

ABSTRACT
Understanding the modalities of interaction of neurotropic viruses with their target cells represents a major challenge that may improve our knowledge of many human neurological disorders for which viral origin is suspected. Borna disease virus (BDV) represents an ideal model to analyze the molecular mechanisms of viral persistence in neurons and its consequences for neuronal homeostasis. It is now established that BDV ensures its long-term maintenance in infected cells through a stable interaction of viral components with the host cell chromatin, in particular, with core histones. This has led to our hypothesis that such an interaction may trigger epigenetic changes in the host cell. Here, we focused on histone acetylation, which plays key roles in epigenetic regulation of gene expression, notably for neurons. We performed a comparative analysis of histone acetylation patterns of neurons infected or not infected by BDV, which revealed that infection decreases histone acetylation on selected lysine residues. We showed that the BDV phosphoprotein (P) is responsible for these perturbations, even when it is expressed alone independently of the viral context, and that this action depends on its phosphorylation by protein kinase C. We also demonstrated that BDV P inhibits cellular histone acetyltransferase activities. Finally, by pharmacologically manipulating cellular acetylation levels, we observed that inhibiting cellular acetyl transferases reduces viral replication in cell culture. Our findings reveal that manipulation of cellular epigenetics by BDV could be a means to modulate viral replication and thus illustrate a fascinating example of virus-host cell interaction.
IMPORTANCE Persistent DNA viruses often subvert the mechanisms that regulate cellular chromatin dynamics, thereby benefitting from the resulting epigenetic changes to create a favorable milieu for their latent and persistent states. Here, we reasoned that Borna disease virus (BDV), the only RNA virus known to durably persist in the nucleus of infected cells, notably neurons, might employ a similar mechanism. In this study, we uncovered a novel modality of virus-cell interaction in which BDV phosphoprotein inhibits cellular histone acetylation by interfering with histone acetyltransferase activities. Manipulation of cellular histone acetylation is accompanied by a modulation of viral replication, revealing a perfect adaptation of this “ancient” virus to its host that may favor neuronal persistence and limit cellular damage.
INTRODUCTION
Long-term persistence in the host cell is a real challenge for viruses, since it requires a tight control of viral replication in order to limit cytopathic effects. This is notably the case for viruses infecting cells with poor renewal capacities, such as neurons. Thus, neurotropic viruses provide a unique opportunity to decipher the molecular mechanisms underlying virus-cell interactions during persistence. Moreover, a better understanding of the physiological consequences of viral persistence in the central nervous system (CNS) may also help to clarify some unknown aspects of neuronal physiology, under normal and pathological conditions (1, 2).
Borna disease virus (BDV) represents an ideal paradigm for investigating the modalities of persistence of a noncytolytic virus in the CNS. BDV exhibits a selective tropism for neurons of the limbic system, in particular the cortex and hippocampus, two structures that govern many cognitive and behavioral functions (3). Although BDV is highly neurotropic, it can also replicate in other cells of the CNS, as well as in many established cell lines in vitro. BDV infects a wide variety of mammals, leading to a large spectrum of neurological disorders, ranging from acute encephalitis to behavioral alterations without inflammation (4, 5). Despite this wide host range, there is still controversy regarding whether BDV can infect humans and be associated with neurological diseases (2). The recent detection of endogenized BDV-like sequences in a wide array of species, including primates, has, however, established that ancient bornaviruses actually infected our ancestors nearly 40 million years ago, thus revealing the long coevolution between bornaviruses and animal species (6).
BDV is an enveloped virus, with a nonsegmented, negative-strand RNA genome of 8.9 kb, the smallest among the members of the Mononegavirales order (7). Its compact genome encodes six proteins, namely, the nucleoprotein (N), phosphoprotein (P), protein X, matrix protein (M), glycoprotein (G), and polymerase (L). Whereas M and G are involved in particle formation, P, N, and L are components of the ribonucleoprotein complex (RNP). One striking feature of BDV infection is that of the location of RNP in the nucleus, where viral transcriptions and replication take place (1). This is an exception for vertebrate RNA viruses, which usually replicate in the cytoplasm of infected cells. The members of the only other family of RNA viruses that replicates in the nucleus, the Orthomyxoviridae, are cytolytic (3). Hence, BDV is the only RNA virus that durably persists in the nucleus of infected cells.
To achieve this efficient intranuclear persistence, BDV closely associates with chromatin and forms viral factories, designated vSPOTs (viral speckles of transcripts), which contain all viral components necessary for viral transcriptions. It was shown that vSPOTs interact in a dynamic manner with host chromosomes, using high-mobility protein group B-1 (HMGB-1) as a scaffold protein between RNP and chromatin (1, 8). Based on these findings, it was tempting to hypothesize that BDV may modify the host cell chromatin or interact with factors controlling chromatin dynamics. Moreover, in a previous proteomic analysis performed using primary neuronal culture protein extracts, we observed that acetylation of histone H2B was changed upon infection, suggesting that epigenetic signaling could indeed be affected by BDV (9).
Among the multiple modes of virus-mediated hijacking of host cell functions, manipulation of epigenetic signaling appears particularly well suited to persistent viruses, especially for DNA viruses. This allows them to benefit from the epigenetic changes that they induce in the nucleus of target cells, thereby producing a favorable environment for their latent/persistent states or for reactivation (10–13). This has been well described for herpesviruses, such as Kaposi's sarcoma-associated herpesvirus, for which the latency-associated nuclear antigen has been shown to interact with host chromatin modifiers and induce epigenetic reprogramming (14, 15). Such a scenario appeared, however, less likely for RNA viruses. The long-lasting nuclear persistence of BDV, together with its close association with chromatin, led us to hypothesize that, similarly to infections by large DNA viruses, BDV infection could be accompanied by virus-mediated changes in the host cell epigenome.
As histone acetylation is a crucial modification allowing fine control of neuronal gene expression and is often perturbed in numerous neurological pathologies (16, 17), we sought to analyze the impact of BDV infection on epigenetic signaling, with a particular focus on histone acetylation patterns. Using both primary neuronal cultures and persistently infected cell lines, we demonstrate that BDV infection leads to a decrease of histone acetylation levels (restricted to specific H2B and H4 lysine residues). We also identify the BDV P as the viral determinant responsible for this inhibition and show that it needs to be phosphorylated by protein kinase C (PKC) to exert this effect. Finally, we show that decreased histone acetylation is due to inhibition of cellular histone acetyl transferases (HATs) and that pharmacological manipulation of cellular acetylation modulates viral replication. This suggests that inhibition of histone acetylation may serve to control viral replication during persistence.
(This research was conducted in part by Emilie M. Bonnaud in partial fulfillment of the requirements for a Ph.D. from Université Toulouse III Paul Sabatier, Toulouse, France, 2015.)
MATERIALS AND METHODS
Ethics statement.
Animal handling and care for the preparation of primary neuronal cultures from rat embryos were performed in accordance with European Union Council Directive 86/609/EEC, and experiments were performed following the French national chart for ethics of animal experiments (articles R 214-87 to -90 of the “Code rural”). Our protocol received approval from the local committee on the ethics of animal experiments (permit number 04-U1043-DG-06). Pregnant rats were deeply anesthetized with CO2 before euthanasia to minimize suffering.
Primary neuronal cultures, virus infection, and plasmid transfection.
Primary cortical neurons were prepared from Sprague-Dawley rat embryos at gestational day 17 by the papain dissociation method as described previously (18). At the end of the procedure, neurons were either directly plated on variable supports (glass coverslips or culture plates, depending on the experiments) or used for plasmid transfection before plating. Before seeding, supports were coated with 500 μg/ml poly-ornithine (Sigma), followed by 5 μg/ml laminin (Roche). Neurons were maintained in Neurobasal medium (Invitrogen) supplemented with 100 μg/ml penicillin-streptomycin, 0.5 mM glutamine, and 2% B-27 supplement (Invitrogen). Half of each culture was infected with cell-free BDV 4 h after plating. Cell-released virus stocks were prepared using Vero cells persistently infected by wild-type BDV (Giessen strain He/80) or different recombinant BDVs (rBDV) as previously described (19). By 14 days postinfection, the efficacy of virus spread into the culture (which routinely attained >95%) was systematically assessed by indirect immunofluorescence for the detection of BDV antigens.
