

ABSTRACT
Dicistroviridae are a family of RNA viruses that possesses a single-stranded positive-sense RNA genome containing two distinct open reading frames (ORFs), each preceded by an internal ribosome entry site that drives translation of the viral structural and nonstructural proteins, respectively. The type species, Cricket paralysis virus (CrPV), has served as a model for studying host-virus interactions; however, investigations into the molecular mechanisms of CrPV and other dicistroviruses have been limited as an established infectious clone was elusive. Here, we report the construction of an infectious molecular clone of CrPV. Transfection of in vitro-transcribed RNA from the CrPV clone into Drosophila Schneider line 2 (S2) cells resulted in cytopathic effects, viral RNA accumulation, detection of negative-sense viral RNA, and expression of viral proteins. Transmission electron microscopy, viral titers, and immunofluorescence-coupled transwell assays demonstrated that infectious viral particles are released from transfected cells. In contrast, mutant clones containing stop codons in either ORF decreased virus infectivity. Injection of adult Drosophila flies with virus derived from CrPV clones but not UV-inactivated clones resulted in mortality. Molecular analysis of the CrPV clone revealed a 196-nucleotide duplication within its 5′ untranslated region (UTR) that stimulated translation of reporter constructs. In cells infected with the CrPV clone, the duplication inhibited viral infectivity yet did not affect viral translation or RNA accumulation, suggesting an effect on viral packaging or entry. The generation of the CrPV infectious clone provides a powerful tool for investigating the viral life cycle and pathogenesis of dicistroviruses and may further understanding of fundamental host-virus interactions in insect cells.
IMPORTANCE Dicistroviridae, which are RNA viruses that infect arthropods, have served as a model to gain insights into fundamental host-virus interactions in insect cells. Further insights into the viral molecular mechanisms are hampered due to a lack of an established infectious clone. We report the construction of the first infectious clone of the dicistrovirus, cricket paralysis virus (CrPV). We show that transfection of the CrPV clone RNA into Drosophila cells led to production of infectious particles that resemble natural CrPV virions and result in cytopathic effects and expression of CrPV proteins and RNA in infected cells. The CrPV clone should provide insights into the dicistrovirus life cycle and host-virus interactions in insect cells. Using this clone, we find that a 196-nucleotide duplication within the 5′ untranslated region of the CrPV clone increased viral translation in reporter constructs but decreased virus infectivity, thus revealing a balance that interplays between viral translation and replication.
INTRODUCTION
The Dicistroviridae are a family of nonenveloped, single-stranded RNA (ssRNA) viruses that infect arthropods (1). Dicistrovirus genomes range from 8 to 10 kb in size, contain a 5′ viral protein cap (VPg) and a 3′ poly(A) tail, and are characterized by a unique dicistronic genome arrangement. Distinct internal ribosome entry sites (IRES) drive translation of each open reading frame (ORF). The 5′ untranslated region (UTR) IRES directs translation of ORF1, which encodes viral nonstructural proteins such as the suppressor of RNA interference (RNAi), RNA helicase, 3C protease, and the RNA-dependent RNA polymerase (RdRp). The intergenic region (IGR) IRES facilitates expression of the viral structural proteins (ORF2) (1).
Dicistroviruses are of economic and medical importance. Studies have linked a subset of bee dicistroviruses to the decline in North American honeybee populations (2, 3). This is especially vital for crops reliant on honeybee pollination, which have an estimated worth of $215 billion worldwide (4). In addition, the causative agent of Taura syndrome in panaeid shrimp, Taura syndrome virus, has devastated the shrimp farming industry throughout the Americas (5, 6). Moreover, the protozoan parasite Trypanosoma cruzi is transmitted through an arthropod vector that can result in Chagas disease, which afflicts 7 to 8 million people in Latin America. The T. cruzi vector is a host of the dicistrovirus, triatoma virus, thus highlighting its possible use as a biopesticide (7).
The type species, cricket paralysis virus (CrPV), was first isolated in 1970 from Australian field crickets and has a wide host range, including Drosophila melanogaster (1, 8). Consequently, many studies have used CrPV and other dicistroviruses as models to delineate translational control mechanisms and antiviral immune mechanisms in insects, such as the RNAi and Imd (immunodeficiency) pathways (9). Furthermore, the unique dicistronic organization of its genome has led to insights into an unprecedented mechanism of translation initiation. The IGR IRES contains domains that functionally mimic a tRNA to recruit ribosomes and initiate translation in a factor-independent manner (10–14). In contrast, the mechanism of the 5′ UTR IRES is, in general, not well understood. It has been reported that translation mediated by the 5′ UTR IRES of the related dicistrovirus, Rhopalosiphum padi virus (RhPV), requires a subset of canonical translation factors (15). Since the dicistrovirus 5′ UTRs do not appear to be well conserved (16, 17), each 5′ UTR IRES may have distinct mechanisms for translation.
Despite the importance of dicistroviruses, the molecular mechanism and replication life cycle during infection are not completely understood. This is primarily due to a lack of an established infectious clone for any member of the Dicistroviridae family. In vitro transcripts of the black queen cell virus (BQCV) were reported to be infectious; however, no infectious clone was obtained (18). The genomic sequence of RhPV was cloned, but it harbored a mutation in its first open reading frame that prohibited infection (19, 20). Here, we report the generation of an infectious full-length clone of the CrPV genome which, compared to the published sequence (21), contains unique nucleotide changes, including a 196-nucleotide (nt) duplication in the 5′ UTR. In vitro-transcribed RNA of the CrPV clone transfected into Drosophila Schneider line 2 (S2) cells recapitulates CrPV infection and produces virions that infect and replicate in Drosophila S2 cells. Furthermore, we demonstrate that the duplication in the 5′ UTR is not an artifact of cloning but occurs within viral stocks and can stimulate translational activity of the 5′ UTR IRES. Using reverse genetics, an infectious clone containing the duplicated element in the 5′ UTR is less fit than one containing a single copy of the element in S2 cells. Finally, we show evidence that infection of virus generated by the CrPV infectious clone causes mortality in adult flies. To our knowledge, this is the first established infectious clone of the Dicistroviridae family.
MATERIALS AND METHODS
Cell culture and virus.
Drosophila Schneider line 2 (S2) cells were maintained and passaged in Shield's and Sang medium (Sigma) supplemented with 10% fetal bovine serum. Kc167 cells were maintained and passaged in Sf-900 III medium (Invitrogen) and supplemented with 10% fetal bovine serum.
Propagation of CrPV in Drosophila S2 cells has been previous described (22). CrPV was isolated from Drosophila S2 cells using an adapted protocol (23) with cesium chloride (CsCl) gradients. Briefly, S2 cells were infected with CrPV and incubated for 24 h. Cells were dislodged into the medium, treated with 0.5% IGEPAL CA-630 (Nonidet P-40) and 0.1% 2-mercaptoethanol, and incubated on ice for 10 min. Cell debris was cleared by centrifugation at 13,800 relative centrifugal force (RCF) for 15 min at 4°C. The supernatant was then treated with RNase A (100 μg/ml) for 30 min at 27°C. Viral particles were then pelleted through a 30% (wt/wt) sucrose cushion in VP buffer (50 mM HEPES, 0.1% 2-mercaptoethanol, and 5 mM CaCl2) by ultracentrifugation at 141,000 RCF for 2.5 h at 11°C. The pellet was resuspended in VP buffer and added to a 50% CsCl gradient in VP buffer. The gradient was ultracentrifuged at 274,000 RCF for 18 h at 4°C. The viral band was extracted and dialyzed with phosphate-buffered saline (PBS) overnight. Viral titers and yield were determined as previously described (22).
Virus was inactivated by UV light for 30 min on ice with a Stratagene 1800 UV cross-linker. Lack of cytopathic effects upon infection verified inactivation of the virus.
CrPV cDNA construction.
