

ABSTRACT
Coronaviruses (CoVs) and low-pathogenicity influenza A viruses (LP IAVs) depend on target cell proteases to cleave their viral glycoproteins and prime them for virus-cell membrane fusion. Several proteases cluster into tetraspanin-enriched microdomains (TEMs), suggesting that TEMs are preferred virus entry portals. Here we found that several CoV receptors and virus-priming proteases were indeed present in TEMs. Isolated TEMs, when mixed with CoV and LP IAV pseudoparticles, cleaved viral fusion proteins to fusion-primed fragments and potentiated viral transductions. That entering viruses utilize TEMs as a protease source was further confirmed using tetraspanin antibodies and tetraspanin short hairpin RNAs (shRNAs). Tetraspanin antibodies inhibited CoV and LP IAV infections, but their virus-blocking activities were overcome by expressing excess TEM-associated proteases. Similarly, cells with reduced levels of the tetraspanin CD9 resisted CoV pseudoparticle transductions but were made susceptible by overproducing TEM-associated proteases. These findings indicated that antibodies and CD9 depletions interfere with viral proteolytic priming in ways that are overcome by surplus proteases. TEMs appear to be exploited by some CoVs and LP IAVs for appropriate coengagement with cell receptors and proteases.
IMPORTANCE Enveloped viruses use their surface glycoproteins to catalyze membrane fusion, an essential cell entry step. Host cell components prime these viral surface glycoproteins to catalyze membrane fusion at specific times and places during virus cell entry. Among these priming components are proteases, which cleave viral surface glycoproteins, unleashing them to refold in ways that catalyze virus-cell membrane fusions. For some enveloped viruses, these proteases are known to reside on target cell surfaces. This research focuses on coronavirus and influenza A virus cell entry and identifies TEMs as sites of viral proteolysis, thereby defining subcellular locations of virus priming with greater precision. Implications of these findings extend to the use of virus entry antagonists, such as protease inhibitors, which might be most effective when localized to these microdomains.
INTRODUCTION
Enveloped viruses require fusion with host cell membranes to deliver viral genetic material and initiate infection. This process is catalyzed by fusion glycoproteins, which project from virion membranes and operate by bringing virion and host cell membranes into proximity, ultimately stimulating their coalescence. Among the host cell factors required for this membrane fusion are receptors and proteases. Receptors tether viruses to host cell membranes, and proteases cleave fusion protein precursors to form the domains that catalyze membrane melding. This proteolytic step is termed “priming,” and depending on the virus type, it may take place in virus-producing cells (1), in extracellular environments (2), or in virus target cells (3). Notably, several protease inhibitors prevent viral fusion protein cleavages, and as such, are antiviral agents (4).
For many respiratory viruses, including several coronaviruses (CoVs) and low-pathogenicity (LP) influenza A viruses (IAVs), the relevant priming proteases operate in virus target cells. These proteases cleave the virion glycoproteins mediating receptor binding and membrane fusion, namely, the spike (S) proteins for CoVs and the hemagglutinin (HA) proteins for IAVs. These proteases include type II transmembrane serine proteases (TTSPs), a relatively large family of plasma membrane-localized glycoproteins that proteolyze numerous extracellular substrates (5). Specifically, the TTSP member transmembrane protease serine 2 (TMPRSS2) primes CoVs, including severe acute respiratory syndrome coronavirus (SARS-CoV) (6, 7) and Middle East respiratory syndrome coronavirus (MERS-CoV) (8, 9). Without TMPRSS2, target cells are significantly less sensitive to these CoVs (8, 10), but they are not entirely CoV resistant, as other host proteases, i.e., cathepsins, can provide for some priming (11, 12). TMPRSS2 and the TTSP human airway trypsin-like (HAT) protease are also sufficient to prime LP IAV, both in vitro (13) and in vivo (14). As there is no evidence for cathepsin priming of IAVs, cell surface proteases may be strictly required to prime LP IAV (15).
The requirement for TTSP-mediated proteolytic processing of CoV and LP IAV glycoproteins is established, but the subcellular location of these cleavage events is not well understood. If these proteases operate during virus entry, then it is likely that target cell virus receptors would coreside with priming proteases to make virus priming feasible (7). One possible location for this coresidence is within tetraspanin-enriched microdomains (TEMs). TEMs are comprised of homo- and heterotypic assemblies of tetraspanins, so named for their four-transmembrane spanning architectures. In TEMs, the tetraspanins form a locally ordered, largely plasma membrane-embedded platform in which projecting integral membrane adhesion receptors and enzymes are interspersed. As dynamically organized membrane protein complexes, TEMs function to modulate cell adhesion, migration, and differentiation (16, 17) as well as pathogen invasion (18).
There is some modest support for the hypothesis that CoV and LP IAV receptors and proteases are concentrated in TEMs and that priming of these viruses is therefore highly localized. First, TEMs contain CoV receptors dipeptidyl-peptidase 4 (DPP4) (19) and aminopeptidase N (APN) (20) and also contain sialic acids (21), the receptors for IAVs. Second, TEMs contain a variety of integral membrane proteases (22). Third, IAV cell entry is both preferentially observed at CD81 tetraspanin-enriched endosomal locations (23) and reduced by CD81 depletion (24).
Since some CoV receptors interact with tetraspanins and since LP IAV infection was reduced by tetraspanin CD81 knockdown, we used both CoVs and IAVs to address the importance of TEMs in cell entry. We evaluated the effects of tetraspanin antibodies and individual CD9 tetraspanin depletion on virus cell entry. We isolated TEMs and analyzed them for the presence of virus receptors and virus-priming proteases. We used the isolated TEMs to extracellularly prime CoVs and IAVs. Our findings supported the hypothesis that these enveloped viruses enter cells through TEMs because these microdomains harbor both virus receptors and virus-priming proteases.
MATERIALS AND METHODS
Cells.
Human embryonic kidney HEK cells 293T and 293β5 (25) and Madin Darby canine kidney (MDCK) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Thermo Scientific) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals), 1× nonessential amino acids, 10 mM HEPES, 1 mM sodium pyruvate, and 100 U/ml penicillin-streptomycin solution (Thermo Scientific). Delayed brain tumor (DBT) cells were maintained in minimal essential medium (MEM) supplemented with 10% tryptose phosphate broth, 5% FBS, 100 U/ml penicillin-streptomycin, and 2 mM l-glutamine. Cells were maintained in a humidified environment at 37°C and 5% CO2.
Plasmids.
Codon-optimized MERS S-containing sequences for a C-terminal C9 epitope tag were purchased from Genscript and subsequently cloned into pcDNA3.1+ between the EcoRI and NotI restriction sites. pcDNA3.1-229E-S-C9 and pcDNA3.1-hAPN plasmids were provided by Fang Li, University of Minnesota. pcDNA3.1-SARS-S-C9 and pcDNA3.1-ACE2-C9 plasmids were provided by Michael Farzan, Scripps Research Institute. pcDNA3.1-HA5-QH-trypsin site was provided by Lijun Rong, University of Illinois—Chicago and was previously described (26). The pHEF-VSV-G plasmid was obtained from BEI Resources. pcDNA3.1-murine carcinoembryonic antigen-related cell adhesion molecule (mCEACAM) was described previously (27). C-terminal Flag-tagged human DPP4 (hDPP4) plasmid pCMV6-Entry-hDPP4 (catalog no. RC209466) (CMV stands for cytomegalovirus) was purchased from OriGene. pCAGGS-TMPRSS2-FLAG and pCAGGS-TMPRSS2-S441A-FLAG were previously constructed (7). TMPRSS11D (HAT) was obtained from Open Biosystems and cloned into pCAGGS between SacI and XhoI restriction sites. pCMVSport6-human CD9 was purchased from Open Biosystems. CD9 and scramble control short hairpin RNA (shRNA) constructs flanked by the U6 promoter and a RNA polymerase III stop sequence were engineered into the pUC57 vector by Genscript. The pNL4.3-HIVluc (luc stands for luciferase) plasmid was provided by the NIH AIDS Research and Reference library. pΔEGFP-S15-mCherry (EGFP stands for enhanced green fluorescent protein) (28) was provided by Edward Campbell, Loyola University Chicago. pEGFP was provided by Chris Wiethoff, Loyola University Chicago.
