol 76:4901–4911. doi: 10.1128/JVI.76.10.4901-4911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samuel MA, Diamond MS. 2005. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol 79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang K, Deubel V. 2011. Mice with different susceptibility to Japanese encephalitis virus infection show selective neutralizing antibody response and myeloid cell infectivity. PLoS One 6:e24744. doi: 10.1371/journal.pone.0024744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han YW, Choi JY, Uyangaa E, Kim SB, Kim JH, Kim BS, Kim K, Eo SK. 2014. Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog 10:e1004319. doi: 10.1371/journal.ppat.1004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takada K, Masaki H, Konishi E, Takahashi M, Kurane I. 2000. Definition of an epitope on Japanese encephalitis virus (JEV) envelope protein recognized by JEV-specific murine CD8+ cytotoxic T lymphocytes. Arch Virol 145:523–534. doi: 10.1007/s007050050043. [DOI] [PubMed] [Google Scholar]
- 50.Libraty DH, Nisalak A, Endy TP, Suntayakorn S, Vaughn DW, Innis BL. 2002. Clinical and immunological risk factors for severe disease in Japanese encephalitis. Trans R Soc Trop Med Hyg 96:173–178. doi: 10.1016/S0035-9203(02)90294-4. [DOI] [PubMed] [Google Scholar]
- 51.Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med 198:1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye J, Jiang R, Cui M, Zhu B, Sun L, Wang Y, Zohaib A, Dong Q, Ruan X, Song Y, He W, Chen H, Cao S. 2014. Etanercept reduces neuroinflammation and lethality in mouse model of Japanese encephalitis. J Infect Dis 210:875–889. doi: 10.1093/infdis/jiu179. [DOI] [PubMed] [Google Scholar]
- 53.Hayasaka D, Shirai K, Aoki K, Nagata N, Simantini DS, Kitaura K, Takamatsu Y, Gould E, Suzuki R, Morita K. 2013. TNF-α acts as an immunoregulator in the mouse brain by reducing the incidence of severe disease following Japanese encephalitis virus infection. PLoS One 8:e71643. doi: 10.1371/journal.pone.0071643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chai Q, He WQ, Zhou M, Lu H, Fu ZF. 2014. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol 88:4698–4710. doi: 10.1128/JVI.03149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Srivastava R, Kalita J, Khan MY, Misra UK. 2012. Status of proinflammatory and anti-inflammatory cytokines in different brain regions of a rat model of Japanese encephalitis. Inflamm Res 61:381–389. doi: 10.1007/s00011-011-0423-5. [DOI] [PubMed] [Google Scholar]
- 56.Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P, Sen E, Basu A. 2007. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55:483–496. doi: 10.1002/glia.20474. [DOI] [PubMed] [Google Scholar]