RNA was extracted from CsCl-purified CrPV using TRIzol (Invitrogen). The construction of the full-length infectious clones was generated as follows. First-strand CrPV cDNA was synthesized through reverse transcription-PCR (RT-PCR) using Superscript III reverse transcriptase (Invitrogen) and an oligo(dT) primer at 50°C for 2 h (see Fig. 3A). The CrPV cDNA was amplified using primers P1 (5′-GAGGGTACCATGTCTTTTCAACAAACAAACAAC-3′; KpnI site is underlined) and P3 (5′- GGAGCTCAGAAAACAACTATTAATCAAAAACCAAATTG-3′; SacI site is underlined), which introduce KpnI and SacI sites. The resulting PCR product, which contains the CrPV genome from the beginning of ORF1 to the end of the 3′ UTR, was cloned into pAC5.1/V5 His B (Invitrogen) using KpnI and SacI sites to create pCrPV-Δ5′ UTR (see Fig. 3A). Amplification of the CrPV genomic cDNA in its entirety was performed using primers P3 and P2 (5′-GAGGGTACCTTTAATAAGTGTTGTGCAGATTAATCTG-3′; KpnI site is underlined). To create the full-length clone of CrPV, a KpnI/HindIII fragment from the P2/P3 PCR product was cloned into the pCrPV-Δ5′ UTR using the same restriction sites. The resulting plasmid was denoted pCrPV-1 (see Fig. 3A). A T7 RNA polymerase promoter flanking the 5′ end of the CrPV genome was incorporated into pCrPV-1 using primer incomplete polymerase extension (PIPE) (24) with primers P4 (5′-GAAGATTAATACGACTCACTATAGGGAGATTTAATAAGTGTTGTGCAGATTAATCTG-3′; T7 promoter is underlined) and P5 (5′-TCTCCCTATAGTGAGTCGTATTAATCTTCTCTCCCTATAGTGAGTCGTATTAATCTTC-3′; T7 promoter is underlined) to create pCrPV-2 (see Fig. 3A). Primers P2 and P6 (5′-TGTAAGCTTTGTTCTTAAGTTCTCTG-3′; HindIII site is underlined) were used to amplify the 5′ UTR without the duplication (5′ UTRΔDup) and part of ORF1 from 2006 CrPV stocks (CrPV-2006) (Fig. 1). This was inserted in between the KpnI and HindII sites of pCrPV-2 to generate pCrPV-3 (see Fig. 3A). Finally, a T7 promoter was added to pCrPV-3 using primers P4 and P5.
FIG 3.

Construction and map of the full-length cDNA clones CrPV-2 and CrPV-3. (A) RNA from CsCl-purified CrPV was reverse transcribed using an oligo(dT) primer. Primers P1 and P3 were used to clone the CrPV genome without the 5′ UTR into pAC5.1/V5 His B while primers P2 and P3 containing KpnI and SacI restriction sites, respectively, were used to amplify the CrPV genome. CrPV contains an endogenous HindIII site in ORF1. Therefore, to clone the 5′ UTR, both the P1/P2 amplicon and pCrPV-Δ5′ UTR were digested with HindII and KpnI. The resulting fragments were ligated together to generate pCrPV-1. A T7 polymerase promoter was added to pCrPV-1 via primer incomplete polymerase extension (PIPE) to create pCrPV-2. To create pCrPV-3, a PCR fragment derived from the 2006 stock of CrPV was inserted into the KpnI and HindIII sites of pCrPV-2 followed by a T7 promoter. (B) At top the organization of the CrPV-2 and CrPV-3 cDNA genomic sequences showing all annotated proteins is shown. Arrowheads indicate either synonymous (black) or nonsynonymous (white) changes that differ from the published CrPV sequence (GenBank accession number NC_003924). Insertion-deletion events are indicated by an asterisk. A gray triangle indicates the location of the 196-nt duplication in CrPV-2. See Table 1 for specific nucleotide changes. The sequences of the 5′ and 3′ ends of the cloned viral genomes are shown at the bottom of the panel. Nucleotides corresponding to the viral genome are bolded. Arrows indicate the beginning and end of the in vitro transcript obtained from using T7 polymerase with Ecl136II-linearized plasmid.
FIG 1.

RT-PCR and sequence of the CrPV 5′ UTRs from different viral stocks. (A) RT-PCR analysis of the CrPV 5′ UTRs from cells that were infected with different CrPV stocks from circa 2006, 2009, or 2013 for 24 h. (B) Sequence of the first 500 nucleotides of the 5′ UTR from CrPV-2013, which contains the tandem 196-nt duplication (the first sequence in italics and the repeated sequence in boldface).
Stop codon mutations in either ORF1 or ORF2 of pCrPV-2 (pCrPV-2-ORF1-STOP and pCrPV-2-ORF2-STOP, respectively) were introduced via PCR-based site-directed mutagenesis. UAA stop codons were introduced by mutating nucleotides C775T (C971T) and G778T (G974T) of ORF1 and nucleotide A6232T (A6428T) of ORF2. The numbering of the nucleotides is based on CrPV-3, with CrPV-2 numbering in parentheses.
Bicistronic and minigenome reporter construction.
For the bicistronic reporter construct, a PCR-derived fragment containing the CrPV or CrPV-2 5′ UTR plus 54 nt of ORF1 was ligated into the EcoRI and NcoI sites of pMC53 upstream of the firefly luciferase (FLuc) open reading frame. Subsequently, the 5′ UTR-firefly luciferase fragment was inserted into EcoRI and XbaI sites of pEJ551 (25). For the minigenome construct, a PCR-derived fragment containing the IGR IRES and a short region of VP2 (nucleotides 6413 to 6571) was inserted between EcoRI and NcoI restriction sites in pEJ551. Subsequently, overlapping PCR was used to insert the CrPV 5′ UTR (without the duplication) in front of the Renilla luciferase (RFluc) open reading frame. Lastly, a synthesized CrPV 3′ UTR (Integrated DNA Technologies) was inserted between XbaI and BamHI sites downstream of the firefly luciferase open reading frame. The resulting plasmid is called pMG-ΔDup. The minigenome containing the 5′ UTR duplicated element (5′ UTR+Dup) was constructed using the Gibson assembly approach (New England Biolabs). The CrPV 5′ UTR containing the duplication was amplified using pCrPV-2 as a template with primers P10 (5′-ACGACTCACTATAGGGTTTAATAAGTGTTGTGCAGATTAATC-3′) and P11 (5′-TAAACTTTCGAAGTGATCACATTGTAAGAATCGG-3′). The minigenome containing the Renilla Luc-IGR IRES-firefly Luc-3′ UTR was amplified with primers P12 (5′-CATGACTTCGAAAGTTTATGATC-3′) and P13 (5′-CCCTATAGTGAGTCGTATTAAAG-3′). The CrPV 5′ UTR amplicon was mixed with the minigenome amplicon and incubated for 15 min at 50°C in the Gibson assembly mix (NEB). The reaction product was transformed into competent Escherichia coli DH5α cells and sequenced to confirm that the 5′ UTR duplication was present.
In vitro transcription and RNA transfection.
Purified plasmid was linearized with either Ecl136II (pCrPV-2 and derivatives) or BamHI (minigenome constructs). RNA was transcribed in a T7 RNA polymerase reaction and subsequently purified with an RNeasy kit (Qiagen). The integrity and purity of the RNA were confirmed on a 1.2% denaturing formaldehyde agarose gel.
Transfection of in vitro-synthesized RNA into S2 cells was performed using Lipofectamine 2000 (Invitrogen) as per the manufacturer's instructions. Three micrograms of RNA derived from either the full-length CrPV clone or its cognate mutants and 1 μg of reporter RNA were used for transfection using 2.5 × 106 cells.
RT-PCR and Northern blotting.
Total RNA was isolated from cells using TRIzol reagent. RT-PCR was performed on 5 μg of RNA using Superscript Reverse Transcriptase III (Invitrogen) at 55°C, followed by DNase I treatment for 30 min at 37°C. For reverse transcription of the negative-sense CrPV viral RNA, tagged primer P7 (5′-CTATGGATCCATGGGAGAAGATCAGCAAAT-3′; tag is underlined) was used. Primers P8 (5′-CTATGGATCCATGGGAGAAG-3′) and P9 (5′-GTGGCTGAAATACTATCTCTGG-3′) were used for PCR amplification of the negative-sense strand of the CrPV genome. The 5′ UTR of CrPV was RT-PCR amplified using primers P2 and P14 (5′-GAGCCATGGCTCAAGGGAGTTGATGTTGTT-3′; NcoI site is underlined). Diagnostic RT-PCRs of Drosophila C virus (DCV), Drosophila X virus (DXV), or flock house virus (FHV) cDNA were performed using the following primers: DCV genome, P15 (5′-TCTGCAGGAGTTCCCGATG-3′) and P16 (5′-CAATGCGCTTCCGGAGAC-3′); segment B of DXV, P17 (5′-TGGACATCGAAACAGGGTACAC-3′) and P18 (5′-CCGCGGATAGAGTTTGGTAACC-3′); FHV RNA1, P19 (5′-CGCCAATGATAAGAAACCAAGCG-3′) and P20 (5′-GGTCCACAAATACACGAGACAGG-3′). Actin was amplified using primers P21 (5′-CGACAACGGCTCTGGCATGTGCA-3′) and P22 (5′-ACGCAGCTCATTGTAGAAGGTG-3′).