To achieve efficient expression of isolated BDV proteins, neurons were transfected using Amaxa technology and a rat neuron Nucleofector kit (Lonza) (5 × 106 neurons with 3 μg of plasmid DNA, using the G-13 program) following the instructions of the manufacturer, allowing routine transfection efficiencies of at least 75% for primary neurons. Neurons were plated for 14 days, the time at which transfection efficiency was systematically verified by indirect immunofluorescence before proceeding to histone preparation. Plasmids used for transfection contained genes encoding BDV N or BDV P proteins under the control of the cytomegalovirus (CMV) early enhancer/chicken β actin (CAG) promoter or an empty vector as a control. In addition, two plasmids encoding different mutants of BDV P were used, one in which the glutamate residue in position 84 was replaced by an asparagine (PE84N) to abolish interaction with HMGB1 (19) and another in which serine residues at positions 26 and 28 were replaced by alanine residues (PAA) to mutate the PKC phosphorylation site of P (19). All plasmids were kind gifts from M. Schwemmle. Plasmid maps are available on request.
Cell lines, production of lentiviral vectors, and transduction experiments.
Vero and HEK-293T cells were obtained from the American Type Culture Collection. We also used Vero cells persistently infected with wild-type BDV (Giessen strain He/80) or recombinant BDV in which the two serine residues in positions 26 and 28 of BDV-P were replaced by alanine residues (rBDV-PAA) or recombinant wild-type BDV (rBDV-Pwt), composed of the canonical sequence of wild-type BDV strain He/80 (19). Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2 mM l-glutamine. For HEK-293T cells, 2 mg/ml Geneticin was added to the medium.
Lentiviral vector plasmids allowing expression of BDV N or wild-type or mutant BDV P under the control of the CAG promoter were engineered by insertion of viral gene sequences into BamHI/XhoI restriction sites of the pTrip-CAG plasmid (kind gift of P. Charneau). Nonreplicative vector particles were produced by transient transfection of HEK-293T cells with each vector plasmid, the second-generation psPAX2 packaging plasmid (Addgene plasmid 12260, provided by D. Trono), and pMD2.G plasmid encoding the vesicular stomatitis virus envelope glycoprotein (VSV-G) (Addgene plasmid 12259, provided by D. Trono). Particles were concentrated from cell culture supernatants by ultracentrifugation. Gene transfer titers were determined on transduced HEK-293T cells by indirect immunofluorescence using anti-BDV N or P protein rabbit antisera.
For the establishment of Vero cell lines stably expressing individual BDV proteins, Vero cells were transduced by lentiviral vectors at a multiplicity of transduction of 10. A lentiviral vector encoding green fluorescent protein (GFP) was used to establish the control cell line. For each cell line, transduction efficacy was assessed by immunofluorescence, to ensure that at least 95% of the cells expressed the protein of interest.
Histone preparation and Western blot analysis.
The procedure for histone preparation was adapted from Shechter et al. (20). Nuclei were isolated upon cell lysis in a hypotonic buffer containing 0.2% Triton X-100, 20 mM β-glycerophosphate, 10 mM HEPES (pH 7.6), 1.5 mM MgCl2, 10 mM KCl, 10 mM sodium butyrate, 1 mM dithiothreitol [DTT], and protease inhibitors (Complete Mini; Roche). After centrifugation and washing, nuclei were incubated in 0.4 N HCl during 1 h at 4°C to allow histone solubilization. After removal of the nuclear debris by centrifugation, histones were precipitated from supernatant by the addition of 4 volumes of acetone and incubation overnight at −20°C. They were then centrifuged at 15,000 × g during 20 min, and histone-enriched pellets were resuspended in Laemmli buffer for Western blot analysis. Western blotting was performed as previously described (19) using either histone preparations or whole-cell extracts and the following primary antibodies: anti-total H2B, anti-H2B acetyl K5, anti-H2B acetyl K12, anti-H2B acetyl K15, anti-H2B acetyl K20, anti-H3 pan acetyl, anti-total H3, and anti-H4 acetyl K8 from Epitomics (rabbit monoclonal antibodies); anti-total H4, anti-H4 tetra-acetyl, anti-H4 acetyl K5, anti-H4 acetyl K12, and anti-H4 acetyl K16 from Active Motif (rabbit antibody); and rabbit antisera anti-BDV N and P proteins (21) and mouse monoclonal anti-actin (Sigma). The secondary antibodies used were anti-rabbit and anti-mouse antibodies coupled to 680-nm-wavelength and 770-nm-wavelength infrared dyes (Biotium). Laser scanning of blots and quantitative analyses were performed using an Odyssey infrared imaging system (Li-Cor). Quantification of the histone acetylation level was carried out by measuring the intensity of the signal corresponding to the acetylated histone normalized by the corresponding signal for total histone. Results were expressed as variations of acetylation compared to the mean levels of control cells (uninfected or mock transfected/transduced), which were set to 1. Data are presented as means ± standard errors of the means (SEM). Statistical significance was determined using paired t tests.
Indirect immunofluorescence analysis.
Cells were fixed with 2% paraformaldehyde at room temperature for 10 min followed by 4% paraformaldehyde for 10 min. They were washed 3 times in phosphate-buffered saline (PBS) (Invitrogen) and permeabilized with PBS–0.1% Triton X-100 during 4 min. Cells were washed twice in PBS and blocked in 2.5% normal goat serum (Invitrogen) for at least 1 h. Primary antibodies (the same as those used for the Western blot analyses described above) were diluted in PBS containing 2.5% normal goat serum and incubated 1 h at room temperature. For controlling infection, transduction, or transfection efficiencies, we used rabbit antisera raised against BDV N or BDV P proteins (21). To assess neuronal integrity, we used a mouse anti-β-tubulin III antibody (Sigma). Upon incubation with primary antibodies, cells were washed 3 times with PBS–0.1% Triton X-100 and incubated with secondary goat anti-rabbit antibody coupled to Alexa Fluor 488 or goat anti-mouse antibody coupled to Alexa Fluor 594, for 1 h at room temperature. After 3 washes, coverslips were mounted onto microscope slides using ProLong Gold (Invitrogen) containing DAPI (4′,6-diamidino-2-phenylindole). To measure histone acetylation levels, images were acquired using an LSM 510 confocal microscope (Zeiss) and fixed acquisition parameters. Imaris software (Bitplane AG, Switzerland) was used to automatically calculate the mean density of fluorescence signals for individual cells, after background subtraction and thresholding. The same parameters were used for the quantification of each picture. To assess BDV N and P protein expression, images were acquired on a Zeiss Apotome microscope.
Analysis of HAT and HDAC activities.
Nuclear extracts were prepared using a commercially available kit (Active Motif), and protein concentrations were determined by Bradford assays. Equal amounts of the extracts were then used to measure HAT and histone deacetylase (HDAC) enzymatic activities, by using a HAT assay kit and an HDAC assay kit (Active Motif). For each reaction, 2 μg of nuclear extracts was used for HAT assays and 4 μg of nuclear extracts for HDAC assays. Reactions were systematically carried out in duplicate for each extract. The assay data were obtained by fluorescence quantification using a Varioskan Flash scanner (Thermo Scientific).
Analysis of BDV multiplication and protein expression by flow cytometry.