Antibodies.
Monoclonal mouse antibodies against CD9 (clone M-L13), CD63 (clone H5C6), and CD81 (clone JS-81) were obtained from BD Pharmingen. Monoclonal mouse antibody against transferrin receptor (clone H68.4) was obtained from Zymed Laboratories. Rabbit anti-Flag and horseradish peroxidase (HRP)-conjugated anti-β-actin antibodies were obtained from Sigma-Aldrich. Mouse antirhodopsin (C9) antibodies were obtained from Millipore. Rabbit anti-CD13 (APN) antibodies were obtained from Abcam. Mouse anticalnexin antibodies were obtained from Cell Signaling. A mouse monoclonal antibody to IAV H1 hemagglutinin (HA) (clone PY102) was provided by Balaji Manicassamy, University of Chicago. Secondary antibodies were purchased from Invitrogen and include Alexa Fluor 488-conjugated goat anti-rabbit, Alexa Fluor 488-conjugated goat anti-mouse, and Alexa Fluor 568-conjugated goat anti-mouse antibodies. Donkey anti-goat, goat anti-mouse, and HRP-conjugated goat anti-rabbit antibodies were purchased from Thermo Scientific.
Viruses.
Influenza A/Puerto Rico/8/1934 H1N1 (PR8) containing a Gaussia luciferase (Gluc) reporter gene (29) was provided by Peter Palese, Mount Sinai School of Medicine. PR8-Gluc stocks were produced using a standard protocol (30). Briefly, MDCK cells were inoculated with PR8-Gluc and incubated in DMEM supplemented with 0.2% bovine serum albumin (BSA). At 30 h postinfection (hpi), the progeny were collected, treated with tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma), and used to infect fresh MDCK cells at a multiplicity of infection (MOI) of 1. Supernatants were then collected, clarified by centrifugation, aliquoted, and stored at −80°C. Two strains of recombinant mouse hepatitis viruses (MHV), MHV-A59 and MHV-JHM, each containing a firefly luciferase (Fluc) reporter gene, were produced, and the titers of virus on DBT cells were determined as described previously (31).
Pseudoviruses.
Vesicular stomatitis virus (VSV)-based pseudovirus particles (pp) were produced by the method of Whitt (32). Briefly, 293T cells were transfected with plasmids encoding viral glycoproteins. Two days later, cells were inoculated for 2 h with VSVΔG-luc (32), rinsed extensively, and incubated for 1 day. Supernatants were collected, centrifuged at 800 × g for 10 min to remove cellular debris, and stored in aliquots at −80°C. HIV-based pp were produced as previously described (28). Briefly, 293T cells were cotransfected with pNL4.3-HIV-luc and pcDNA plasmids encoding the appropriate glycoproteins, and where indicated, pΔEGFP-S15-mCherry was also cotransfected. After 2 days, supernatants were collected, centrifuged at 1,000 × g at 4°C for 10 min to remove cell debris, and stored in aliquots at −80°C.
CD9 knockdowns.
Two shRNA constructs were used, one designed to target CD9 and the other a scrambled control. 293β5 cells were cotransfected with 0.05 μg/106 cells of pCDNA3.1-hDPP4 along with 1 μg/106 cells of the indicated shRNA plasmid or a pUC57 construct lacking the shRNA. Stable transfectants were selected in DMEM supplemented with 10% FBS (DMEM–10% FBS) containing 1.2 mg/ml of G418 (Thermo Scientific) for neomycin resistance on the DPP4 plasmid. Cells underwent selection for at least 7 days before being used in assays.
Infection in the presence of tetraspanin antibodies.
DBT cells or 293β5 cells were transfected with appropriate plasmids encoding viral receptors or proteases, divided into 96-well cluster plates, and incubated for 30 min at 37°C with the antibodies indicated in the figures at 0.12 μg/μl (∼107 antibodies/cell). The viruses indicated in the figures were then added for 2 h at 37°C, and then the cells were rinsed, incubated at 37°C for 6 h (MHV and PR8), 16 h (VSV), or 48 h (HIV). For PR8, cells were not lysed, and media were analyzed for secreted Gluc. For the other viruses, cells were lysed in passive lysis buffer (Promega). Luciferase levels in media or lysates were measured after the addition of either Fluc substrate (Promega) or Gluc substrate (New England BioLabs) using a Veritas microplate luminometer (Turner BioSystems).
Flow cytometry.
To measure antibody binding, 293β5 cells were lifted with Accutase (Millipore), pelleted, and resuspended to 106 cells/ml in phosphate-buffered saline (PBS) supplemented with 2% FBS (PBS–2% FBS) containing the antibodies indicated in the figures at 0.12 μg/μl. After 30 min at 37°C, cells were rinsed three times by pelleting and resuspension in PBS–2% FBS and then incubated for 30 min at 4°C with Alexa Fluor 488-conjugated donkey anti-mouse IgG. After sequential rinsing, cell fluorescence was detected using a BD C6 Accuri flow cytometer. To measure HIV pp binding, 293β5 cells, transfected with empty pCMV6 or with pCMV6-Entry-hDPP4, were suspended in PBS–2% FBS. The cells were divided, and aliquots were incubated for 30 min at 37°C with tetraspanin antibodies at 0.12 μg/μl. The cells were chilled and then incubated for 1 h on ice with HIV-mCherry-MERS S. The cells were rinsed three times by pelleting and resuspension, and mCherry fluorescence was detected using a BD C6 flow cytometer or a BD LSRFortessa flow cytometer as indicated. All flow cytometric data were analyzed using FlowJo software.
Fluorescence-activated cell sorting (FACS).
DBT cells were transfected with 0.5 μg of pEGFP, and a total of 4 μg of a pCAGGS empty vector or TMPRSS2 plasmid per 106 cells. Twenty-four hours after transfection, the cells were lifted with trypsin, washed three times with cold PBS supplemented with 2% FBS, and sorted using a BD FACSAria cell sorter. Live, green fluorescent protein (GFP)-positive (GFP+) cells were plated and incubated at 37°C overnight before antibody blockade experiments were performed as described above.
Immunofluorescence microscopy.
293β5 cells were transfected with the indicated plasmid DNAs, incubated for 2 days, and then cooled to room temperature (RT). Antibodies and HIV-mCherry pp were added, and the cells were incubated for 30 min at RT and then for 10 min at 37°C and returned to RT. Alexa Fluor-conjugated secondary antibodies were applied for 10 min at RT, along with Hoechst 33258 (Molecular Probes). The cells were rinsed with PBS, fixed with 3.7% paraformaldehyde in 100 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) buffer (pH 6.8), mounted using PermaMount, and imaged with a DeltaVision microscope (Applied Precision) equipped with a digital camera (CoolSNAP HQ; Photometrics), using a 1.4-numerical-aperture 60× lens objective. Images were deconvolved with SoftWoRx deconvolution software (Applied Precision). Colocalization was measured and quantified using Imaris version 6.3.1 (Bitplane Scientific Solutions).
Isolation of tetraspanin-enriched microdomains (TEMs).
Adherent 293β5 cells (∼105/cm2) were rinsed with ice-cold PBS, incubated for 30 min at 4°C with 1 mg/ml EZ-Link Sulfo-NHS-LC-Biotin (Pierce) in PBS, rinsed, and then incubated for 20 min at 4°C with 100 mM glycine in PBS. The cells were rinsed with PBS, then incubated for 20 min at 4°C in morpholineethanesulfonic acid (MES) buffer (25 mM MES [pH 6.0], 125 mM NaCl, 1 mM CaCl2, 1 mM MgCl2) containing 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) detergent (catalog no. 220201; Calbiochem) or 1% Triton X-100 detergent (Sigma). Cell lysates (107/ml) were removed from plates and emulsified by 20 cycles of extrusion through 27-gauge needles. The nuclei were removed by centrifugation, and the lysates were mixed with equal volumes of 80% (wt/vol) sucrose in MES buffer, placed into Beckman SW60 tubes, and overlaid with 3 ml of 30% (wt/vol) sucrose and then with 0.5 ml of 5% (wt/vol) sucrose, both in MES buffer. Samples were centrifuged with a Beckman SW60 rotor at 370,000 × g for 18 h at 4°C. Fractions were collected from air-gradient interfaces. Biotinylated proteins in gradient fractions were bound to streptavidin agarose beads (Pierce). Nonreducing dot blotting and Western blotting procedures were used to identify the distributions of proteins in gradient fractions, as described previously (33).