Northern blotting was performed by loading 5 μg of RNA on a denaturing agarose gel which was subsequently transferred to Zeta probe blotting membrane (Bio-Rad). DNA probes were radiolabeled with a DecaLabel DNA labeling kit (Fermentas) and hybridized overnight. Radioactive bands were detected via phosphorimager analysis (Typhoon; GE Healthcare).
Western blotting.
Equal amounts of S2 protein lysates (20 μg) were resolved on a 12% SDS-PAGE gel and then transferred to a polyvinylidene difluoride Immobilon-FL membrane (Millipore). Membranes were blocked for 30 min at room temperature with 5% skim milk in Tris-buffered saline plus Tween (TBST). Blots were incubated for 1 h at room temperature with either CrPV ORF1 (raised against CrPV RdRp) rabbit polyclonal (1:10,000) or CrPV ORF2 (raised against CrPV VP2) rabbit polyclonal (1:10,000) (22) antibody. Membranes were washed three times with TBST and incubated with either IRDye 800CW goat anti-rabbit IgG (1:20,000; Li-Cor Biosciences) or donkey anti-rabbit IgG-horseradish peroxidase (1:20,000; GE Healthcare) for 1 h at room temperature. An Odyssey imager (Li-Cor Biosciences) and enhanced chemiluminescence (Thermo Scientific) were used for detection.
EM.
Negative-stained specimens were prepared by adsorbing samples to glow-discharged carbon-coated copper grids and subsequent staining with uranyl formate. A Tecnai Spirit transmission electron microscope (EM; FEI) operated at an accelerating voltage of 120 kV was used to examine these specimens. Images were acquired using a 4,000- by 4,000-pixel Eagle charge-coupled-device (CCD) camera (FEI) at a nominal magnification of ×49,000.
Transwell assays.
S2 cells were transfected with in vitro-transcribed viral genomic RNA for 24 h at 25°C. Untreated cells were seeded onto a coverslip pretreated with concanavalin A (0.5 mg/ml). Transfected cells were washed three times with PBS and then seeded onto a transwell insert within the six-well plate. The coverslips were washed with PBS and treated with 3% paraformaldehyde for 15 min, followed by methanol treatment for 10 min. The cells were washed with PBS and incubated with anti-ORF2 antibody (1:500 in 5% bovine serum albumin [BSA] in PBS) for 1 h at room temperature. Cells were washed three times with PBS and incubated with goat anti-rabbit Texas Red IgG (1:200 in 5% BSA in PBS; Invitrogen) for 1 h at room temperature, followed by 0.5 μg/ml Hoechst to stain the nuclei. Slides were imaged using a confocal microscope (Olympus FV1000 using Olympus Fluoview, version 2.0a) with a 60× oil immersion lens. Images shown represent a single Z-section and were processed in Photoshop CS6.
In vitro and in vivo translation assays.
S2 cell translation extracts were obtained using an adapted protocol (26, 27). Briefly, 2.0 × 109 cells were resuspended in PBS and either mock infected or infected with CrPV (multiplicity of infection [MOI] of 10). Cells were then diluted to 1.0 × 107 cells/ml and incubated at 25°C for 6 h. Cells were pelleted (1,000 × g for 8 min), washed once with PBS, and resuspended in hypotonic buffer (10 mM HEPES-KOH [pH 7.4], 10 mM potassium acetate [KOAc], 0.5 mM magnesium acetate [MgOAc], 1 mM dithiothreitol [DTT]) and subsequently incubated on ice for 5 min. Cells were lysed by extrusion 25 times through a 23-gauge needle. Resulting extracts were adjusted with 50 mM KOAc. Cell debris was cleared by centrifugation (16,000 RCF for 5 min at 4°C), and the supernatant was stored at −80°C.
Bicistronic or minigenome RNA (90 nM) was incubated in either mock-infected or CrPV-infected S2 cell extracts for 30 min at 30°C. Luciferase activity was measured using a dual-luciferase assay (Promega). In vitro translation of the full-length viral RNA genome (uncapped) was performed in Spodoptera frugiperda 21 (Sf-21) cell extract (Promega) in the presence of [35S]methionine-cysteine. Reaction mixtures were loaded on SDS-PAGE gels. Gels were dried, and radioactive bands were monitored by phosphorimager analysis.
For in vivo translation assays, mock- or CrPV-infected S2 cells were incubated with [35S]methionine-cysteine for the last 30 min of the infection. Equal amounts of lysate (10 μg) were then loaded on SDS-PAGE gels. Gels were dried, and radioactive bands were monitored by phosphorimager analysis. For luciferase assays, S2 cells (1.5 × 106 cells) were transfected with in vitro-transcribed RNA for 2 h, followed by mock or CrPV infection (MOI of 10) for 6 h. Cells were harvested and lysed, and luciferase activity was measured (Promega) using a microplate luminometer (Centro LB 960; Berthold Technologies).
Fly stocks and viral injections.
Isogenic w1118 flies were maintained on standard cornmeal food at 25°C and 70% humidity with a 12-h light-dark cycle. Freshly eclosed virgin males and females were separated and collected in five groups of 10 each. Flies (10 males and 10 females) were injected with 200 nl of PBS, CrPV-2, or CrPV-3 (5,000 FFU) using a Neurophore BH-2 injection system (Harvard Apparatus) and transferred to standard food and flipped to fresh food every 3 days.
Nucleotide sequence accession numbers.
The sequences of pCrPV-2 and pCrPV-3 have been deposited in GenBank under accession numbers KP974706 and KP974707, respectively.
RESULTS
Selection of a 196-nucleotide element within the CrPV 5′ UTR that augments translation.
In general, the mechanism underlying CrPV 5′ UTR IRES translation is poorly understood. To address this, we initially used RT-PCR to clone the 5′ UTR from CrPV stocks that were propagated in our S2 tissue culture cells. Using a CrPV stock from 2006 (CrPV-2006), an RT-PCR product corresponding to the published predicted size of the CrPV 5′ UTR plus 54 nt of ORF1 (763 nucleotides) was detected (Fig. 1A). Interestingly, using a CrPV stock from 2013 (CrPV-2013), RT-PCR analysis detected a band corresponding to the 5′ UTR that increased in size by roughly 200 bp (Fig. 1A). Moreover, a CrPV stock from 2009 (CrPV-2009) produced a product that is consistent with the size of the 5′ UTR of CrPV-2006 as well as a very weak band consistent with that of the 5′ UTR of CrPV-2013 (Fig. 1A). Sequencing analysis revealed that CrPV-2013 acquired a tandem duplication of nucleotides 76 to 271 (196 nt) in the 5′ UTR that was not previously described in the original CrPV sequence (Fig. 1B) (GenBank accession number NC_003924). As RNA viruses largely exist as quasispecies (28), we surmised that the duplication in the 5′ UTR likely arose due to selection during infection, which presumably increased viral fitness.