Vero cells (4 × 105) were plated and infected with cell-free BDV at a multiplicity of infection of 0.5. They were treated with 1 mM sodium butyrate (Sigma) or 10 μM anacardic acid (Tocris Bioscience) on the day of infection and again 48 h later. After 4 days of incubation, cells were dissociated with trypsin, harvested, and fixed in 2% paraformaldehyde for 15 min at room temperature. After fixation, cells were permeabilized during 20 min with fluorescence-activated cell sorter (FACS) buffer containing 0.5% saponin, 1% bovine serum albumin, and 3 mM EDTA. Cells were then incubated in blocking buffer (3% preimmune rabbit serum diluted in FACS buffer) for 1 h before addition of 5 μg/ml of antibodies for 1 h at 4°C. Anti-BDV P and anti-isotype control antibodies were purified from rabbit sera (Abselect serum antibody purification system; Innova Biosciences) and preconjugated with an Atto-488 fluorescent label (Lightning-Link conjugation system; Innova Biosciences). After extensive washes, flow cytometry data were collected on a FACSCalibur (BD Biosciences) and analyzed using FlowJo software (TreeStar, version 8.8.6).
RT and qPCR for measurement of vRNA.
Vero cells (105) were plated and infected with cell-free BDV at a multiplicity of infection of 0.5. At different times postinfection, total RNAs were prepared from cells using a NucleoSpin kit (Macherey-Nagel) following the manufacturer's instructions. RNA (1 μg) was subjected to reverse transcription (RT) using SuperScript III reverse transcriptase (Life Technologies) with a primer specific for genomic viral RNA (vRNA) (primer sequence, 5′GACACTACGACGGGAACGAT3′). The resulting cDNAs were used for quantitative PCR (qPCR) experiments with primers specific for the BDV genome (forward primer, 5′CGGTAGACCAGCTCCTGAAG3′; reverse primer, 5′GAGGTCCACCTTCTCCATCA3′). In parallel, 1 μg of RNA was subjected to reverse transcription using random hexamers to amplify total cDNAs. These cDNAs were used for qPCR performed with primers specific for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) for normalization (forward primer, 5′CAATGACCCCTTCATTGACC3′; reverse primer, 5′GACAAGCTTCCCGTTCTCAG3′). qPCRs were prepared with LightCycler 480 DNA SYBR green I Master reaction mix (Roche Diagnostics) and run in duplicate on a LightCycler 480 instrument (Roche Diagnostics). Viral genome quantification was expressed in arbitrary units according to the following threshold cycle (CT) formula: amount of viral RNA = 2−(CT BDV − CT GAPDH).
RESULTS
BDV infection decreases histone acetylation of specific lysine residues.
To assess the impact of BDV infection on histone acetylation patterns, primary neuronal cultures were infected by BDV or left uninfected. Neurons were cultured for 14 days, a time sufficient to allow BDV spreading to all neurons, as verified by indirect immunofluorescence analysis (data not shown) and in agreement with our previous findings (21, 22). There was no overt impact of infection on the morphology or viability of the cultures, which is consistent with the noncytolytic replication strategy of BDV. From these neurons, we prepared histone fractions (20), which were analyzed by Western blotting using antibodies targeting each known acetylatable lysine of H2B (Fig. 1A, left panel). We observed that BDV infection led to a significant decrease of H2B acetylation on K5 and K20 (33% and 26% decreases, respectively) (Fig. 1A), consistent with our previous proteomics findings (9). In addition, infection also induced a decrease of acetylation on K15 but not on K12. We next asked whether this acetylation defect was restricted to H2B or whether it could be extended to two other core histones, H3 and H4, for which acetylation modifications have been extensively described (23). While global levels of H3 acetylation were unchanged by infection, a significant decrease of the global acetylation level of H4 was observed in infected neurons (Fig. 1B) which was attributable to decreased acetylation of K5, K8, and K16 but not of K12 (Fig. 1C).
FIG 1.
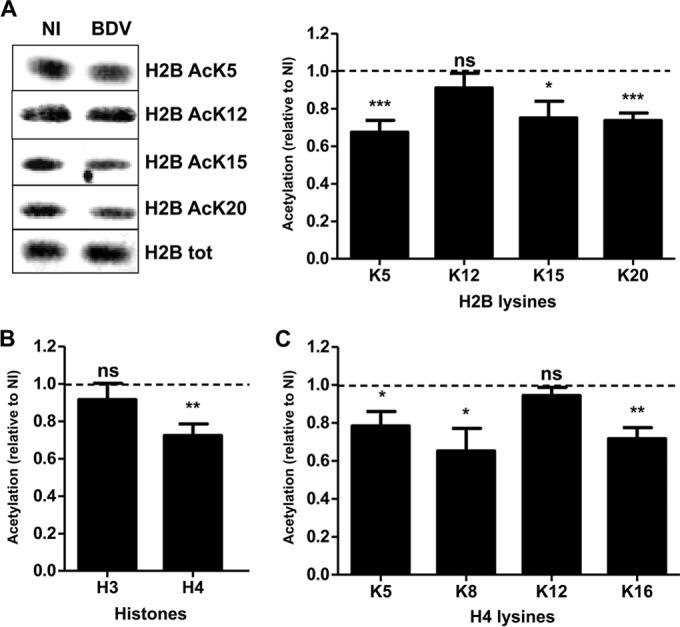
BDV infection decreases histone acetylation levels in primary neuronal cultures. (A) Western blot analysis of H2B acetylation levels. (Left panel) Histone fractions were prepared from parallel noninfected (NI) or infected (BDV) neuronal cultures and analyzed by Western blotting with the indicated antibodies. Total (tot) H2B levels were assessed for each sample and used to normalize acetylation signals. (Right panel) Quantification of H2B acetylation. Acetylation values (normalized for total H2B) obtained for infected neurons were normalized to the values obtained for noninfected neurons, which were set to 1 and which are represented by the dashed lines on the graphs. Data are expressed as means ± SEM of the results from at least nine independent sets of neuronal cultures. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns = nonsignificant (by paired t test). (B) Analysis of global H3 and H4 acetylation by Western blotting, done as described for panel A. (C) Analysis of acetylation on each H4 lysine residue by Western blotting, done as described for panel A.
As BDV is able to infect a wide variety of cell types in vitro, even those of nonneuronal origin, we next asked whether this histone acetylation decrease was also observed in other cell types. To this end, histone acetylation levels were evaluated in Vero cells persistently infected by BDV (Vero-BV [24]). We observed that histone acetylation levels were also decreased in Vero-BV cells and that, remarkably, the histone acetylation occurred on exactly the same lysine residues as in the primary neuronal cultures (Fig. 2). Interestingly, the changes were even more pronounced, reaching a decrease of around 40% to 50% depending on the lysine, presumably resulting from the fact that Vero-BV cells had been infected for numerous passages. As for the neurons, there was no change in acetylation of H2B K12 or H4 K12 or in global H3 acetylation, suggesting that BDV may impact a general process conserved in several cell types that selectively targets the acetylation status of specific lysine residues of H2B and H4.
FIG 2.