Virus priming assays.
PR8 or MERS pp were incubated at 37°C for 30 min with equal volumes of low-density (LD) or high-density (HD) sucrose gradient fractions or with 2.5 U trypsin/reaction (in 50 μl total) (Sigma). Treated PR8 and MERS pp were divided, and proteins in one set of aliquots were precipitated with trichloroacetic acid and analyzed by Western blotting. The other set of aliquots was used to transduce 293β5 cells. Cells transduced with MERS pp were pretreated for 1 h with 10 μM leupeptin (Sigma) or without leupeptin, inoculated for 2 h, rinsed, and incubated without leupeptin for 18 h. The cells were then lysed, and luciferase levels were measured. Cells infected with PR8 viruses were infected at an MOI of 1, rinsed after 2 h, and incubated for an additional 6 h. Media were collected, and Gluc levels were measured.
RESULTS
Tetraspanin antibodies inhibit coronavirus and influenza A virus infections.
Tetraspanins facilitate the entry of many viruses, including hepatitis C virus (34), human papillomavirus (35), and IAV (23). To determine whether entering CoVs also utilize tetraspanins, we evaluated the effects of tetraspanin antibodies on infection by mouse hepatitis virus (MHV) strains A59 and JHM. To this end, murine DBT cells were incubated for 30 min with mouse monoclonal antibodies against the CD9, CD63, and CD81 tetraspanins or with an equimolar mixture of the three (anti-Tspan). A monoclonal antibody against transferrin receptor (anti-TfR) was used as an isotype-matched control for general cell coating by antibodies. The cells were inoculated with luciferase-expressing recombinant MHV-A59 or MHV-JHM for 2 h, and then unadsorbed viruses and antibodies were rinsed away. As measured by luciferase levels at 8 hpi, the two viruses were significantly inhibited by all three tetraspanin antibodies, with the antibody combination (anti-Tspan) inhibiting the A59 and JHM strains by ∼50% and ∼90%, respectively (Fig. 1A). The TfR antibodies did not block the viruses. None of the antibodies interfered with transduction by VSV pp bearing VSV G proteins (VSV pp), indicating that tetraspanin antibodies do not generally suppress virus entry or reporter gene expression.
FIG 1.

Effects of tetraspanin antibodies on MHV and IAV infection. (A) DBT cells were treated with monoclonal antibodies to CD9 (αCD9), CD63, and CD81 or an equimolar mixture of the three tetraspanin antibodies (αTspan). After 30 min at 37°C, cells were infected with recombinant MHV-A59 or MHV-JHM viruses containing a firefly luciferase (Fluc) reporter gene. Following a 2-h entry period, unadsorbed antibody and virus were removed. At 8 hpi, infection levels were measured by quantifying Fluc reporter gene products and were normalized to the untreated controls. A VSV-G pseudovirus particle (pp) reporter virus was also used. Mouse immunoglobulin G and monoclonal antibody against transferrin receptor (TfR) were used as controls for antibody subtype and irrelevant cell binding, respectively. Results are representative of three independent experiments. Values that are significantly different (P < 0.05) from the value for no antibody (No Ab) are indicated by an asterisk. (B) Cells transfected with an empty vector (EV), 0.1 μg TMPRSS2/106 cells, or increasing amounts of HAT plasmid were infected with a PR8 influenza virus containing a Gaussia luciferase (Gluc) reporter gene. Unadsorbed virus was removed after a 2-h entry period. At 8 hpi, the medium was removed from the cells and analyzed for Gluc expression. (C) 293β5 cells transfected with 0.001 μg HAT/106 cells were treated with antibodies as described above for panel A. This experiment also included a nonspecific mouse IgG control antibody (M Ig). The medium was collected and analyzed for secreted Gluc 8 hpi. *, P < 0.05 compared to the no-antibody value.
While previous studies have found that CD81 knockdown inhibits IAV entry (23), it is not known whether antibodies to CD81 or other tetraspanins also inhibit IAV infection. Thus, we determined whether the tetraspanin antibodies inhibit influenza A/Puerto Rico/8/1934 (H1N1), also known as PR8 IAV. For ease of analysis, we used PR8 containing a Gaussia luciferase (Gluc) reporter gene (29). Many host cells are resistant to PR8 infection, because they do not express proteases that prime viral HA proteins (13). Therefore, we transfected 293β5 cells with plasmids encoding HAT or TMPRSS2 and then infected the cells with PR8 1 day later. HAT will make target cells susceptible to LP IAV, while TMPRSS2 will not (15). By measuring Gluc accumulation in culture media, we determined that transfecting cells with 0.001 μg/well of HAT was sufficient to render cells susceptible to PR8 infection and that increasing HAT transfection generally led to increased infection (Fig. 1B). Knowing this, we examined whether tetraspanin antibodies might block PR8 infection of the HAT-expressing cells. Indeed PR8 infection was significantly inhibited by all three tetraspanin antibodies, with the antibody combination (anti-Tspan) effecting ∼50% blockade. The tetraspanin antibodies did not inhibit VSV pp transductions (Fig. 1C).
Tetraspanin antibodies inhibit coronaviruses at the cell entry stage.
To limit our analyses of tetraspanin antibodies to single-cycle infections, we produced several VSV-based pp, each encoding Fluc reporter genes and each containing the S proteins of a relevant human CoV. The virus preparations were designated according to their S proteins (MERS pp, SARS pp, and 229E pp). Their transduction into cells was taken to reflect features of the authentic MERS-CoV, SARS-CoV, and 229E CoV entry processes (7, 33). Transduction-susceptible target cells were established by transfecting 293β5 cells with genes encoding virus receptors: human APN (hAPN) for human coronavirus 229E (HCoV-229E) (36), hDPP4 for MERS-CoV (37), and human angiotensin-converting enzyme 2 (hACE2) for SARS-CoV (38). These target cells were then inoculated with the pp preparations in the absence or presence of tetraspanin antibodies, as was done with authentic viruses. After 1-h inoculation periods, unadsorbed pp and antibodies were removed, and transduction levels were measured the next day by quantifying Fluc gene expression.
The tetraspanin antibodies impaired transduction by all three CoV pp preparations, with MERS pp and SARS pp most notably inhibited (Fig. 2A). TfR antibodies did not affect transductions. Using MERS pp, we further determined that tetraspanin antibodies were inhibitory only at the pp entry stage, with no effects on transduction when added 30 min following virus inoculation (Fig. 2B). Furthermore, inhibition by the tetraspanin antibodies was not related to the viral particle core, as HIV-based MERS pp were blocked equally to VSV-based MERS pp (data not shown). All of these findings were consistent with inhibition of virus entry at TEMs.
FIG 2.

Effects of tetraspanin antibodies on CoV pp cell entry. (A) 293β5 cells overexpressing appropriate receptors (APN for 229E CoV, DPP4 for MERS-CoV, and ACE2 for SARS-CoV) were treated with monoclonal antibodies to CD9, CD63, or CD81 or an equimolar mixture of the three tetraspanin antibodies (αTspan). After 30 min at 37°C, the indicated VSV pp bearing the spike proteins from 229E, MERS, or SARS virus were inoculated for 2 h at 37°C, and unadsorbed virus and antibody were then removed from cells. Transduction levels were measured by quantifying Fluc reporter gene products and normalized to the untreated controls. Mouse IgG (M Ig) and a monoclonal antibody against transferrin receptor (TfR) were used as controls for antibody subtype and irrelevant cell binding, respectively. Results are representative of three independent experiments. Values that are significantly different (P < 0.05) from the control value with no antibody (No Ab) are indicated by an asterisk. (B) 293β5 cells were incubated without antibodies, with control anti-transferrin receptor antibodies (αTfR), or with a mixture of antitetraspanin antibodies (αTspan) for 30-min periods immediately before (−30) or after (+30) a 60-min VSV-MERS S pp inoculation period. Transduction levels were measured by quantifying luciferase accumulation and normalized to the controls in which antibodies were not applied. *, P < 0.05 compared to the no-antibody value.