We next investigated the mechanism by which the 196-nucleotide duplication affects CrPV infection. As the duplication occurred within a known IRES sequence, we hypothesized that the duplication might influence the translational activity of the 5′ UTR IRES. To address this, we generated a minigenome reporter construct that encodes Renilla and firefly luciferase genes in lieu of CrPV ORF1 and ORF2, respectively (Fig. 2A). Consequently, expression of Renilla luciferase (RLuc) is driven by the 5′ UTR IRES while firefly luciferase (FLuc) expression is directed by the IGR IRES (Fig. 2A). In vitro-transcribed minigenome RNA with (5′ UTR+Dup) or without (5′ UTRΔDup) the 5′ UTR duplication was incubated in translation extracts derived from mock- or CrPV-infected S2 cells, and luciferase activities were measured. In both mock- and CrPV-infected cell extracts, the 5′ UTR+Dup-containing reporter RNA resulted in greater than 4-fold more Renilla luciferase activity than the 5′ UTRΔDup RNA (Fig. 2B). In all cases, IGR IRES-mediated translation levels of the firefly luciferase ORF were similar in mock- and CrPV-infected lysates. To determine whether this effect is similar in vivo, the same minigenome RNAs were transfected into S2 cells followed by mock or CrPV infection. Cells were harvested at 6 h postinfection (p.i.), and luciferase activities were measured. We previously determined that the luciferase activities increased linearly from 1 to 6 h after transfection of the reporter RNAs, suggesting that the RNA is intact and engaged in translation during this time (25). Similar to the in vitro results, the 5′ UTR containing the duplication resulted in higher Renilla luciferase activity than that without the duplication (Fig. 2C), indicating that the duplication within the 5′ UTR stimulates translation in the context of the minigenome reporter RNA.
FIG 2.

Translational control mediated by the duplication in the CrPV-2 5′ UTR. (A) Schematics of reporter constructs. Minigenome reporter RNAs (top) contain Renilla and firefly luciferase genes in lieu of the CrPV ORF1 and ORF2, respectively, but maintain the untranslated regions of the CrPV genome. The bicistronic reporter RNA (bottom) contains a 5′ m7G cap structure and a 3′ poly(A) tail. Renilla luciferase is translated via 5′-end scanning-dependent translation while firefly luciferase is IRES dependent. Gray triangles represent the duplicated sequence element. (B) In vitro translation. In vitro-transcribed minigenome RNA containing the 5′ UTR with (5′ UTR+Dup) or without (5′ UTRΔDup) the 196-bp duplication was incubated in mock- or CrPV-infected S2 cell extracts for 30 min at 30°C. The Renilla and firefly luciferase activities monitor 5′ UTR- and IGR IRES-dependent translation, respectively. Luciferase activity is normalized to that of the minigenome reporter construct containing the 5′ UTRΔDup. (C) Translational activity in S2 cells. S2 cells, transfected with the indicated minigenome RNAs for 2 h at 25°C, were either mock or CrPV infected (MOI of 10) for 6 h. Luciferase activity is normalized to that of the minigenome reporter construct containing the 5′UTRΔDup. (D) IRES activity. Bicistronic RNAs containing either the CrPV 5′ UTR+Dup or 5′ UTRΔDup or an empty intergenic region were incubated in mock- or CrPV-infected S2 cell extracts for 30 min at 30°C. Renilla luciferase measures scanning-dependent translation while firefly luciferase measures IRES-mediated translation. The ratios of firefly to Renilla luciferase activities were normalized to the IGR IRES activity. Shown are averages from at least three independent experiments (± standard deviations).
The 5′ UTR of dicistroviruses has been reported to contain IRES activity (29–31). To determine whether the duplication has an effect on IRES translation, we inserted the 5′ UTR with (5′ UTR+Dup) or without (5′ UTRΔDup) the duplication into the intergenic region of a bicistronic reporter construct (Fig. 2A). Here, the 5′ UTR directs translation of firefly luciferase while Renilla luciferase translation occurs by a 5′ end-dependent scanning mechanism (25). In vitro-transcribed bicistronic RNAs were incubated in S2 cell extracts, and the ratio of FLuc/RLuc activities was measured. As shown previously, the IGR IRES directed translation of the luciferase ORF in contrast to the activity of a no-IRES control (Fig. 2D). Translation driven by both the 5′ UTR+Dup and 5′ UTRΔDup was approximately 20% and 10% of IGR IRES translation in mock- and CrPV-infected lysates, respectively (Fig. 2D), indicating that the duplication within the 5′ UTR does not affect IRES activity. This result is in contrast to that observed in the minigenome reporter RNA where the 5′ UTR+Dup stimulated translation compared to activity of the 5′ UTR without the duplication (Fig. 2B and C). These results show an important distinction between the location of the 5′ UTR within the reporter RNA and the mechanism of translation via the 5′ UTR.
Construction and characterization of a CrPV infectious clone.
To determine the role of the 5′ UTR duplication during CrPV infection, we constructed an infectious clone of CrPV with (pCrPV-2) and without (pCrPV-3) the 5′ UTR duplication (Fig. 3A). Viral RNA isolated from pure CrPV virions was used to amplify by RT-PCR a partial CrPV cDNA lacking the 5′ UTR using primers P1 and P2. The resulting PCR product was cloned into the pAC5.1/V5 His B plasmid creating pCrPV-Δ5′ UTR with KpnI and SacI restriction sites flanking the genomic sequence (Fig. 3A). Attempts to clone the full-length CrPV cDNA using a primer that hybridizes to the 5′ end of the CrPV genome were unsuccessful. Therefore, we took advantage of a unique endogenous HindIII site in the CrPV genome in order to construct a full-length clone. An RT-PCR product of CrPV cDNA that contains the 5′ UTR was ligated into the KpnI-HindIII sites of pCrPV-Δ5′ UTR, thus generating pCrPV-1 (Fig. 3A). A T7 RNA polymerase promoter was inserted between the KpnI site and the 5′ end of the CrPV genome to create the construct pCrPV-2 (Fig. 3A). To construct pCrPV-3, a PCR fragment from CrPV-2006 was inserted into the HindIII and KpnI sites of pCrPV-2 (Fig. 3A).
Propagation of pCrPV-2 and pCrPV-3 plasmids was performed in E. coli DH5α cells at 30°C. The entire sequence of each CrPV genomic clone was confirmed by sequencing. Previous reports have shown that clones of viral genomic sequences can be highly unstable (32–35). This is attributed to the fact that propagation of viral cDNA clones in E. coli can result in toxic products that prevent cloning efforts or accumulation of mutations in the genomic sequence, causing clones to become defective (36, 37). Accordingly, the CrPV genome lacking the 5′ UTR was stable (pCrPV-Δ5′ UTR) (Fig. 3A); however, during propagation of the full-length clones (pCrPV-2 and pCrPV-3) (Fig. 3A) in E. coli DH5α at 37°C, it acquired large nucleotide insertions (up to ∼1 kb) within the genome (data not shown). To overcome this issue, we found that growth of E. coli cells harboring pCrPV-2, pCrPV-3, or other clones at 30°C permitted propagation with no apparent aberrations.
Sequence analysis of CrPV-2 and CrPV-3 revealed 34 nucleotide substitutions that differ from the previously published CrPV sequence (GenBank accession number NC_003924) (21) (Table 1). Of these, two nucleotide changes are located in the 5′ and 3′ UTRs, and 32 are within ORF1 and ORF2. No changes were found within the IGR IRES, reflecting the importance of this element for viral IRES activity. Inspection of ORF1 and ORF2 nucleotide changes revealed that 14 are synonymous while 19 are nonsynonymous (Table 1; Fig. 3B). In addition to the nucleotide substitutions, insertion and deletion events occurred at four positions, nucleotides 3671 (insertion of a G), 3678 (deletion of an A), 5245 (insertion of an A), and 5270 (deletion of a C), which resulted in an alternative amino acid sequence but maintained the reading frame (Table 1; Fig. 3B). The numbering refers to the nucleotides within the published CrPV sequence (GenBank accession number NC_003924) (21). Altogether, the changes within CrPV-2 and CrPV-3 resulted in 31 alterations of the amino acid sequence, of which 26 occurred in ORF1 and 5 occurred in ORF2, with no differences occurring in the predicted conserved motifs of the nonstructural proteins (Table 1; Fig. 3B). As mentioned previously, a tandem duplication of nucleotides A76 to A271 (196 nt) is present in the 5′ UTR of CrPV-2 that is not in CrPV-3 or the previously published CrPV genome (Table 1; Fig. 3B).
TABLE 1.