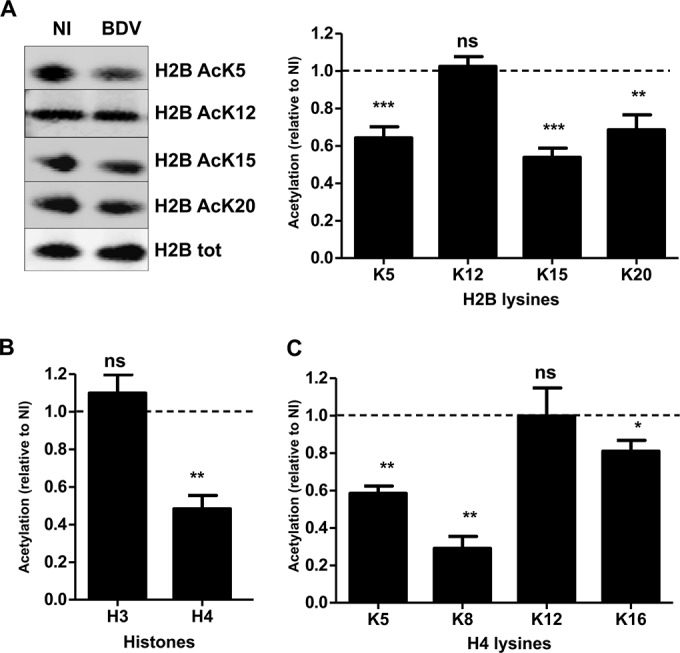
BDV infection decreases histone acetylation levels in persistently infected Vero cells. (A) Analysis of H2B acetylation. (Left panel) Histone fractions were prepared from parallel noninfected (NI) or persistently infected (BDV) Vero cells and analyzed by Western blotting with the indicated antibodies. Total H2B levels were assessed for each sample and used to normalize acetylation signals. (Right panel) Quantification of H2B acetylation. Acetylation values (normalized for total H2B) obtained for infected cells were normalized to the values obtained for noninfected cells, which were set to 1 and which are represented by the dashed lines on the graphs. Data are expressed as means ± SEM of the results from at least seven independent experiments, i.e., using cultures of Vero cells that were independently grown after the original split during 5 to 20 passages. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ns = nonsignificant (by paired t test). (B) Analysis of H3 and H4 global acetylation by Western blotting, done as described for panel A. (C) Western blot analysis of acetylation on each H4 lysine residues, done as described for panel A.
To confirm our findings concerning the impact of BDV infection on histone acetylation using a different approach, we performed indirect immunofluorescence analysis of histone acetylation using Vero and Vero-BV cells. As shown in Fig. 3, this analysis revealed a general decrease of fluorescence levels in infected cells when we assessed H2B acetylation for K5, K15, and K20, but not for K12, in perfect agreement with our Western blot results. This experiment also revealed that the decreases of acetylation were similar in all cells (Fig. 3, right panels) and thus represented a global effect resulting from BDV infection.
FIG 3.

Histone acetylation decrease due to BDV affects all infected cells. Immunofluorescence analysis was performed on noninfected (NI, left column) or persistently infected (BDV, right column) Vero cells, in order to visualize acetylation on H2B lysine residues, as indicated on the left of the figure. Bar = 25 μm. Fluorescence quantification is presented to the right of each corresponding image. The mean signal density per nucleus is expressed in arbitrary units (A. U.), and each spot corresponds to one nucleus. Analysis data represent the sum of the results from three cultures of Vero cells that were independently grown after the original split during 5 to 20 passages, with more than 100 cells being analyzed for each condition. ***, P ≤ 0.0001; ns = nonsignificant (by Mann-Whitney test).
BDV P is responsible for histone acetylation decreases.
We next assessed which of the BDV-encoded proteins could be responsible for this acetylation decrease. Among these, BDV N and BDV P represent potential candidates for decreasing histone acetylation. Indeed, they belong to the RNP complex and are abundant in the nucleus of infected cells, where they localize in close association with chromatin (1). To identify which of these two proteins could account for decreased histone acetylation, plasmids encoding BDV N or BDV P were transfected using Amaxa technology that allows a high rate of transfection for primary neuronal cultures without altering viability. Fourteen days after transfection, we used immunofluorescence analysis for ß-tubulin III to verify that neuronal integrity was indeed preserved. Moreover, at least 75% of neurons expressed the transfected BDV protein (Fig. 4A). We also observed that both BDV N and BDV P were expressed predominantly in the nucleus of transfected neurons, consistent with the presence of nuclear localization signals in both proteins (25, 26) (Fig. 4A). The results of analysis of histone acetylation levels in transfected neurons were compared to levels in neurons transfected with an empty plasmid. As shown in Fig. 4B for the acetylation of H2B K20, we observed that the singled-out expression of BDV P into primary neurons led to decreased histone acetylation levels, a result that was not observed for BDV N-expressing neurons (Fig. 4B and C). This experiment was reproduced for each lysine residue analyzed previously, and results are summarized in Table 1. Strikingly, isolated expression of BDV P in neurons had the same impact on histone acetylation as BDV infection, with the same lysine specificity and the same level of decrease. In order to verify whether the same findings were observed in nonneuronal cells, we generated stable Vero cell lines expressing each of the viral candidate proteins, or GFP as a control, and analyzed histone acetylation (Table 2). Acetylation of the different lysine residues of H2B and H4 was drastically and significantly reduced only in the cell line expressing BDV P, confirming that BDV P is responsible for decreased histone acetylation in a nonneuronal cell line. These data indicate that BDV P is the key viral determinant responsible for the decrease of histone acetylation during BDV infection.
FIG 4.

The BDV P protein is responsible for decreased histone acetylation levels. (A) Indirect immunofluorescence analysis of primary neurons 14 days posttransfection. Neurons were stained for DAPI, the viral BDV P or N proteins (green), and the neuronal marker β-tubulin III (βIII tubulin [red]). Merged images are shown on the right panels. Bars = 50 μm. (B) Western blot analysis, performed on nuclear extracts to visualize expression levels of BDV P or N proteins (top panel; Empty = neurons transfected with an empty vector) and on histone extracts to assess the levels of K20-acetylated H2B and total H2B (lower panels). (C) Quantification of H2B K20 acetylation levels in transfected neurons. Acetylation levels (relative to total H2B) were normalized to the values obtained for neurons transfected by an empty plasmid, which were set to 1. Data are expressed as means ± SEM of the results from at least five independent sets of transfected neuronal cultures. **, P ≤ 0.01 (by paired t test).
TABLE 1.
Histone acetylation levels for each lysine in primary neuronal cultures expressing isolated viral proteinsa
| Histone | Lysine | BDV P |
BDV N |
||
|---|---|---|---|---|---|
| n | Acetylation level (mean ± SEM) | n | Acetylation level (mean ± SEM) | ||
| H2B | K5 | 7 | 0.63 ± 0.06*** | 3 | 1.02 ± 0.03 |
| K15 | 7 | 0.55 ± 0.08** | 3 | 0.93 ± 0.04 | |
| K20 | 7 | 0.62 ± 0.06** | 5 | 0.97 ± 0.04 | |
| H4 | Pan Ac | 7 | 0.70 ± 0.08** | 4 | 0.97 ± 0.03 |
| K5 | 5 | 0.64 ± 0.07** | 4 | 1.00 ± 0.08 | |
| K8 | 4 | 0.63 ± 0.06** | 2 | 1.00 ± 0.03 | |
| K12 | 4 | 0.94 ± 0.04 | 3 | 1.04 ± 0.04 | |
| K16 | 4 | 0.77 ± 0.06* | 3 | 1.01 ± 0.13 | |
Acetylation levels are expressed as variations compared to the mean levels of control cells (uninfected or mock transfected/transduced), which were set to 1. n, number of independent sets of neuronal cultures; Pan Ac, pan-acetylated. *, P≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
TABLE 2.
Histone acetylation levels for each lysine in Vero cell lines expressing isolated viral proteinsa
| Histone | Lysine | BDV P |
BDV N |
||
|---|---|---|---|---|---|
| n | Acetylation level (mean ± SEM) | n | Acetylation level (mean ± SEM) | ||
| H2B | K5 | 7 | 0.69 ± 0.07** | 4 | 1.11 ± 0.10 |
| K15 | 7 | 0.66 ± 0.08** | 4 | 1.05 ± 0.10 | |
| K20 | 7 | 0.69 ± 0.06** | 4 | 1.05 ± 0.09 | |
| H4 | Pan Ac | 6 | 0.72 ± 0.61** | 3 | 0.98 ± 0.05 |
| K5 | 6 | 0.69 ± 0.04*** | 3 | 1.13 ± 0.11 | |
| K8 | 6 | 0.80 ± 0.04** | 3 | 1.16 ± 0.06 | |
| K12 | 6 | 0.97 ± 0.02 | 3 | 1.05 ± 0.04 | |
| K16 | 6 | 0.74 ± 0.07* | 3 | 1.09 ± 0.07 | |
n, number of replicates (i.e., number of experiments performed using cultures of transduced Vero cells that were independently grown after the original split during 5 to 20 passages). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
BDV P binding to HMGB-1 is not required for decreased histone acetylation.