Tetraspanin antibodies do not block virus-cell binding.
To determine whether the antibodies used for virus blockades bound similarly to target cells, we subjected antibody-coated cells to flow cytometry. The inert TfR antibodies and inhibitory tetraspanin antibodies bound similarly (Fig. 3A), indicating that virus blockades do not arise simply from high levels of antibodies on cells. Next, to determine whether antibodies on cells interfere with virus-cell binding, we used fluorescently labeled, HIV-based MERS pp, which we manufactured according to prescribed methods (28). mCherry MERS pp were adsorbed at 4°C to 293β5 cells overexpressing hDPP4 receptors, either in the absence or presence of tetraspanin antibodies. Subsequent flow cytometric analyses revealed that the fluorescent HIV-based MERS pp bound abundantly to the ∼35% of cells that were overexpressing hDPP4 (Fig. 3B, left). Tetraspanin antibodies did not reduce this percentage of cells bound by MERS pp (Fig. 3B, right). Tetraspanin antibodies did slightly reduce the fluorescence intensities of the MERS pp-bound cells (Fig. 3B, compare left red and right gold lines). We conclude that the tetraspanin antibodies only modestly interfered with virus binding to cells.
FIG 3.

Immunofluorescence analysis of pp binding to cells in the presence of tetraspanin antibodies. (A) Flow cytometric analysis of the binding efficiencies of the antibodies used in tetraspanin blockade experiments. 293β5 cells were incubated without antibodies (No Abs) or with the indicated antibodies. Following a 30-min incubation, the cells were washed and incubated with an Alexa Fluor 488 (AF488)-conjugated anti-mouse secondary antibody. Flow cytometry was performed to detect the intensity of AF488 of treated cells. (B) 293β5 cells overexpressing DPP4 (+DPP4) or an empty vector (−DPP4) were incubated with HIV-mCherry MERS-CoV pp for 1 h at 4°C. Following incubation, cells were washed of unbound virus and analyzed by flow cytometry to detect the level of mCherry. The percentage of mCherry-positive cells is indicated above the gate (left panel). DPP4-overexpressing cells were treated with anti-Tspan antibodies. After 30 min at 37°C, HIV-mCherry-MERS-CoV pp were inoculated for 120 min at 4°C. Following washing of unadsorbed virus and antibody, flow cytometry was performed to detect bound HIV-based pp (right panel). (C) HIV-mCherry pp without protruding glycoproteins (Bald) or with VSV G, IAV HA, or MERS-CoV S, were mixed with anti-CD81 antibodies and inoculated onto DPP4-overexpressing cells for 30 min at 4°C. After a 10-min, 37°C patching period, cells were fixed and analyzed by confocal microscopy to determine the locations of the pp (red) and CD81 (green). Colocalization of CD81 and HIV-positive puncta were quantified using Imaris software. Data were plotted as the percentage of HIV-based pp that were localized to CD81.
These data strongly suggested that tetraspanin antibodies interfere with virus entry after virus-receptor binding. To explain how the antibodies block viruses, we posited that viruses might associate with TEMs after binding to cells and that the tetraspanin antibodies disrupt some TEM-associated process(es) facilitating virus entry. To determine whether the mCherry MERS pp do indeed appear at TEMs during their cell entry, we incubated chilled 293β5 cells with the fluorescent pp along with tetraspanin CD81 antibodies and then shifted the cells to 37°C for 10 min to permit “patching,” i.e., antibody-mediated tetraspanin cross-linking into larger TEM structures (39). Quantitative confocal microscopy revealed that CD81 colocalized with ∼20% of MERS pp but with only ∼10% of VSV pp (Fig. 3C, right). Absence of “bald,” i.e., viral glycoprotein-free fluorescent pp binding (Fig. 3C, left) confirmed viral glycoprotein-dependent interactions with cells. Similar, but less compelling copatching of IAV pp and CD81 were also observed in these experiments (Fig. 3C). These data indicate that, shortly after binding to cells, some MERS and IAV pp will be present at TEM locations.
Proteases eliminate the antiviral activities of tetraspanin antibodies.
TEMs are known to contain a variety of cell surface proteases (22), among which may be one or several CoV- and IAV-priming proteases (7, 8, 13, 14). This led us to hypothesize that tetraspanin antibodies interfere with TEM-associated proteolytic priming, possibly by preventing TEM proteases from accessing receptor-bound viruses. By this hypothesis, overexpression of proteases, or addition of proteases to media, should provide viral access to priming and thereby reduce or eliminate the antiviral activities of the tetraspanin antibodies. Thus, we overexpressed priming proteases in target cells and performed antibody blockade experiments. Using the frequently cited CoV-priming protease TMPRSS2 (8, 40), we found that MHV infections of DBT cells were augmented ∼20-fold by TMPRSS2 overexpression, indicating that this protease is utilized by MHV and is limiting in the DBT cell context. We supplied DBT cells with graded doses of TMPRSS2-encoding plasmids, along with small constant amounts of a GFP reporter plasmid. Following expression, GFP+ cells were isolated by FACS and then used in antibody blockade experiments as described above. The results indicated that tetraspanin antibodies blocked MHV infection of normal DBT cells (Fig. 1A and 4A), but not of DBT cells expressing TMPRSS2 (Fig. 4A). Of note, 106 cells transfected with 0.004 μg of TMPRSS2 plasmid contained TMPRSS2 protein levels that were far below our Western blot and immunofluorescence assay detection limits, making it clear that even small amounts of priming proteases nullify the antiviral effects of tetraspanin antibodies (Fig. 4A).
FIG 4.
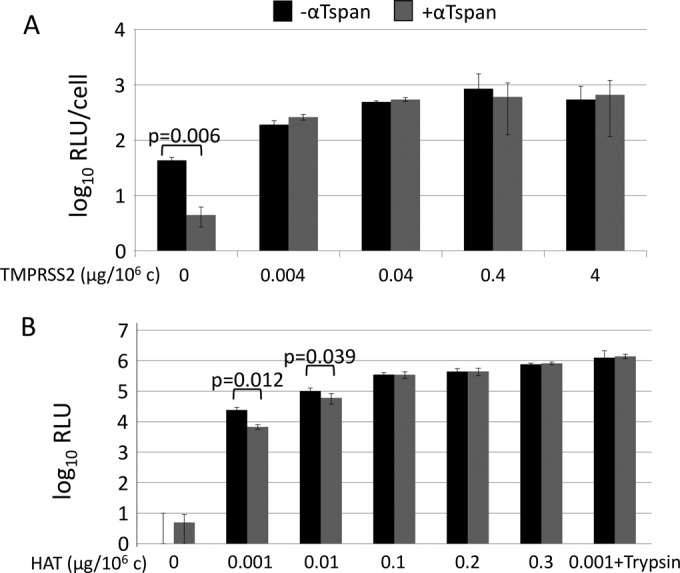
Effects of proteases on tetraspanin antibody-blocked infections. (A) DBT cells were transfected with increasing amounts of TMPRSS2 plasmid and a small amount of GFP reporter. Following isolation of transfected cells by FACS, cells were treated with either a mixture of tetraspanin antibodies (+αTspan) or media (−αTspan). After 30 min, cells were infected with MHV-JHM. At 2 hpi, the cells were washed to removed unadsorbed virus and antibody. At 8 hpi, the cells were lysed and analyzed for Fluc reporter expression. Data were graphed as Fluc relative light units (RLU) per cell. (B) 293β5 cells were transfected with the indicated amounts of HAT before being exposed to the same Tspan antibody blockade described above for panel A. The cells were then infected with PR8-Gluc for 2 h and washed, and secreted Gluc was measured at 8 hpi. PR8 viruses pretreated with trypsin (+Trypsin) were also used to infect cells transfected with 0.001 μg HAT/106 cells (rightmost columns).