Nucleotide and amino acid sequence differences between the CrPV-2 and CrPV sequencesa
| 5′ UTR nucleotide change | ORF1 |
ORF2 |
3′ UTR nucleotide change | ||||
|---|---|---|---|---|---|---|---|
| Nucleotide change | Amino acid change | VPb | Nucleotide change | Amino acid change | VP | ||
| A76–A271, A272–A467c | C1545T | 2B | C6279A | VP2 | G9005A | ||
| G312A | A1719G | 2C | A6316G | I34V | VP2 | ||
| T1979C | V424A | 2C | G6426A | G70R | VP2 | ||
| C2127T | 2C | C6707G | T164R | VP2 | |||
| T2128C | F474L | 2C | C6860T | T215I | VP2 | ||
| T3190A | F828I | 3A | C6882T | VP2 | |||
| G3671d | A988G | 3C | C7359A | VP3 | |||
| A3678e | T989N, A990S | 3C | |||||
| A4189G | N1161D | 3C | T7896C | VP3 | |||
| A4255G | P1183A | 3C | A8019T | VP3 | |||
| C4745T | T1346I | 3D | C8355T | VP1 | |||
| T4814C | F1369S | 3D | A8870G | E885G | VP1 | ||
| A5245d | F1513I | 3D | |||||
| G5262T | F1514L | 3D | |||||
| C5270e | G1515W, Q1516A, S1517I, C1518F, G1519W, K1520E, S1521V | 3D | |||||
| T5693A | F1662Y | 3D | |||||
| C5696G | P1663R | 3D | |||||
| C5706A | 3D | ||||||
| C5715G | H1669Q | 3D | |||||
| A5724G | 3D | ||||||
| A5726G | K1673R | 3D | |||||
| A5728G | R1674G | 3D | |||||
| C5745G | H1679Q | 3D | |||||
| T5784A | 3D | ||||||
| A5971G | K1755E | 3D | |||||
Nucleotide numbers correspond to those of the published CrPV sequence. The first amino acid letter corresponds to CrPV (GenBank accession number NC_003924) while the second is the amino acid in the same position in CrPV-2.
VP, viral protein.
Duplicated sequence.
Insertion.
Deletion.
Transfection of in vitro-transcribed CrPV-2 and CrPV-3 RNA resembles CrPV infection.
To validate the infectious clones of CrPV, we first determined whether viral proteins are expressed using in vitro translation in Sf-21 extracts. Incubation of in vitro-transcribed RNA from pCrPV-2 or pCrPV-3 in the presence of [35S]methionine-cysteine resulted in the expression of proteins ranging from <25 kDa to >170 kDa (Fig. 4, lane 4). If unprocessed, ORF1 and ORF2 polyproteins are 1,771 and 895 amino acids in length with predicted molecular masses of 203.8 and 100.4 kDa, respectively. To determine whether the proteins are translated from ORF1 or ORF2, we mutated stop codons into the CrPV-2 cDNA. Stop codons at nucleotides C971T and G974T within ORF1 of CrPV-2 (CrPV-2-ORF1-STOP) should prevent the synthesis of viral nonstructural proteins, including the viral protease, the RNA helicase, and RNA-dependent RNA polymerase (RdRp). We inserted two stop codons to prevent translational read-through. Conversely, a stop codon at nucleotide C6428T within ORF2 (CrPV-2-ORF2-STOP) should halt synthesis of viral structural proteins. In vitro translation using Sf-21 extracts demonstrated that the stop codons introduced into ORF1 or ORF2 of CrPV-2 prevented synthesis of the viral nonstructural or structural proteins (Fig. 4, lanes 5 and 6). Specifically, compared to reaction mixtures containing CrPV-2 or CrPV-3, the CrPV-2-ORF1-STOP resulted in loss of expression of all proteins except for a protein that migrated at ∼110 kDa (Fig. 4, lanes 4 and 5). Because CrPV-2-ORF1-STOP prevents expression of all ORF1 proteins and, in particular, the 3C-like protease, the ∼110-kDa protein is presumably the unprocessed ORF2 polyprotein. Furthermore, the proteins expressed in reaction mixtures containing CrPV-2-ORF2-STOP likely represent a combination of processed and unprocessed ORF1 proteins (Fig. 4, lane 6). Moreover, the lack of expression of proteins at 70 and 35 kDa in the CrPV-2-ORF2-STOP reaction products confirms that these proteins represent ORF2 structural proteins. Importantly, we annotated the viral proteins expressed in vitro based on 35S-labeled lysates from CrPV-infected cells and previous reports showing the migration of CrPV structural proteins (Fig. 4, lane 2) (21, 22, 38). Thus, we conclude that CrPV-2 and CrPV-3 can express all known CrPV proteins in vitro.
FIG 4.
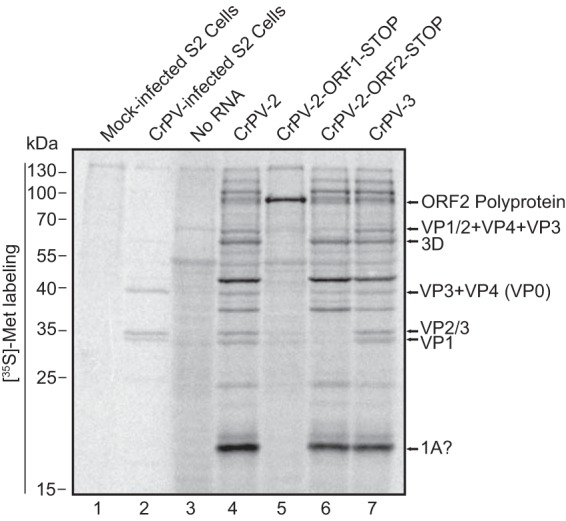
CrPV-2 and CrPV-3 RNAs produce viral proteins in vitro. In vitro-synthesized RNA from CrPV-2, CrPV-3, CrPV-2-ORF1-STOP, or CrPV-2-ORF2-STOP was incubated in Sf-21 translation extracts for 2 h at 30°C in the presence of [35S]methionine-cysteine. Reaction products were resolved by SDS-PAGE, and radioactive proteins were visualized by phosphorimager analysis. As a control, lysates from [35S]methionine-cysteine-labeled mock- and CrPV-infected S2 cells were resolved by SDS-PAGE in parallel (lanes 1 and 2). Shown are representative gels from at least three independent experiments.
We next asked whether CrPV-2 and CrPV-3 are infectious. Transfection of S2 cells with in vitro-transcribed CrPV-2 and CrPV-3 RNA resulted in cytopathic effects (CPE) that include membrane blebbing, detachment of cells from the substratum, cell clumping, and cell lysis at 48 h posttransfection (hpt), which are phenotypes also observed in cells infected with CrPV (Fig. 5A). Northern blot analysis showed CrPV-2 and CrPV-3 viral RNA accumulated over time, strongly suggesting that the infectious clones can replicate in S2 cells. Interestingly, transfection of CrPV-3 RNA reproducibly accumulated at an earlier time point than that of CrPV-2 (Fig. 5B). Furthermore, RT-PCR analysis using tagged primers specific to the negative strand followed by nested PCR using a primer specific to the primer tag demonstrated that cells transfected with CrPV-2 or CrPV-3 RNA or infected with CrPV result in synthesis of negative-sense viral RNA at 24 hpt (Fig. 5C). The use of the tagged-primer RT-PCR approach was necessary as the infectious CrPV-2 and CrPV-3 RNA led to false priming in the RT reaction in the absence of a primer, thus producing an RT-PCR product (data not shown), which is similar to that observed for dengue virus, hepatitis C virus, and alphavirus RNA (39–41). Finally, expression of both nonstructural (3CD) and structural (VP2) viral proteins was detected by immunoblot analysis at 48 hpt (Fig. 5D). In summary, these results collectively demonstrate that CrPV-2 and CrPV-3 can replicate and produce viral proteins in S2 cells.
FIG 5.

Transfection of in vitro-transcribed CrPV-2 and CrPV-3 RNAs in S2 cells. (A) Phase-contrast images of S2 cells mock transfected, CrPV infected (MOI of 0.4), or transfected with in vitro-transcribed CrPV-2, CrPV-3, CrPV-2-ORF1-STOP, or CrPV-2-ORF2-STOP RNA at 48 h posttransfection (hpt). (B) Northern blots of CrPV RNA genome from RNA isolated from CrPV-infected S2 cells or cells transfected with the indicated CrPV genomic RNA. Methylene blue staining of blots are shown below. (C) RT-PCR analysis of CrPV negative-strand (−) synthesis from RNA of cells transfected with the indicated CrPV genomic RNA or CrPV-infected at 24 hpt. Results of reactions in the presence (+RT) or absence (−RT) of reverse transcriptase are shown. (D) Western blots of viral 3CD (ORF1) and VP2 (ORF2) from lysates of cells transfected with the indicated CrPV genomic RNA or CrPV infected at 48 hpt.