One feature of BDV P is its ability to bind chromatin via its interaction with HMGB-1, a nonhistone architectural protein of the chromatin (1, 27). Given the important roles of HMGB-1 in chromatin dynamics (28), we next reasoned that the impact of BDV P on histone acetylation might be dependent on its interaction with HMGB-1. To test this hypothesis, we used a mutant form of BDV P, BDV PE84N, which no longer binds to HMGB-1 due to a mutation in its interaction domain (1). As described above, primary neuronal cultures were transfected with plasmids encoding wild-type BDV P or the BDV PE84N mutant or an empty plasmid, and we next analyzed histone acetylation levels (Fig. 5). Indirect immunofluorescence analysis indicated that the transfection efficiency was around 80% and that the mutant BDV PE84N still localized into the nucleus (Fig. 5A). Using H2B K20 acetylation as a readout, we observed that acetylation was still inhibited in neurons expressing the BDV PE84N mutant (Fig. 5B). Thus, interaction of BDV P with chromatin via its binding to HMGB-1 appears dispensable for the protein to exert its action on histone acetylation.
FIG 5.

BDV P binding to HMGB-1 is dispensable for its impact on histone acetylation. (A) Indirect immunofluorescence analysis of primary neurons 14 days posttransfection. Neurons were stained for DAPI, the wild-type BDV P or PE84N mutant protein (green), and the neuronal marker β-tubulin III (red). Merged images are shown on the right panel. Bar = 50 μm. (B) Quantification of H2B K20 acetylation levels. Acetylation levels (relative to total H2B) were normalized to the values obtained for neurons transfected by an empty plasmid, which were set to 1. Data are expressed as means ± SEM of the results from six independent sets of transfected neuronal cultures. *, P ≤ 0.05; **, P ≤ 0.01 (by paired t test).
BDV P phosphorylation by PKC is essential for its action on histone acetylation.
Similarly to other nonsegmented negative-strand RNA viruses, BDV P is a subunit of the viral polymerase complex and its activity is regulated upon phosphorylation by host kinases. BDV P is preferentially phosphorylated at serine residues 26 and 28 by PKC and, to a lesser extent, at serine residues 70 and 86 by casein kinase II (29, 30). PKC phosphorylation is of particular interest for BDV pathogenesis, since we previously showed that BDV P acts as a PKC decoy substrate and impairs synaptic plasticity by diverting PKC from its natural targets in neurons (21, 22). Interestingly, PKC also regulates histone acetylation levels, notably by controlling the activity of cellular histone acetyl transferases (HATs) (31, 32). Thus, we next asked whether BDV P phosphorylation by PKC was required for its impact on histone acetylation.
To this end, we used the PKC nonphosphorylatable BDV PAA mutant, in which the two serine residues at positions 26 and 28 of BDV P had been replaced by alanine (A) residues (22). We used transfected primary neuronal cultures (Fig. 6A), as well as Vero cell lines stably expressing wild-type or mutant BDV P (Fig. 6B). In addition, we also used Vero cells persistently infected by recombinant BDV (rBDV) expressing wild-type or mutant PAA (Fig. 6C) (22). In each case, BDV PAA subcellular localization was assessed by indirect immunofluorescence and was similar to that of the wild-type protein (Fig. 6, left panels). Histone preparations from these different cells were analyzed for acetylation levels by Western blotting (Fig. 6, right panels for H2B K20 acetylation). In all experimental models, the BDV PAA mutant had lost the ability to inhibit histone acetylation. Moreover, results were similar when measurement of acetylation was done with anti-acetylated H2B K5 and anti-pan-acetylated H4 antibodies (data not shown). Therefore, phosphorylation of BDV P by PKC is mandatory to mediate decreases of histone acetylation.
FIG 6.

Mutation of the PKC phosphorylation sites of BDV P abrogates its action on histone acetylation. This analysis was performed in transfected primary neuronal cultures (control, empty vector) (A), Vero cells stably expressing isolated proteins (control, cell line expressing GFP) (B), and Vero cells persistently infected by a recombinant BDV (rBDV) wild-type strain or strain encoding the mutant PAA protein (control, noninfected cells) (C). (Left panels) Indirect immunofluorescence analysis of BDV wild-type P (Pwt) or PAA expression in the different cells. Cells were stained for DAPI and for wild-type BDV P or PAA mutant protein (green). Merged images are shown on the right. Bar = 50 μm. (Right panels) Comparative analysis of H2B K20 acetylation levels in cells expressing wild-type BDV P or the BDV PAA mutant. In all cases, acetylation levels were normalized to levels obtained for control conditions, which were set to 1 and which are indicated by dashed lines on the graphs. Data are expressed as means ± SEM of the results from at least four independent experiments (i.e., independent sets of cultures of transfected neurons or cultures of Vero cells that were independently grown after the original split during 5 to 20 passages). **, P ≤ 0.01 (by paired t test).
Impaired histone acetylation due to BDV infection results from inhibition of neuronal HAT activities.
Histone acetylation is a dynamic, enzyme-regulated process which results from the balance between the activities of HATs and histone deacetylases (HDACs), two widespread families of enzymes acting in concert (33, 34). To examine whether BDV infection perturbs the equilibrium between these two families, enzymatic assays were performed to evaluate cellular HAT and HDAC activities, using nuclear extracts prepared from noninfected or infected primary neuronal cultures. Levels of HDAC activities were similar in nuclear preparations from infected and noninfected neurons (Fig. 7A). In contrast, HAT activities were significantly decreased in nuclear extracts from BDV-infected neurons, reaching a level of about a 25% decrease (Fig. 7B). The reduced histone acetylation levels consecutive to BDV infection are thus due to inhibition of acetylation rather than to increased deacetylation. We next assessed whether the BDV P expressed alone in primary cortical neurons could also inhibit the cellular HAT enzymatic activities. Nuclear extracts prepared from mock-transfected or BDV P-transfected neurons were used to perform HAT enzymatic assays as described above (Fig. 7C). Isolated expression of wild-type BDV P in neurons led to an inhibition of HAT activities at a level similar to that seen with infection (around 25%). This effect was partly, although not entirely, restored in neurons expressing the nonphosphorylatable BDV PAA mutant (Fig. 7C). Thus, the BDV P protein mediates inhibition of cellular HAT enzymatic activities and its phosphorylation by PKC may be implicated in this inhibition.
FIG 7.

BDV infection and BDV P inhibit neuronal HAT activities. Enzymatic assays for dosing HDAC (A) and HAT (B and C) activities were performed using nuclear extracts prepared from noninfected (NI) or infected (BDV) primary neuronal cultures (A and B) and from neurons transfected with an empty plasmid or plasmids encoding wild-type BDV P or BDV PAA mutant (C). Enzymatic activities measured as described in Materials and Methods are expressed as arbitrary units (A. U.). Data are the means ± SEM of the results from at least four independent sets of neuronal cultures. **, P ≤ 0.01; *, P ≤ 0.05; ns = nonsignificant (by paired t test).
HAT and HDAC inhibitors modulate BDV replication.