In similar experiments, PR8 and its priming protease HAT were evaluated in entry assays. Here, 293β5 cells were transfected with graded doses of HAT-encoding plasmids and then infected with PR8, either in the absence or presence of tetraspanin antibodies. The results indicated that HAT bypassed the antibody blockades in a dose-dependent manner (Fig. 4B). Furthermore, trypsin-pretreated viruses also bypassed the antibody blockades (Fig. 4B, rightmost columns). Therefore, the hypothesis that proteases mitigate the antiviral activities of tetraspanin antibodies applies to TMPRSS2, HAT, and trypsin proteases and to MHV and PR8 viruses.
CD9 knockdown inhibits MERS pp transductions.
Antibodies binding to CD9, CD81, and CD63 inhibited CoV (MHV) and IAV (PR8) entry (Fig. 1). Of these three, CD9 stood out as a candidate for further evaluation, in part because of reports that sperm-egg fusion requires CD9 (41) and that HIV-induced fusion requires endogenous CD9 levels (42). Therefore, we evaluated how different CD9 levels might affect susceptibility to the VSV-based MERS pp transductions. We produced 293β5 cells stably expressing hDPP4 and a shRNA against CD9. Western blotting confirmed that these cells expressed hDPP4 but expressed only 9% of the CD9 in control cells (Fig. 5A). Notably, the reduced CD9 did not affect any change in the levels of DPP4, unlike that observed by Okamato et al. (43). In comparison with the vector control and scrambled shRNA control cells, the CD9 knockdown (KD) cells were 77% less susceptible to MERS pp transduction. Overexpression of CD9 restored transduction susceptibility to the CD9 KD cells (Fig. 5A). The under- and overexpression of CD9 had no effects on VSV G-mediated transductions (data not shown), indicating a MERS S-specific effect of the CD9 knockdown. All of these results demonstrated that CD9 supports MERS at the level of S-protein-mediated virus entry.
FIG 5.

Effect of CD9 knockdown on MERS pp entry. (A) 293β5 cells stably expressing DPP4 and either an empty shRNA vector (empty vector [EV]), a scrambled shRNA, or an shRNA specific for CD9 were transfected with an empty vector (−CD9 cDNA) or a vector containing CD9 cDNA (+CD9 cDNA). These cells were transduced with MERS-CoV pp, and transduction levels were measured by Fluc reporter gene expression. Cell lysates were analyzed by Western blotting for CD9 and β-actin (below graph). (B) Cells stably expressing an empty vector, a scrambled shRNA, or a CD9 shRNA were transfected with increasing amounts of TMPRSS2 before transduction with MERS-CoV pp. Transduction levels were measured by Fluc reporter gene expression.
Proteases render CD9 knockdown cells susceptible to MERS pp transductions.
To determine whether CD9 knockdown inhibits MERS pp entry at the proteolytic priming stage of entry, we forced protease overexpression by introducing graded doses of TMPRSS2-encoding plasmids, then transduced cells with MERS pp, and quantified transduction levels with Fluc measurements. The results of these experiments indicated that excess TMPRSS2 restored transduction sensitivity to CD9 knockdown cells, up to the levels of the CD9-replete cells (Fig. 5B). Specifically, 0.3 μg TMPRSS2 plasmid/106 cells increased MERS pp transductions by 4 (±1)-fold in CD9-positive cells and 11 (±3)-fold in CD9 KD cells (Fig. 5B). These CD9-specific differences were statistically significant (P < 0.0001). Thus, the impediments to virus entry brought about by CD9 knockdown were overcome by excess proteases.
TEMs contain coronavirus receptors and priming proteases.
Our findings fit with the hypothesis that infecting viruses encounter cell receptors and priming proteases at TEM locations. Antibody binding to TEMs, or omission of particular tetraspanins, interferes with these encounters, reducing infection. We used a biochemical approach to determine whether cell receptors and priming proteases are indeed within TEMs. Surface-biotinylated 293β5 cells were lysed in buffers containing CHAPS, a zwitterionic detergent that emulsifies cell membranes without disrupting primary or secondary TEM interactions (44). After sucrose density gradient fractionation, CHAPS-soluble proteins remained near the bottom of sucrose gradients, designated the high-density (HD) regions, while CHAPS-insoluble protein-lipid complexes floated to the top, low-density (LD) regions. Dot blotting revealed that ∼20% of biotinylated (plasma membrane) proteins were in the LD region (Fig. 6A, top left). Streptavidin pulldown of the isolated HD and LD fractions revealed that all of the detectable cell surface CD9, CD63, and CD81 were present in the LD material (Fig. 6A, bottom left), indicating that the LD fraction includes the TEMs. Notably, cells lysed by Triton X-100 (TX-100), a detergent known to solubilize TEMs, also generated an LD fraction that comprised ∼20% of plasma membrane proteins (Fig. 6A, top right) but was devoid of any cell surface tetraspanins (Fig. 6A, bottom right). Thus, we designated the CHAPS LD subcellular fractions as “TEMs” and the TX-100 LD fractions as “lipid rafts.”
FIG 6.

Isolation and analysis of tetraspanin-enriched microdomain (TEM) fractions. (A) The surfaces of 293β5 cells were biotinylated before lysis with CHAPS or TX-100. (Top) Following differential centrifugation, 10 fractions were collected from each tube and analyzed for total cell surface and biotinylated proteins. (Bottom) Following collection of the HD and LD fractions, streptavidin pulldown was performed, and each fraction was analyzed for cell surface CD9, CD63, and CD81. (B) 293β5 cells overexpressing epitope-tagged CoV receptors ACE2, APN, CEACAM, and DPP4 or Flag-tagged TTSP TMPRSS2, TMPRSS2-S441A, or HAT. Transfected cells were subjected to CHAPS lysis and density gradient centrifugation as described above for panel A. Western blotting was used to determine separation of the indicated proteins into HD and LD fractions. β-Actin and calnexin were used as controls for complete cell lysis and proteins not present in CHAPS LD fractions.
To determine whether CoV receptors and priming proteases partition into TEMs, it was necessary that 293β5 cells were first transfected to overexpress ACE2, APN, CEACAM (the receptor for MHV [45]), or DPP4. The transfected cells were then lysed in CHAPS, and TEMs were isolated by two sequential cycles of flotation on sucrose gradients. Western immunoblotting revealed that ∼90% of ACE2, APN, and CEACAM and ∼50% of the DPP4 were partitioned into TEM fractions (Fig. 6B). Cells overexpressing the virus-priming proteases TMPRSS2 and HAT were similarly fractionated, and roughly 50% of these proteases were found in the TEM fractions (Fig. 6B). Mature TMPRSS2 and HAT exist as linked cleavage products, and some of the TMPRSS2 and HAT that was not found in TEM (LD) fractions may come from shedding of the Flag-tagged TMPR ectodomain fragments into soluble (HD) material. An uncleavable TMPRSS2S441A mutant more prominently partitioned into the TEM (LD) fractions. This uncleaved integral-membrane TMPRSS2S441A zymogen may more accurately represent the predominant TEM localization of TMPRs. Some β-actin (<10%) partitioned with TEMs, consistent with known TEM-cytoskeleton interactions (46). Calnexin, a transmembrane protein abundant in the endoplasmic reticulum (47), was excluded from the TEMs, indicating complete cell solubilization by CHAPS detergent. As the TEM fractions contained only ∼20% of the total plasma membrane proteins, these results indicated that the CoV receptors and priming proteases were at least 5- to 50-fold more abundant in TEMs than elsewhere on cell surfaces. While this is significant microdomain localization, we speculate that these results may be underestimating the extent of receptors and proteases naturally within TEMs, as there are upper limits to accommodating overexpressed receptors and proteases in the TEMs.
TEM localization of one CoV receptor (DPP4) and one priming protease (TMPRSS2) was validated by immunofluorescence microscopy. DPP4 and TMPRSS2 were both found near or within CD81-enriched cell surface puncta (Fig. 7). Similar partitioning of CD81 with a catalytically inactive mutant TMPRSS2S441A was also observed (Fig. 7), indicating that enzymatic activity has no effect on subcellular localization. Thus, there are recognizable proportions of CoV receptors and priming proteases residing within TEMs.
FIG 7.