To determine whether replication by the infectious clones is dependent on the synthesis of viral proteins, we transfected the CrPV-2 stop codon mutant RNAs and assessed the formation of CPE, viral RNA replication, and viral protein synthesis. As expected, S2 cells transfected with CrPV-2-ORF1-STOP RNA failed to display CPE as this mutation prevents expression of nonstructural proteins (Fig. 4, 5A). Interestingly, transfection of CrPV-2-ORF2-STOP RNA resulted in CPE, suggesting that expression of ORF1 proteins can still induce CPE (Fig. 5A). To confirm these results, Northern blot analysis showed no accumulation of viral RNA over time for CrPV-2-ORF1-STOP. However, transfection of the CrPV-2-ORF2-STOP led to a constant level of viral RNA, presumably due to the ongoing translation of the RdRp within ORF1 (Fig. 5B). Accordingly, RT-PCR analysis detected negative-sense viral RNAs in CrPV-2-ORF2-STOP-transfected cells (Fig. 5C). In contrast, CrPV-2-ORF1-STOP RNA-transfected cells produced very low levels of negative-strand viral RNA (Fig. 5C). The faint negative-sense band observed in CrPV-2-ORF1-STOP reaction products is likely attributed to the fact that negative-sense RNA is also detected by RT-PCR using in vitro-transcribed RNA only as a template (data not shown). Thus, these results suggest that replication is not occurring in CrPV-2-ORF1-STOP-transfected cells. Viral nonstructural (3CD antibody) and structural proteins (VP2 antibody) were not detected by Western blotting in either CrPV-2-ORF1-STOP- or CrPV-2-ORF2-STOP-transfected cells (Fig. 5D). The observation that the nonstructural protein RdRp (3CD) is absent in CrPV-ORF-2-STOP RNA-transfected cells despite displaying negative-strand synthesis and CPEs indicates that multiple rounds of infection after transfection are needed to detect viral proteins whereas RT-PCR analysis is sensitive enough for detection. In fact, in cells transfected with CrPV-2 or CrPV-3, viral proteins are not detected until 48 hpt (Fig. 5D). In summary, transfection of the CrPV-2 and CrPV-3 clones in S2 cells can lead to CPE, viral negative-strand synthesis, and viral protein expression.
S2 cells transfected with CrPV-2 or CrPV-3 RNA produce infectious virions.
To determine whether virions are produced, particles were isolated from CrPV-2- or CrPV-3-transfected S2 cells at 24 h posttransfection. Transmission electron microscopy revealed the presence of virus-like particles in both cases with a diameter of approximately 26 nm and an icosahedral shape, which is similar to characteristics of particles isolated from CrPV-infected S2 cells (Fig. 6A).
FIG 6.

Transfected CrPV RNA clones produce infectious viral particles. (A) Negatively stained electron micrographs of viral particles purified from CrPV-infected S2 cells or from S2 cells transfected with CrPV-2 or CrPV-3 RNA at 48 h posttransfection. Scale bar, 100 nm. (B) Transwell assay. S2 cells transfected with the indicated in vitro-transcribed CrPV genomic RNA or infected with CrPV at an MOI of 0.4 for 24 h were seeded on a 0.4-μm transwell insert, which overlays naive S2 cells at the bottom of the well. Cells were then incubated for 24 or 48 h. Cells were analyzed by indirect immunofluorescence using anti-CrPV VP2, and the nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI). Shown are representative images from at least three independent experiments.
To address whether the CrPV-2 and CrPV-3 viral particles produced by cells are infectious, we employed a two-pronged approach. First, a transwell assay coupled with immunofluorescence was used to determine if cells transfected with CrPV-2 or CrPV-3 RNA can infect naive cells. Transfected or infected cells were seeded on the top of the transwell insert, which overlays naive S2 cells at the bottom of the well. Particles produced by the transfected cells will traverse the transwell membrane and infect the naive cells. We monitored infection of naive cells by expression of the structural VP2 protein using immunofluorescence analysis. As expected, cells transfected with CrPV-2 RNA or CrPV-3 RNA or infected with CrPV displayed VP2 expression, whereas mock-transfected cells or cells transfected with CrPV-2-ORF1-STOP or CrPV-2-ORF2-STOP RNA did not (Fig. 6B). Second, we measured viral titers over time of cells transfected with CrPV-2 or CrPV-3 RNA (Fig. 7). As expected, clones containing stop codons in either ORF1 or ORF2 produced no detectable titer up to 48 hpt. In contrast, infectious virions could be detected as early as 24 hpt for cells transfected with CrPV-2 or CrPV-3 RNA. Interestingly, a significantly larger amount of CrPV-3 was detected than of CrPV-2 at both 24 and 48 hpt (Fig. 7). This may reflect the earlier accumulation of CrPV-3 viral RNA observed previously by Northern blotting (Fig. 5B). Altogether, these results strongly demonstrate that the CrPV-2 and CrPV-3 RNAs are infectious in S2 cells.
FIG 7.
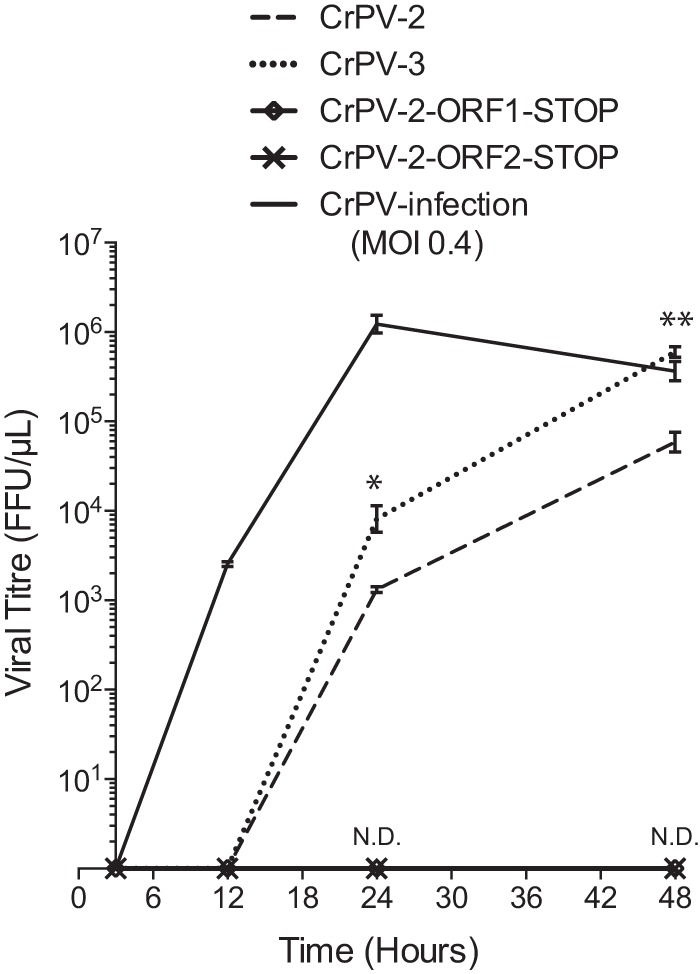
S2 Cells transfected with in vitro-transcribed CrPV-2 and CrPV-3 RNAs accumulate infectious virions over time. A total of 2.5 × 106 S2 cells were transfected with 3 μg of the indicated in vitro-transcribed genomic RNA or infected with CrPV (MOI of 0.4). Titers were measured as described in Materials and Methods at 3, 12, 24, and 48 hpt. Shown are averages from at least three independent experiments (± standard deviations). *, P < 0.05; **, P < 0.005; ND, not detected.
Infection by CrPV-2 and CrPV-3 clones is not dependent on helper viruses.