Having shown that BDV infection modifies the histone acetylation pattern by inhibiting cellular HAT activities, we next investigated whether such modifications would help to create a more favorable milieu for viral replication. To examine the interplay between cellular acetylation levels and BDV replication, HAT and HDAC activities were pharmacologically modulated and the resulting consequences on BDV replication were followed at different times postinfection. We used sodium butyrate, an inhibitor of HDAC activities (35), and anacardic acid, an inhibitor of HAT activities (36), to treat Vero cells that were concomitantly infected by BDV. We first checked that the two drugs did not affect cell viability at the concentrations used during the experiments (data not shown). We also verified that both inhibitors indeed modulated histone acetylation levels in noninfected cells compared to untreated cells (Fig. 8A). Four days after the onset of treatment, anacardic acid induced a 50% decrease of H2B K20 acetylation compared to control cell results (Fig. 8A), a level similar to that observed during BDV infection (Fig. 2). In contrast, sodium butyrate treatment led to a large (7-fold) increase of H2B K20 acetylation.
FIG 8.

HAT and HDAC inhibitors interfere with BDV replication. Vero cells were infected by BDV at a multiplicity of infection of 0.5, treated with 1 mM sodium butyrate (NaBu) or 10 μM anacardic acid, and followed daily for 4 days. Six independent sets of de novo infections of Vero cells with cell-free BDV were performed. Of these, three were used for flow cytometry analysis and three to prepare protein/RNA extracts for Western blots and RT-qPCR. (A) Impact of a 4-day treatment on H2B K20 acetylation levels using noninfected cells. Acetylation levels (relative to total H2B) measured in treated cells were normalized to the values obtained for nontreated cells, which were set to 1. (B) Flow cytometry-based analysis of BDV spread in cell culture. Cells were stained with either control rabbit IgG antibodies (black line) or rabbit anti-BDV P polyclonal IgG antibodies (gray full histogram). Percentages of BDV P-positive cells and mean fluorescence intensities (MFI) of this population are reported on the top and in the right bottom corner of the graphs, respectively. Results of one representative experiment of three giving similar results are shown. (C) Time course Western blot analysis (from day 1 [d1] to day 4 [d4]) of viral protein levels in cells that were either left nontreated (NT) or treated with NaBu or anacardic acid (AnA). The different antibodies used are indicated on the right side of the Western blots. The blots are from one representative experiment of three that gave similar results. (D) Time course (d1 to d4) RT-qPCR analysis of vRNA levels using cells that were either left nontreated (NT) or treated with NaBu or anacardic acid. Data are as means ± SEM of the results from three independent experiments. P ≤ 0.01 (by two-way analysis of variance [ANOVA]).
To measure the impact of drug treatment on viral replication, Vero cells were harvested 4 days after infection and percentages of infected cells were determined by flow cytometry upon cell staining with an anti-BDV P antibody. The data presented in Fig. 8B, representative of the results of three independent experiments, indicate that, without drug treatment, 75% of cells were positive for BDV P (Fig. 8B, top panel). The percentage of infected cells dropped to 62% upon treatment with anacardic acid, indicating that viral dissemination into the culture was reduced compared to that seen with control cells (Fig. 8B, bottom panel). Moreover, the mean fluorescence intensity (MFI) value, reflecting the amount of synthetized BDV P, was lower in anacardic acid-treated cells than in untreated cells (8 versus 14), suggesting that expression of viral proteins was also decreased following this treatment. In contrast, sodium butyrate-treated cells displayed strongly enhanced viral dissemination, with 94% infected cells at 4 days postinfection (Fig. 8B, middle panel) and very efficient expression of BDV P in infected cells as indicated by the MFI value of 41. In parallel, we also assessed expression of BDV N and P proteins in nontreated or treated cells by Western blot analysis. A progressive reduction of BDV N and P protein expression was observed in anacardic acid-treated cells, in agreement with flow cytometry data. In contrast, BDV protein expression was enhanced in sodium butyrate-treated cells. Finally, we also conducted qRT-PCR assays to measure vRNA production. vRNA levels were measured each day postinfection during 4 days, in nontreated or sodium butyrate- or anacardic acid-treated Vero cells (Fig. 8D). Inhibition of cellular HATs by anacardic acid led to a 50% reduction in the level of BDV vRNA 4 days postinfection, whereas HDAC inhibition by sodium butyrate allowed a 2.5-fold enhancement of vRNA amounts at the same time point. Altogether, these results indicate that replication of BDV is positively correlated with histone acetylation levels, a lower level of viral replication being observed when histone acetylation levels are reduced.
DISCUSSION
The objective of this study was to gain insight into the outcome of the intranuclear persistence of a RNA virus, such as BDV, with respect to host cell chromatin dynamics. Here, we demonstrate that BDV infection leads to a significant decrease of histone acetylation on selected lysine residues. We also show that this decrease is due to inhibition of cellular HAT activities, mediated by the viral phosphoprotein, and that P phosphorylation by PKC plays a role in its effect. Finally, we show that manipulation of host cell epigenetics leads to modulation of viral replication (Fig. 9). These data illustrate a novel and fascinating example of virus-host cell interaction which could be a strategy to favor long-lasting BDV persistence. This strategy, different from classical latency, would rely on “self-restriction” of replication. This would prevent exhaustion of the host cell consecutive to an excessive recruitment of host factors, thus limiting cellular damage.
FIG 9.
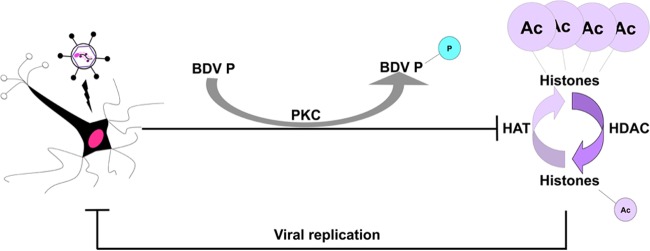
A hypothetical model of BDV P-mediated manipulation of epigenetic signaling. During infection, BDV P decreases cellular HAT activities, partly upon its phosphorylation by PKC, thereby resulting in lowered histone acetylation levels. Such reduced histone acetylation levels would, in turn, lead to reduced viral replication.
This acetylation decrease, which could seem moderate at first, was, however, consistently observed in both primary neurons and established cell lines. The observed levels of reduction (around 30%) are actually close to levels observed in neurons from CREB binding protein (CBP) HAT knockout mice, which display clear memory defects (37, 38). Interestingly, our observation that the exact same lysine residues were affected in all cell types argues for a conserved mechanism of inhibition in neuronal and nonneuronal cells. Also, decreases of histone acetylation were more important in persistently infected Vero cells than in neurons infected for 14 days, suggesting that BDV persistence in the host cell may potentiate the inhibition of histone acetylation. A recent stable isotope labeling for cell culture (SILAC)-based analysis of the impact of BDV infection on the proteome of human oligodendroglial cells also revealed changes in histone lysine acetylation patterns (39). That study reported similar decreased values for certain lysine residues but also many differences, in particular, for H2B K15, for which an increase of acetylation was observed. It would be interesting to determine whether such differences are due to divergences in cell types or viral strains.
In our study, the decrease of histone acetylation due to BDV infection appeared to be restricted to specific H2B and H4 lysine residues. It would be interesting in the future to determine their genome-wide distribution, similarly to what has been reported for the adenovirus E1a protein (40). In this case, chromatin immunoprecipitation (ChIP) sequencing experiments revealed that the global decrease of acetylation on H3 K18 observed during infection correlated with a redistribution of acetylation at the genome scale, with enrichment in the promoter regions of the genes needed for cell replication.
Our results unambiguously demonstrated that BDV P, by inhibiting cellular HAT activities, was responsible for decreased histone acetylation, even when expressed alone, thereby pointing to a new role for this multifunctional protein that lies at the crossroads between BDV and numerous cellular pathways (22, 41, 42). Interestingly, the impact of BDV P on histone acetylation appears to be dissociated from its interaction with chromatin, in particular, from its binding to HMGB-1, as revealed by the use of the BDV PE84N mutant, although we did not formally test whether this was true for all lysine residues affected by BDV and used only H2B K20 as a readout. This fact is consistent with our recent results determined using real-time live-cell imaging, which showed that BDV P is actually highly mobile in vSPOTs, suggesting a labile and transitory interaction with chromatin that may not be sufficient to directly interfere with chromatin dynamics (8).