Localization of MERS-CoV entry factors DPP4 and TMPRSS2 in relation to tetraspanin CD81. 293β5 cells were transfected with plasmids encoding the indicated Flag-tagged proteins. Twenty-four hours later, live cells were coincubated with anti-Flag and anti-CD81 antibodies, with a 10-min incubation at 37°C to induce patching. Fluorescent secondary antibodies were applied to mark the positions of Flag-tagged proteins and CD81, and Hoechst 33258 (blue) marks the positions of cell nuclei. Images show 0.5-μm-thick confocal slices through the middle section of cells.
TEMs prime coronaviruses and influenza A viruses for entry.
To determine whether the isolated TEMs have virus-priming activities, we mixed them with MERS pp, inoculated the mixtures onto susceptible target cells, and then measured transduction efficiencies. To ensure that the transduction measurements reflected proteolytic priming by the TEMs, and not by endogenous target cell proteases, we suppressed target 293β5 cell priming proteases with leupeptin, a broad-spectrum protease inhibitor. Leupeptin-treated 293β5 cells were profoundly resistant to MERS pp transduction (Fig. 8A, top), indicative of requirements for the host proteases. However, MERS pp that were exposed to TEM fractions transduced the leupeptin-treated cells (Fig. 8A, top), indicating priming. Of note, bypass of leupeptin was pronounced when MERS pp were exposed to trypsin or to TEMs containing overexpressed TMPRSS2 but did not reach the levels observed in the absence of leupeptin. In addition, substantial bypass of leupeptin was not achieved by CHAPS HD fractions (data not shown), even though the HD fractions included all soluble proteins as well as ∼80% of the plasma membrane proteins (Fig. 6A). These findings indicated that MERS entry-priming activities were greatly concentrated in the TEMs. MERS pp that had been exposed to TEMs were also evaluated to assess the extent of S-protein cleavage. Western immunoblots indicated that the TEMs affected cleavage of S proteins, generating proteolytic patterns that were indistinguishable from those generated by trypsin (Fig. 8A, bottom). The apparent molecular sizes of the N-linked glycoprotein products were consistent with cleavages at three multibasic sites, one at positions 626 to 629 (RQQR) to create the minor 130-kDa fragment, one at positions 884 to 887 (RSAR) to create the major 70-kDa fragment, and one at positions 1110 to 1113 (QSKR) to create the minor 40-kDa fragment. The molecular size of the major 70-kDa fragment is equivalent to that of the previously documented S2′ (1).
FIG 8.

CoV-priming activity of TEMs. (A) MERS-CoV pp were used to transduce 293β5 cells treated with leupeptin or a medium control. Prior to transduction, MERS-CoV pp were treated with trypsin, TEMs were isolated from untransfected cells, or TEMs were isolated from TMPRSS2-overexpressing cells. (Top) Transduction levels were measured by luciferase reporter gene expression. (Bottom) These MERS pp were also concentrated and analyzed by Western blotting with an antibody to detect a C-terminal C9 tag on the MERS-CoV spike. (B) 293β5 cells were infected with PR8 that was treated with trypsin, TEMs isolated from untransfected cells, or TEMs from HAT-overexpressing cells. (Top) Infection was measured by Gluc expression. (Bottom) Viruses were concentrated and analyzed by Western blotting. The molecular sizes (M) (in kilodaltons) are indicated to the left of the blots. *, P < 0.05 compared to the value for no incubation.
Analogous experiments were completed with IAV PR8 viruses. In these assays, however, inactivation of host proteases by leupeptin was not required, as PR8 did not respond to endogenous levels of 293 cell proteases. The results of these experiments demonstrated that TEM fractions activated PR8 infectivities, nearly as much as TEMs with overexpressed HAT (Fig. 8B, top). The fact that the TEMs were isolated from IAV-resistant 293β5 cells yet were capable of priming IAV for infection are potentially explained by the significant concentration of cell proteases achieved through TEM isolation.
As with the MERS S on viral pp, PR8-associated HA proteins were analyzed for cleavage status by Western immunoblotting. Here, the TEMs effected cleavage of full-length HA0 into HA1 fragments, irrespective of whether HAT was overexpressed and equal to that achieved by trypsin (Fig. 8B, bottom). Thus, the TEM fractions harvested from 293 cells have proteases that cleave and prime both MERS-CoV S and PR8 IAV HA proteins.
DISCUSSION
We evaluated TEMs for host cell entry factors and determined that they contain both CoV receptors and transmembrane serine proteases (TMPRs) and were capable of cleaving and priming CoV and IAV fusion proteins. We did not determine whether IAV sialic acid receptors are concentrated in TEMs, but there is evidence of sialylated proteins associating with tetraspanins (21, 48, 49). The findings suggested that, in natural infections, the CoVs and LP IAVs encounter TEMs during their cell entry and in doing so become proteolytically primed. This suggestion was consistent with virus entry blockades by tetraspanin antibodies and with reduced virus entry upon depletion of the tetraspanin CD9. From these results, we have come to the view that TEMs are platforms for several CoV and for LP IAV proteolytic priming events.
Previous investigators have hypothesized that TEMs are more flexible or curved than other membrane regions and therefore provide a platform that is more favorable for membrane melding. For example, in mouse oocytes, TEMs facilitate cell membrane wrinkling, which hypothetically lowers the kinetic barrier to fusion with sperm cell membranes (41). However, in the virus-cell membrane fusions that we have evaluated, it seems that TEMs do not facilitate the membrane fusions per se but rather facilitate virus proteolytic priming. This claim arises in part because tetraspanin antibodies blocked MHV and PR8 infections (Fig. 1) but not when transmembrane protease concentrations were elevated (Fig. 4). With respect to the mechanisms by which tetraspanin antibodies block virus entry, one possibility is that the bivalent antibodies hold tetraspanins together, rigidifying TEMs and impeding membrane protein movements. Reduced diffusion of virus-receptor complexes might therefore increase the time required for viruses to encounter proteases. Increasing protease concentrations might ease this supposed requirement for lateral mobility of receptor-bound viruses. Another possibility is that transmembrane protease activities depend on precise embedment into TEMs, with tetraspanin antibodies interfering with this hypothetical positioning. This notion has some support from our finding that CD9 depletion reduced MERS pp transduction at the level of MERS S-protein proteolytic priming.
Even with a restricted focus on TEMs as virus entry portals, it remains challenging to identify which cellular proteases are utilized for CoV S and IAV HA priming. For LP IAV, the relevant priming proteases are known to include TMPRSS2 (14) and HAT (15). The CoVs appear to be less restricted in their protease requirements, and members may utilize many or all of the ∼19 TMPRs (5), as well as the ∼25 membrane metalloproteinases (50). TMPRSS2, however, stands out as a key CoV-priming protease (3, 6–9, 51). Perhaps TMPRSS2 is more promiscuous with substrates than the other TTSPs, although evidence for this is lacking. Alternatively, TMPRSS2 may act as a “master” protease that cleaves nearby zymogens, activating them in proteolytic cascades (52). Other TMPRSS2-activated proteases may then cleave CoV S proteins. In addition, some CoVs may bypass TEM-associated proteases, undergoing cleavage-priming after endocytosis. A key example here is with MHV-2, which utilizes endosomal cathepsins to prime S proteins for entry (53). There is no evidence that cathepsins are localized to TEMs. Thus, the requirements for TEM-associated cleavage events may depend on the CoV strain and on the particular combinations of virus-priming proteases in target cells. Analyses of clinical CoV isolates for their entry into cells reflecting in vivo infection environments may be necessary to assess the importance of TEM-associated S proteolysis in natural infection and disease, for example, by using transgenic mice lacking TMPRSS2 (TMPRSS2−/−) (54) and HAT (HAT−/−) (55).
The human CoVs use the transmembrane ectopeptidases ACE2 (38), APN (36), and DPP4 (37) as host cell receptors. These receptors do not share any obvious structural similarities, and while they do share ectopeptidase activities, these enzymatic functions are dispensable for virus entry (36–38). Localization in TEMs, therefore, may be a shared feature that is relevant to the selection of these ectopeptidases as CoV receptors. One possibility is that the CoVs evolved to use TEM-associated receptors so that, once bound to cells, the viruses are poised for cleavage by TEM-resident proteases. It is, however, also possible that the viruses, adapted to TEM-associated receptors for yet unknown reasons, evolved to utilize nearby proteases for cleavage and priming. This evolution of viruses for particular receptors and proteases, when viewed in a dynamic context, posits that receptor binding elicits structural changes in viral spikes that transiently expose proteolytic substrates. Without proteases nearby, uncleaved intermediate S conformers might continue through unproductive folding pathways that are incompatible with virus entry. Conceivably, the proteolytic TEM environment is the preferred location for receptor-induced conformational changes of S proteins, rapid proteolytic cleavage of the intermediate S conformations, and possibly the subsequent refolding to postfusion forms, in spatial and temporal patterns that foster efficient virus entry.