Insect RNA viruses are notoriously difficult to clone, as illustrated in a recent review (42). A main challenge is that insect cell lines are commonly persistently infected with RNA viruses, such as flock house virus (FHV) and Drosophila X virus (DXV), which can lead to artifacts in the development of infectious viral clones (42). To determine if CrPV-2 and CrPV-3 can recapitulate infection in the absence of other viruses, we first tested for the presence of three viruses commonly found in S2 cells lines, DXV, FHV, and Drosophila C virus (DCV; another dicistrovirus) (43–46). RT-PCR analysis of S2 cells within our lab (University of British Columbia [UBC] S2 cells) against the DCV genome, DXV genome segment B, or the FHV genome segment RNA1 tested positive for the presence of DXV and FHV (Fig. 8A). In contrast, another Drosophila cell line, Kc167, tested positive for only DXV while S2 cells from Invitrogen were negative for all three viruses, as previously reported (Fig. 8A) (45). We next tested the capacity for CrPV-2 and CrPV-3 to replicate in Kc167 and Invitrogen S2 cells. In both cases, cells transfected with either CrPV-2 or CrPV-3 RNA accumulated viral RNA over time as analyzed by Northern blotting, albeit the accumulation was delayed compared to that in our stock of S2 cells (UBC S2 cells) (compare Fig. 5B to 8B). Conversely, cells transfected with CrPV-ORF1-STOP RNA did not accumulate viral RNA over time for either Kc167 or Invitrogen S2 cells (Fig. 8B). Viral titers of Kc167 and Invitrogen S2 cells transfected with CrPV-2 or CrPV-3 RNA produced infectious virions (Table 2). In agreement with UBC S2 cells, CrPV-3 had a reproducibly higher titer than CrPV-2. Taken together, the replication of CrPV-2 and CrPV-3 in S2 cells is not dependent on these viruses.
FIG 8.

CrPV infectious clones can infect Drosophila S2 cells devoid of Drosophila C virus, flock house virus, and Drosophila X virus. (A) Detection of Drosophila C virus (DCV), flock house virus (FHV), and Drosophila X virus (DXV) by RT-PCR analysis in UBC S2 cells, Kc167 cells, Invitrogen S2 cells, and S2 cells from the Drosophila Genomics Resource Center. (B) Northern blots of CrPV RNA genome from RNA isolated from Kc167 cells and S2 cells from Invitrogen that were transfected with the indicated CrPV genomic RNA. The presence (+) or absence (−) of DCV, FHV, and DXV is indicated for each cell line. Methylene blue staining of blots is shown below.
TABLE 2.
Titers of CrPV-2, CrPV-3, and CrPV-2-ORF1-STOP in Invitrogen S2 cells and Kc167 cells
| Cell type (time point) | Transfected RNA | Titer (FFU/μl)a |
|---|---|---|
| Invitrogen S2 (48 hpt) | CrPV-2 | 2.34 × 104 |
| CrPV-3 | 5.47 × 104 | |
| CrPV-2-ORF1-STOP | ND | |
| Kc167 (96 hpt) | CrPV-2 | 1.05 × 105 |
| CrPV-3 | 2.09 × 106 | |
| CrPV-2-ORF1-STOP | ND |
Data are the average titers determined from duplicate experiments. ND, not detected.
CrPV-2 virions are infectious in Drosophila melanogaster.
CrPV can infect Drosophila, which has served as a model system to study virus-host interactions (45, 47–50). To determine whether CrPV-2 and CrPV-3 are infectious in adult Drosophila other than in tissue culture cells, we first propagated CrPV-2 and CrPV-3 virus after transfection in S2 cells. We also used CrPV that has been propagated in S2 cells. A total of 5,000 fluorescent focus-forming units (FFU) of CrPV, CrPV-2, CrPV-3, or UV-inactivated viruses or PBS was injected intrathoracically into D. melanogaster (iso-w1118). Mortality was observed in adult flies injected with CrPV, CrPV-2, and CrPV-3 at 2 days postinjection (Fig. 9). By day 4, all flies injected with either CrPV, CrPV-2, or CrPV-3 succumbed to death; however, those flies injected with PBS or UV-inactivated virus survived and remained healthy (Fig. 9). These results strongly suggest that, like CrPV, CrPV-2 and CrPV-3 virions are infectious to adult D. melanogaster.
FIG 9.
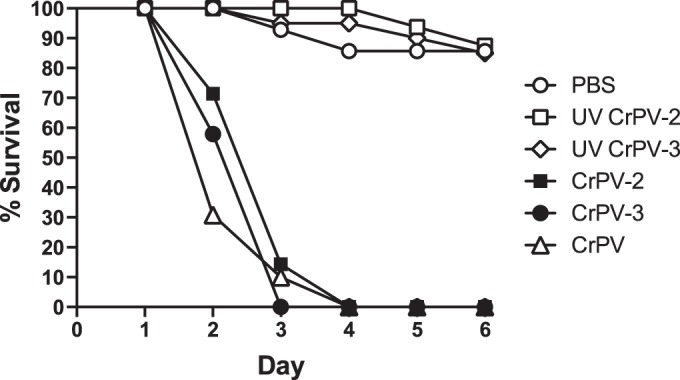
Injection of CrPV-2 and CrPV-3 virions into adult Drosophila melanogaster flies. Virgin Iso w1118 flies (10 males and 10 females) were injected intrathoracically with 5,000 FFU of CrPV, CrPV-2, CrPV-3, or UV-inactivated CrPV-2 and CrPV-3 or with PBS. Subsequently, flies were flipped onto standard medium, and survival was monitored daily.
The duplication in the 5′ UTR of CrPV-2 is stable and reduces viral fitness.
The observation that the 5′ UTR containing the duplication stimulated translation prompted us to test the hypothesis that CrPV may have acquired this duplication to influence viral fitness. Strikingly, cells transfected with CrPV-3 RNA accumulated viral RNA much earlier and had significantly higher viral titers than cells transfected with CrPV-2 RNA (Table 2; Fig. 5B and 7). Altogether, the duplication within the 5′ UTR of CrPV-2 retarded viral RNA synthesis and overall production of infectious virions compared to CrPV-3 virus production despite having a stimulatory effect on translation.
It is possible that the fitness loss seen in CrPV-2 is due to either a defect in viral RNA replication, translation, egress, or entry. To determine if the fitness loss is due to a disruption in the first replication cycle of CrPV, we infected S2 cells with CrPV-2 or CrPV-3 at an MOI of 10 and monitored viral protein and RNA synthesis (Fig. 10A). Viral protein synthesis levels were similar for both CrPV-2 and CrPV-3 as detected by [35S]Met-Cys pulse labeling (Fig. 10A). Despite the enhanced translation from the 5′ UTR+Dup observed with reporter constructs, there was no visible enhancement in the production of ORF1 proteins during CrPV-2 infection by immunoblot analysis (Fig. 10A). Furthermore, there was no detectable difference in levels of RNA accumulation between CrPV-2 and CrPV-3 as visualized by Northern blotting (Fig. 10A). Altogether, this suggests that there is no defect in viral RNA replication or translation between CrPV-2 and CrPV-3 infections in S2 cells. To determine if the phenotype exhibited by CrPV-2 occurs during the subsequent rounds of infection, we infected cells at an MOI of 1 and measured viral titers over time (Fig. 10B). At 6 and 9 h p.i., titers for CrPV-2 and CrPV-3 were not significantly different; however, by 12 h p.i., the titer was reproducibly higher for CrPV-3 than for CrPV-2 and continued to generate 1-log-fold more virus than CrPV-2 by 24 h p.i., consistent with previous observations of cells transfected with CrPV infectious clones (compare Fig. 7 and 10B). To ensure that the 5′ UTR duplication in CrPV-2 is stable and is not simply lost during the course of infection, we passaged CrPV-2 and CrPV-3 in S2 cells for 5 passages and performed RT-PCR analysis of the 5′ UTR. Our results showed that the 5′ UTR duplication is stable in CrPV-2 and that CrPV-3 does not gain the duplication during these passages (Fig. 10C). Overall, we conclude that the duplication in the 5′ UTR of CrPV-2, despite having increased translational activity in the context of reporter constructs, reduces viral fitness compared to that of a virus lacking this extra element. Furthermore, this fitness loss likely does not occur at the replication step of the CrPV life cycle but may occur at another step, such as viral packaging or entry into uninfected cells.
FIG 10.