In contrast, invalidation of the PKC phosphorylation sites on BDV P completely abrogated its impact on histone acetylation and, at least partially, on HAT activities. This may be due to a direct interference by phosphorylated P in HAT activities. Alternatively, BDV P could serve as a PKC decoy substrate, in agreement with our previous findings (21, 22), and divert PKC from one or several HATs. Indeed, it is known that the enzymatic activities of several HATs can be positively or negatively regulated by PKC (31, 43–46). Such a mechanism would differ from what has been described for other viruses such as adenovirus, for which the viral protein E1a directly binds the CBP/p300 HAT (10). Another example is influenza virus, for which it was shown that nonstructural protein-1 possesses a histone-like sequence that serves as mimic substrate for Tip60 HAT (47). Since the restoration of the HAT activities seen with the PAA mutant was not complete, we thus cannot formally exclude the possibility that other functions controlled by P may also play a role.
Characterization of which HAT(s) is actually impacted by BDV P will be challenging. Only a little is known about the posttranslational modifications of most HATs, which are often very large proteins with many potentially modifiable residues (48). Moreover, although PKC can phosphorylate several HATs (44, 45), the resulting impact on their activities, combined with other posttranslational modifications, is poorly understood. The fact that the patterns of impaired histone lysine acetylation were remarkably similar among cell types could provide some clues for a future identification of the HATs, although most of them present broad specificity for their substrates and could thus acetylate virtually the vast majority of histone lysine residues (23).
The role of BDV P phosphorylation in viral replication is still incompletely understood. Phosphorylation mutants of BDV P display increased polymerase activity in minireplicon assays, whereas they induce cell-type-dependent impaired viral spread, leading to the hypothesis that P might play multiple and contrasting roles in the viral cycle (19, 29). Similarly, it has been shown that phosphorylation of Ebola virus VP30 protein plays opposite roles in viral replication and transcription (49). Here, we demonstrate that the PAA mutant protein has lost the capacity to reduce histone acetylation, whereas it has been shown to increase viral RNA polymerase activity (19, 29). Those results are thus in agreement with our finding that BDV replication is favored when the level of cellular histone acetylation is higher.
Finally, epigenetic modifications, in particular, histone acetylation, have now emerged as central regulators of cognitive processes during learning and memory (16, 50). Hence, impaired histone acetylation occurring in the brain of BDV-infected animals may participate in the behavioral perturbations observed in BDV-infected hosts (4). We are currently assessing the impact on cognition of the isolated expression of BDV P in neurons in order to expand the results obtained in transgenic mice expressing BDV P in astrocytes (51). These data may reveal for the first time that an animal virus, or a singled-out viral protein, can perturb cognitive functions by interfering with PKC-dependent and neuronal epigenetic signaling pathways.
ACKNOWLEDGMENTS
We thank the staff of the different facilities of our research center (imaging and flow cytometry platforms) for excellent assistance that was instrumental in the proper realization of this project. We thank D. Trono and P. Charneau for lentiviral vector plasmids. We are grateful to Stéphane Chavanas, Roland Liblau, and Abdelhadi Saoudi for critical readings of the manuscript and insightful comments.
This work was supported by grants from ANR (ANR-10-Blanc-1322 to D.G.-D.) and the Fondation pour la Recherche Médicale (Equipe FRM 2009 to D.G.-D.), a grant from the Université Toulouse III Paul Sabatier Research Council (to C.E.M.), and institutional grants from INSERM and CNRS (to D.G.-D.). E.M.B. was supported by a doctoral fellowship from the Ministère de l'Enseignement Supérieur et de la Recherche.
E.M.B., M.S., D.G.-D., and C.E.M. conceived and designed the experiments. E.M.B., M.S., A.B., A.T., C.F., and C.E.M. performed the experiments. E.M.B., M.S., and C.E.M. analyzed the data. D.G.-D. and C.E.M. wrote the paper.
REFERENCES
- 1.Matsumoto Y, Hayashi Y, Omori H, Honda T, Daito T, Horie M, Ikuta K, Fujino K, Nakamura S, Schneider U, Chase G, Yoshimori T, Schwemmle M, Tomonaga K. 2012. Bornavirus closely associates and segregates with host chromosomes to ensure persistent intranuclear infection. Cell Host Microbe 11:492–503. doi: 10.1016/j.chom.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Lipkin WI, Briese T, Hornig M. 2011. Borna disease virus—fact and fantasy. Virus Res 162:162–172. doi: 10.1016/j.virusres.2011.09.036. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Dunia D, Volmer R, Mayer D, Schwemmle M. 2005. Borna disease virus interference with neuronal plasticity. Virus Res 111:224–234. doi: 10.1016/j.virusres.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Pletnikov MV, Moran TH, Carbone KM. 2002. Borna disease virus infection of the neonatal rat: developmental brain injury model of autism spectrum disorders. Front Biosci 7:d593–d607. doi: 10.2741/pletnik. [DOI] [PubMed] [Google Scholar]
- 5.Carbone KM, Rubin SA, Nishino Y, Pletnikov MV. 2001. Borna disease: virus-induced neurobehavioral disease pathogenesis. Curr Opin Microbiol 4:467–475. doi: 10.1016/S1369-5274(00)00237-X. [DOI] [PubMed] [Google Scholar]
- 6.Horie M, Honda T, Suzuki Y, Kobayashi Y, Daito T, Oshida T, Ikuta K, Jern P, Gojobori T, Coffin JM, Tomonaga K. 2010. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 463:84–87. doi: 10.1038/nature08695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Torre JC. 2002. Molecular biology of Borna disease virus and persistence. Front Biosci 7:d569–d579. doi: 10.2741/torre. [DOI] [PubMed] [Google Scholar]
- 8.Charlier CM, Wu YJ, Allart S, Malnou CE, Schwemmle M, Gonzalez-Dunia D. 2013. Analysis of Borna disease virus trafficking in live infected cells by using a virus encoding a tetracysteine-tagged p protein. J Virol 87:12339–12348. doi: 10.1128/JVI.01127-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suberbielle E, Stella A, Pont F, Monnet C, Mouton E, Lamouroux L, Monsarrat B, Gonzalez-Dunia D. 2008. Proteomic analysis reveals selective impediment of neuronal remodeling upon Borna disease virus infection. J Virol 82:12265–12279. doi: 10.1128/JVI.01615-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. 2008. Adenovirus small e1a alters global patterns of histone modification. Science 321:1084–1085. doi: 10.1126/science.1155544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paschos K, Allday MJ. 2010. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol 18:439–447. doi: 10.1016/j.tim.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels M, Parks RJ, Kristie TM. 2013. Snapshots: chromatin control of viral infection. Virology 435:141–156. doi: 10.1016/j.virol.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gómez-Díaz E, Jorda M, Peinado MA, Rivero A. 2012. Epigenetics of host-pathogen interactions: the road ahead and the road behind. PLoS Pathog 8:e1003007. doi: 10.1371/journal.ppat.1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KY, Huerta SB, Izumiya C, Wang DH, Martinez A, Shevchenko B, Kung HJ, Campbell M, Izumiya Y. 2013. Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J Virol 87:6782–6793. doi: 10.1128/JVI.00011-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J, Yang Y, Turner PC, Jain V, McIntyre LM, Renne R. 2014. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog 10:e1004240. doi: 10.1371/journal.ppat.1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peixoto L, Abel T. 2013. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 38:62–76. doi: 10.1038/npp.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsankova N, Renthal W, Kumar A, Nestler EJ. 2007. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 18.Chevalier G, Suberbielle E, Monnet C, Duplan V, Martin-Blondel G, Farrugia F, Le Masson G, Liblau R, Gonzalez-Dunia D. 2011. Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PloS Pathog 7:e1002393. doi: 10.1371/journal.ppat.1002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmid S, Metz P, Prat CM, Gonzalez-Dunia D, Schwemmle M. 2010. Protein kinase C-dependent phosphorylation of Borna disease virus P protein is required for efficient viral spread. Arch Virol 155:789–793. doi: 10.1007/s00705-010-0645-9. [DOI] [PubMed] [Google Scholar]