Distinctions between TEMs and lipid rafts are noteworthy. TEMs are operationally distinguished from classical lipid rafts by their insolubility in zwitterionic detergents, such as CHAPS and Brij 98 (56), and by their complete disruption by nonionic detergents, such as TX100 (44) (Fig. 6). However, both TEM and lipid rafts are enriched in cholesterol, sphingolipids, and glycosylphosphatidylinositol (GPI)-linked surface proteins (16, 57–59), and cholesterol chelators such as cyclodextrins will disrupt both TEM and lipid raft architectures (60, 61). These common features of TEMs and lipid rafts can make it difficult to determine which microdomain serves as a virus entry site. For example, cholesterol depletion decreases CoV-cell entry, and it is resupplementation that restores entry (27, 62). Similar results were obtained in studies of IAV entry (63). The TEM-disrupting effects of cyclodextrins, in conjunction with our observations of TEM-associated virus receptors and virus-priming proteases, raise the possibility that cholesterol depletion blocks virus entry by separating receptors from priming proteases in TEMs. This suggestion can be addressed by determining whether preprimed viruses are resistant to cholesterol starvation. Revisiting previous studies with greater attention paid to distinguishing TEMs and lipid rafts may yield additional insights into the subcellular locations of CoV and IAV cell entry.
Determining the subcellular location of virus-priming events has implications for development of antiviral drugs, including antiviral proteases. Currently, broad-spectrum protease inhibitors can be used to prevent viral infection and spread both in vitro (64) and in vivo (65), but these treatments are not approved for human use and there is little data on their efficacy or side effects. By targeting protease inhibitors to TEMs, one might increase inhibitor potencies, and also elicit antiviral activity without causing undesired reductions of total lung proteolytic activity. To achieve this targeting, inhibitors might be conjugated to TEM-binding motifs, such as those found on the hepatitis C virus E2 protein (66) or to components of TEMs, such as cholesterol. With respect to cholesterol, it has already been demonstrated that inhibitors of virus entry are potentiated by linkage to cholesterol moieties (67). These cholesterol-conjugated inhibitors are helical peptides that target transient folding intermediates of viral glycoproteins, preventing their ability to catalyze membrane fusion and thus blocking virus entry. We suggest, at least for the CoVs, that these intermediates are formed subsequent to proteolytic priming in TEMs. Helical peptides targeting these CoV intermediates are well described (68, 69), and targeting these peptides to the TEM locus of priming may increase their antiviral efficacies.
ACKNOWLEDGMENTS
We thank Lijun Rong, Fang Li, and Michael Farzan for plasmid DNAs, Michael Whitt for recombinant VSVs, Peter Palese for recombinant IAV, and Balaji Manicassamy for anti-IAV antibodies. We especially thank Edward Campbell for expert support with immunofluorescence microscopy experiments and Stanley Perlman for manuscript comments.
This work was supported by NIH grants R56AI092128 and P01AI060699.
REFERENCES
- 1.Millet JK, Whittaker GR. 2014. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A 111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kido H, Yokogoshi Y, Sakai K, Tashiro M, Kishino Y, Fukutomi A, Katunuma N. 1992. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J Biol Chem 267:13573–13579. [PubMed] [Google Scholar]
- 3.Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, Lucas JM, Nelson PS, Pohlmann S, Soilleux EJ. 2012. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One 7:e35876. doi: 10.1371/journal.pone.0035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhirnov OP, Klenk HD, Wright PF. 2011. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res 92:27–36. doi: 10.1016/j.antiviral.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Bugge TH, Antalis TM, Wu Q. 2009. Type II transmembrane serine proteases. J Biol Chem 284:23177–23181. doi: 10.1074/jbc.R109.021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pohlmann S. 2011. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol 85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. 2011. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol 85:873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87:12552–12561. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Kramer-Kuhl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, von Hahn T, Simmons G, Hofmann H, Pohlmann S. 2013. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol 87:5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann-Winkler H, Thiel V, Pohlmann S. 2013. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87:6150–6160. doi: 10.1128/JVI.03372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawase M, Shirato K, Matsuyama S, Taguchi F. 2009. Protease-mediated entry via the endosome of human coronavirus 229E. J Virol 83:712–721. doi: 10.1128/JVI.01933-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A 102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pohlman S, Schughart K. 2013. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog 9:e1003774. doi: 10.1371/journal.ppat.1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bottcher-Friebertshauser E, Freuer C, Sielaff F, Schmidt S, Eickmann M, Uhlendorff J, Steinmetzer T, Klenk HD, Garten W. 2010. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J Virol 84:5605–5614. doi: 10.1128/JVI.00140-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charrin S, le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. 2009. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J 420:133–154. doi: 10.1042/BJ20082422. [DOI] [PubMed] [Google Scholar]
- 17.Yanez-Mo M, Barreiro O, Gordon-Alonso M, Sala-Valdes M, Sanchez-Madrid F. 2009. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol 19:434–446. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 18.van Spriel AB, Figdor CG. 2010. The role of tetraspanins in the pathogenesis of infectious diseases. Microbes Infect 12:106–112. doi: 10.1016/j.micinf.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Le Naour F, Andre M, Boucheix C, Rubinstein E. 2006. Membrane microdomains and proteomics: lessons from tetraspanin microdomains and comparison with lipid rafts. Proteomics 6:6447–6454. doi: 10.1002/pmic.200600282. [DOI] [PubMed] [Google Scholar]
- 20.Rana S, Claas C, Kretz CC, Nazarenko I, Zoeller M. 2011. Activation-induced internalization differs for the tetraspanins CD9 and Tspan8: impact on tumor cell motility. Int J Biochem Cell Biol 43:106–119. doi: 10.1016/j.biocel.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Ono M, Hakomori S. 2004. Glycosylation defining cancer cell motility and invasiveness. Glycoconj J 20:71–78. doi: 10.1023/B:GLYC.0000018019.22070.7d. [DOI] [PubMed] [Google Scholar]
- 22.Andre M, Le Caer JP, Greco C, Planchon S, El Nemer W, Boucheix C, Rubinstein E, Chamot-Rooke J, Le Naour F. 2006. Proteomic analysis of the tetraspanin web using LC-ESI-MS/MS and MALDI-FTICR-MS. Proteomics 6:1437–1449. doi: 10.1002/pmic.200500180. [DOI] [PubMed] [Google Scholar]
- 23.He J, Sun E, Bujny MV, Kim D, Davidson MW, Zhuang X. 2013. Dual function of CD81 in influenza virus uncoating and budding. PLoS Pathog 9:e1003701. doi: 10.1371/journal.ppat.1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen EK, Nemerow GR, Smith JG. 2010. Direct evidence from single-cell analysis that human alpha-defensins block adenovirus uncoating to neutralize infection. J Virol 84:4041–4049. doi: 10.1128/JVI.02471-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rumschlag-Booms E, Guo Y, Wang J, Caffrey M, Rong L. 2009. Comparative analysis between a low pathogenic and a high pathogenic influenza H5 hemagglutinin in cell entry. Virol J 6:76. doi: 10.1186/1743-422X-6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorp EB, Gallagher TM. 2004. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J Virol 78:2682–2692. doi: 10.1128/JVI.78.6.2682-2692.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell EM, Perez O, Melar M, Hope TJ. 2007. Labeling HIV-1 virions with two fluorescent proteins allows identification of virions that have productively entered the target cell. Virology 360:286–293. doi: 10.1016/j.virol.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heaton NS, Levya-Grado VH, Tan GS, Eggink D, Hai R, Palese P. 2013. In vivo bioluminescent imaging of influenza A virus infection and characterization of novel cross-protective monoclonal antibodies. J Virol 87:8272–8281. doi: 10.1128/JVI.00969-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szretter KJ, Balish AL, Katz JM. 2006. Influenza: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15:Unit 15G.1. doi: 10.1002/0471729256.mc15g01s3. [DOI] [PubMed] [Google Scholar]