- 20.Shechter D, Dormann HL, Allis CD, Hake SB. 2007. Extraction, purification and analysis of histones. Nat Protoc 2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 21.Volmer R, Monnet C, Gonzalez-Dunia D. 2006. Borna disease virus blocks potentiation of synaptic activity through inhibition of protein kinase C signaling. PloS Pathog 2:e19. doi: 10.1371/journal.ppat.0020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prat CM, Schmid S, Farrugia F, Cenac N, Le Masson G, Schwemmle M, Gonzalez-Dunia D. 2009. Mutation of the protein kinase C site in Borna disease virus phosphoprotein abrogates viral interference with neuronal signaling and restores normal synaptic activity. PLoS Pathog 5:e1000425. doi: 10.1371/journal.ppat.1000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Volmer R, Bajramovic JJ, Schneider U, Ufano S, Pochet S, Gonzalez-Dunia D. 2005. Mechanism of the antiviral action of 1-beta-d-arabinofuranosylcytosine on Borna disease virus. J Virol 79:4514–4518. doi: 10.1128/JVI.79.7.4514-4518.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shoya Y, Kobayashi T, Koda T, Ikuta K, Kakinuma M, Kishi M. 1998. Two proline-rich nuclear localization signals in the amino- and carboxyl-terminal regions of the Borna disease virus phosphoprotein. J Virol 72:9755–9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi T, Shoya Y, Koda T, Takashima I, Lai PK, Ikuta K, Kakinuma M, Kishi M. 1998. Nuclear targeting activity associated with the amino terminal region of the Borna disease virus nucleoprotein. Virology 243:188–197. doi: 10.1006/viro.1998.9049. [DOI] [PubMed] [Google Scholar]
- 27.Kamitani W, Shoya Y, Kobayashi T, Watanabe M, Lee BJ, Zhang G, Tomonaga K, Ikuta K. 2001. Borna disease virus phosphoprotein binds a neurite outgrowth factor, amphoterin/HMG-1. J Virol 75:8742–8751. doi: 10.1128/JVI.75.18.8742-8751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bustin M, Reeves R. 1996. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol 54:35–100. doi: 10.1016/S0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 29.Schmid S, Mayer D, Schneider U, Schwemmle M. 2007. Functional characterization of the major and minor phosphorylation sites of the P protein of Borna disease virus. J Virol 81:5497–5507. doi: 10.1128/JVI.02233-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwemmle M, De B, Shi LC, Banerjee A, Lipkin WI. 1997. Borna disease virus P-protein is phosphorylated by protein kinase c-epsilon and casein kinase II. J Biol Chem 272:21818–21823. doi: 10.1074/jbc.272.35.21818. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Weaver IC, Gauthier-Fisher A, Wang H, He L, Yeomans J, Wondisford F, Kaplan DR, Miller FD. 2010. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell 18:114–125. doi: 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 32.Clarke DL, Sutcliffe A, Deacon K, Bradbury D, Corbett L, Knox AJ. 2008. PKCbetaII augments NF-kappaB-dependent transcription at the CCL11 promoter via p300/CBP-associated factor recruitment and histone H4 acetylation. J Immunol 181:3503–3514. doi: 10.4049/jimmunol.181.5.3503. [DOI] [PubMed] [Google Scholar]
- 33.Struhl K. 1998. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 34.Grunstein M. 1997. Histone acetylation in chromatin structure and transcription. Nature 389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 35.Candido EP, Reeves R, Davie JR. 1978. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14:105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 36.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. 2003. Small molecule modulators of histone acetyltransferase p300. J Biol Chem 278:19134–19140. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 37.Barrett RM, Malvaez M, Kramar E, Matheos DP, Arrizon A, Cabrera SM, Lynch G, Greene RW, Wood MA. 2011. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology 36:1545–1556. doi: 10.1038/npp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen G, Zou X, Watanabe H, van Deursen JM, Shen J. 2010. CREB binding protein is required for both short-term and long-term memory formation. J Neurosci 30:13066–13077. doi: 10.1523/JNEUROSCI.2378-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Zhao L, Yang Y, Bode L, Huang H, Liu C, Huang R, Zhang L, Wang X, Zhang L, Liu S, Zhou J, Li X, He T, Cheng Z, Xie P. 2014. Human Borna disease virus infection impacts host proteome and histone lysine acetylation in human oligodendroglia cells. Virology 464–465:196–205. doi: 10.1016/j.virol.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrari R, Su T, Li B, Bonora G, Oberai A, Chan Y, Sasidharan R, Berk AJ, Pellegrini M, Kurdistani SK. 2012. Reorganization of the host epigenome by a viral oncogene. Genome Res 22:1212–1221. doi: 10.1101/gr.132308.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Planz O, Pleschka S, Wolff T. 2009. Borna disease virus: a unique pathogen and its interaction with intracellular signalling pathways. Cell Microbiol 11:872–879. doi: 10.1111/j.1462-5822.2009.01310.x. [DOI] [PubMed] [Google Scholar]
- 42.Peng G, Yan Y, Zhu C, Wang S, Yan X, Lu L, Li W, Hu J, Wei W, Mu Y, Chen Y, Feng Y, Gong R, Wu K, Zhang F, Zhang X, Zhu Y, Wu J. 2008. Borna disease virus P protein affects neural transmission through interactions with gamma-aminobutyric acid receptor-associated protein. J Virol 82:12487–12497. doi: 10.1128/JVI.00877-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, Radovick S, Wondisford FE. 2009. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 137:635–646. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD. 2012. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 11:23–35. doi: 10.1016/j.stem.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 45.Yuan LW, Soh JW, Weinstein IB. 2002. Inhibition of histone acetyltransferase function of p300 by PKCdelta. Biochim Biophys Acta 1592:205–211. doi: 10.1016/S0167-4889(02)00327-0. [DOI] [PubMed] [Google Scholar]
- 46.Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. 2011. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci 31:2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marazzi I, Ho JS, Kim J, Manicassamy B, Dewell S, Albrecht RA, Seibert CW, Schaefer U, Jeffrey KL, Prinjha RK, Lee K, Garcia-Sastre A, Roeder RG, Tarakhovsky A. 2012. Suppression of the antiviral response by an influenza histone mimic. Nature 483:428–433. doi: 10.1038/nature10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Legube G, Trouche D. 2003. Regulating histone acetyltransferases and deacetylases. EMBO Rep 4:944–947. doi: 10.1038/sj.embor.embor941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biedenkopf N, Hartlieb B, Hoenen T, Becker S. 2013. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J Biol Chem 288:11165–11174. doi: 10.1074/jbc.M113.461285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. 2008. Decoding the epigenetic language of neuronal plasticity. Neuron 60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamitani W, Ono E, Yoshino S, Kobayashi T, Taharaguchi S, Lee BJ, Yamashita M, Okamoto M, Taniyama H, Tomonaga K, Ikuta K. 2003. Glial expression of Borna disease virus phosphoprotein induces behavioral and neurological abnormalities in transgenic mice. Proc Natl Acad Sci U S A 100:8969–8974. doi: 10.1073/pnas.1531155100. [DOI] [PMC free article] [PubMed] [Google Scholar]