- 31.Shulla A, Gallagher T. 2009. Role of spike protein endodomains in regulating coronavirus entry. J Biol Chem 284:32725–32734. doi: 10.1074/jbc.M109.043547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitt MA. 2010. Generation of VSV pseudotypes using recombinant DeltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J Virol Methods 169:365–374. doi: 10.1016/j.jviromet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barlan A, Zhao J, Sarkar MK, Li K, McCray PB Jr, Perlman S, Gallagher T. 2014. Receptor variation and susceptibility to Middle East respiratory syndrome coronavirus infection. J Virol 88:4953–4961. doi: 10.1128/JVI.00161-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset F-L. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem 278:41624–41630. doi: 10.1074/jbc.M305289200. [DOI] [PubMed] [Google Scholar]
- 35.Scheffer KD, Gawlitza A, Spoden GA, Zhang XA, Lambert C, Berditchevski F, Florin L. 2013. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J Virol 87:3435–3446. doi: 10.1128/JVI.02906-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV. 1992. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357:420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, Thiel V, Drosten C, Rottier PJ, Osterhaus AD, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254. doi: 10.1038/nature12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziyyat A, Rubinstein E, Monier-Gavelle F, Barraud V, Kulski O, Prenant M, Boucheix C, Bomsel M, Wolf JP. 2006. CD9 controls the formation of clusters that contain tetraspanins and the integrin alpha 6 beta 1, which are involved in human and mouse gamete fusion. J Cell Sci 119:416–424. doi: 10.1242/jcs.02730. [DOI] [PubMed] [Google Scholar]
- 40.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84:12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Runge KE, Evans JE, He ZY, Gupta S, McDonald KL, Stahlberg H, Primakoff P, Myles DG. 2007. Oocyte CD9 is enriched on the microvillar membrane and required for normal microvillar shape and distribution. Dev Biol 304:317–325. doi: 10.1016/j.ydbio.2006.12.041. [DOI] [PubMed] [Google Scholar]
- 42.Symeonides M, Lambele M, Roy NH, Thali M. 2014. Evidence showing that tetraspanins inhibit HIV-1-induced cell-cell fusion at a post-hemifusion stage. Viruses 6:1078–1090. doi: 10.3390/v6031078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okamoto T, Iwata S, Yamazaki H, Hatano R, Komiya E, Dang NH, Ohnuma K, Morimoto C. 2014. CD9 negatively regulates CD26 expression and inhibits CD26-mediated enhancement of invasive potential of malignant mesothelioma cells. PLoS One 9:e86671. doi: 10.1371/journal.pone.0086671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claas C, Stipp CS, Hemler ME. 2001. Evaluation of prototype transmembrane 4 superfamily protein complexes and their relation to lipid rafts. J Biol Chem 276:7974–7984. doi: 10.1074/jbc.M008650200. [DOI] [PubMed] [Google Scholar]
- 45.Dveksler GS, Pensiero MN, Cardellichio CB, Williams RK, Jiang GS, Holmes KV, Dieffenbach CW. 1991. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. J Virol 65:6881–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sala-Valdes M, Ursa A, Charrin S, Rubinstein E, Hemler ME, Sanchez-Madrid F, Yanez-Mo M. 2006. EWI-2 and EWI-F link the tetraspanin web to the actin cytoskeleton through their direct association with ezrin-radixin-moesin proteins. J Biol Chem 281:19665–19675. doi: 10.1074/jbc.M602116200. [DOI] [PubMed] [Google Scholar]
- 47.Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ II, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJ. 1991. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem 266:19599–19610. [PubMed] [Google Scholar]
- 48.Cannon KS, Cresswell P. 2001. Quality control of transmembrane domain assembly in the tetraspanin CD82. EMBO J 20:2443–2453. doi: 10.1093/emboj/20.10.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toledo MS, Suzuki E, Handa K, Hakomori S. 2005. Effect of ganglioside and tetraspanins in microdomains on interaction of integrins with fibroblast growth factor receptor. J Biol Chem 280:16227–16234. doi: 10.1074/jbc.M413713200. [DOI] [PubMed] [Google Scholar]
- 50.Bourboulia D, Stetler-Stevenson WG. 2010. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion. Semin Cancer Biol 20:161–168. doi: 10.1016/j.semcancer.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. 2014. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, Hood L, Nelson PS. 1999. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res 59:4180–4184. [PubMed] [Google Scholar]
- 53.Qiu Z, Hingley ST, Simmons G, Yu C, Das Sarma J, Bates P, Weiss SR. 2006. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J Virol 80:5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim TS, Heinlein C, Hackman RC, Nelson PS. 2006. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol Cell Biol 26:965–975. doi: 10.1128/MCB.26.3.965-975.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sales KU, Hobson JP, Wagenaar-Miller R, Szabo R, Rasmussen AL, Bey A, Shah MF, Molinolo AA, Bugge TH. 2011. Expression and genetic loss of function analysis of the HAT/DESC cluster proteases TMPRSS11A and HAT. PLoS One 6:e23261. doi: 10.1371/journal.pone.0023261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hemler ME. 2005. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 57.Dietrich C, Volovyk ZN, Levi M, Thompson NL, Jacobson K. 2001. Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc Natl Acad Sci U S A 98:10642–10647. doi: 10.1073/pnas.191168698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Murakami Y, Yasui T, Wakana S, Kikutani H, Kinoshita T, Maeda Y. 2013. Significance of GPI-anchored protein enrichment in lipid rafts for the control of autoimmunity. J Biol Chem 288:25490–25499. doi: 10.1074/jbc.M113.492611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown DA, London E. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 275:17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 60.Gomez-Mouton C, Abad JL, Mira E, Lacalle RA, Gallardo E, Jimenez-Baranda S, Illa I, Bernad A, Manes S, Martinez AC. 2001. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc Natl Acad Sci U S A 98:9642–9647. doi: 10.1073/pnas.171160298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Charrin S, Manié S, Thiele C, Billard M, Gerlier D, Boucheix C, Rubinstein E. 2003. A physical and functional link between cholesterol and tetraspanins. Eur J Immunol 33:2479–2489. doi: 10.1002/eji.200323884. [DOI] [PubMed] [Google Scholar]
- 62.Nomura R, Kiyota A, Suzaki E, Kataoka K, Ohe Y, Miyamoto K, Senda T, Fujimoto T. 2004. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J Virol 78:8701–8708. doi: 10.1128/JVI.78.16.8701-8708.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun X, Whittaker GR. 2003. Role for influenza virus envelope cholesterol in virus entry and infection. J Virol 77:12543–12551. doi: 10.1128/JVI.77.23.12543-12551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. 2012. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol 86:6537–6545. doi: 10.1128/JVI.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bahgat MM, Blazejewska P, Schughart K. 2011. Inhibition of lung serine proteases in mice: a potentially new approach to control influenza infection. Virol J 8:27. doi: 10.1186/1743-422X-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Owsianka AM, Timms JM, Tarr AW, Brown RJ, Hickling TP, Szwejk A, Bienkowska-Szewczyk K, Thomson BJ, Patel AH, Ball JK. 2006. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J Virol 80:8695–8704. doi: 10.1128/JVI.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee KK, Pessi A, Gui L, Santoprete A, Talekar A, Moscona A, Porotto M. 2011. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J Biol Chem 286:42141–42149. doi: 10.1074/jbc.M111.254243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bosch BJ, Martina BE, Van Der Zee R, Lepault J, Haijema BJ, Versluis C, Heck AJ, De Groot R, Osterhaus AD, Rottier PJ. 2004. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc Natl Acad Sci U S A 101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu L, Liu Q, Zhu Y, Chan KH, Qin L, Li Y, Wang Q, Chan JF, Du L, Yu F, Ma C, Ye S, Yuen KY, Zhang R, Jiang S. 2014. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat Commun 5:3067. doi: 10.1038/ncomms4067. [DOI] [PMC free article] [PubMed] [Google Scholar]