The CrPV-2 5′ UTR reduces viral fitness. (A) S2 cells were mock infected or infected with CrPV-2 or CrPV-3 (MOI of 10). Cells were metabolically labeled with [35S]methionine-cysteine for the last 30 min of infection. Lysates were subjected to SDS-PAGE, immunoblotting, or Northern blot analysis. Accumulation of viral RNA was monitored by probing for CrPV RNA genome. Expression of CrPV ORF1 was assessed by anti-RdRp antibody. (B) S2 cells were infected with CrPV-2 or CrPV-3 (MOI of 1), and viral titers were measured as described in Materials and Methods at 6, 9, 12, and 24 h p.i. **, P < 0.005; ***, P < 0.0005. Shown are averages from at least three independent experiments (± standard deviations). (C) RT-PCR analysis of the CrPV-2 and CrPV-3 5′ UTRs after serial passage in S2 cells. Cells were transfected with either CrPV-2 or CrPV-3 RNA and virus was harvested (passage 0 [P0]). Cells were then infected at an MOI of 10 and passaged in S2 cells 5 times (P1 to P5). At each passage, RNA was isolated and subjected to RT-PCR analysis.
DISCUSSION
Despite the identification of several members of the Dicistroviridae family (21, 51–56), the establishment of an infectious clone has been elusive (42). In this study, we generated infectious molecular clones of CrPV, termed CrPV-2 and CrPV-3 (Fig. 3A). Several lines of evidence support this conclusion. Transfection of the CrPV-2 or CrPV-3 RNA in Drosophila S2 cells results in (i) cytopathic effects, (ii) expression of viral nonstructural and structural proteins as detected by Western blotting, (iii) synthesis of negative-strand viral RNA as detected by tagged RT-PCR analysis, and (iv) accumulation of viral RNA and infectivity over time as detected by Northern blotting and viral titers. Furthermore, (v) particles produced from CrPV-2- and CrPV-3-transfected S2 cells are infectious in naive cells, as determined using a transwell assay, and (vi) the CrPV-2 and CrPV-3 infectious particles resemble the shape and size of natural CrPV virions as determined by EM. (vii) Importantly, insertion of stop codons within the viral coding regions of CrPV-2 attenuated virus infectivity. (viii) Finally, both CrPV-2 and CrPV-3 virions produced from S2 cells are infectious, resulting in mortality when injected into adult Drosophila. In summary, we have conclusively demonstrated the generation of a robust dicistrovirus molecular clone that can be propagated in Drosophila S2 cells. Given that many studies use dicistroviruses as a model system, the CrPV-2 and CrPV-3 clones should provide useful tools to study the life cycle of dicistroviruses and host-virus interactions in insects.
During infection, RNA viruses largely exist as quasispecies (28); thus, the accumulation of nucleotide changes that deviate from the published sequence is not unexpected. CrPV-2 and CrPV-3 contain several nucleotide changes that likely evolved during viral propagation in S2 cells (Table 1; Fig. 3B). None of the changes observed in either CrPV-2 or CrPV-3 are found in known active sites although the majority occur within the viral RdRp (3D), which may reflect changes in the RdRp for optimal RNA replication in S2 cells.
The most striking difference between CrPV-2 and the published CrPV sequence is the presence of a 196-nucleotide duplication within the 5′ UTR (Table 1; Fig. 3B). How did this duplication come about within the CrPV 5′ UTR? RNA recombination occurs frequently in some positive-sense ssRNA viruses during genome replication and can drive viral evolution (57). The 5′ UTR duplication may have arisen from a template-switching event during genome replication and may have been subsequently selected for as a fitness gain. To our surprise, viral infectivity of CrPV-2 is reduced, not enhanced, compared to the infectivity of a clone of CrPV that lacks the 5′ UTR duplication (CrPV-3) even though the 5′ UTR+Dup stimulates translation (Fig. 2, 7, 8B, and 10B). CrPV-2 produced 1-log-fold fewer infectious particles than CrPV-3 upon RNA transfection and infection at a low MOI, supporting the notion that infection is reduced with the duplication in the 5′ UTR (Fig. 7 and 10B). In transfected S2 cells, CrPV-3 RNA accumulated earlier than CrPV-2 RNA, suggesting that there is a reduction in viral replication when the duplication is present (Fig. 5B). Strikingly, in cells that are infected with CrPV-2 at a low MOI, replication appears to halt at 12 h p.i., which is in contrast to that observed in cells that are transfected with CrPV-2 RNA, where replication is seen up to 48 hpt (Fig. 7 and 10B). The reason for this difference is not clear; however, it may be due to the number of rounds of infection that have occurred in each experiment. Interestingly, infection at a high MOI did not produce any observable differences in replication or viral protein synthesis in cells infected with CrPV-2 or CrPV-3 (Fig. 10A). Altogether, these results suggest that the rate-limiting step for CrPV-2 replication may occur at viral packaging or entry. A recent report suggests that the spatial organization of packaging signals within the RNA sequence of ssRNA viruses is important to ensure efficient capsid assembly (58). It is possible that the duplication in the 5′ UTR of CrPV-2 may disrupt the proper binding of capsomers to the viral RNA, thus hindering efficient assembly. Alternatively, entry of some RNA viruses into host cells is dependent on conformational changes in their capsids and RNA. For example, upon exposure to acidic pH in the endosome, rhinovirus capsids undergo a conversion to the porous subviral A particle, which is then followed by a conformational change in the RNA and exit of the genome in the 3′-to-5′ direction into the host cytosol (59, 60). The duplication in the 5′ UTR of CrPV-2 may impede release by interfering with either RNA-protein or RNA-RNA contacts in the capsid. These hypotheses need to be examined further. Finally, the 5′ UTR of CrPV-2 may simply be the consequence of the founder effect. The viral population is subjected to bottlenecks (i.e., inoculum size), allowing founder viruses with suboptimal fitness levels to succeed due to random sampling (61). This effect may explain how a low-fitness strain of CrPV could emerge over a high-fitness strain (e.g., CrPV-2 versus CrPV-3).
Viral infection in S2 cells was established by transfecting in vitro-transcribed CrPV-2 and CrPV-3 RNAs that lacked a 5′ linked VPg and a 3′ poly(A) tail, both of which are naturally present on the CrPV RNA genome (1). It is not surprising that CrPV-2 and CrPV-3 RNAs can replicate in the absence of these elements as they are also dispensable for replication of other RNA viruses (62, 63). However, the absence of a 5′ linked VPg and a poly(A) tail may explain the delay in viral replication in CrPV-transfected cells compared to replication in infected cells (compare Fig. 7 and 10). It remains to be investigated if inclusion of these elements in the CrPV infectious clones facilitates infection.
The 5′ UTR of CrPV contains an IRES that facilitates the translation of viral nonstructural proteins (21). Our results showed that the duplication element within the CrPV-2 5′ UTR stimulated translation only when it was positioned within the 5′ UTR of reporter mRNAs (Fig. 2B and C). This result may not be surprising as it reflects the natural location of this element within the viral genome. How does the duplication element stimulate 5′ UTR translation? In general, the mechanism of translation for the majority of dicistrovirus 5′ UTR IRES is poorly understood. Moreover, the dicistrovirus 5′ UTRs vary in length, and the nucleotide sequences are not well conserved (16, 17). The lack in conservation may be due to the unique translation factor requirements of each dicistrovirus 5′ UTR IRES to drive viral translation in its specific host cells. Indeed, not all 5′ UTR IRES function in all assays: the 5′ UTR IRES of the CrPV and the related dicistroviruses Plautia stali intestine virus and RhPV can each mediate translation in a subset of species-specific translation extracts (21, 29, 31, 64). Reconstitution experiments indicated that the RhPV 5′ UTR IRES absolutely requires the canonical translation factors, eIF1, eIF2, and eIF3, for efficient 48S complex formation and translation initiation and that this complex is stimulated by eIF1A, eIF4A, and eIF4F (15). It is possible that the enhanced translational activity observed with the CrPV 5′ UTR duplication may be due to either an increase in initiation factor or ribosome recruitment or modulation in the flexibility of its RNA structure in order to facilitate translation, the latter of which would be in agreement with previous reports that the RhPV 5′ UTR is unstructured (15, 64). However, the enhanced translation observed from the 5′ UTR in the context of the minigenome reporter appears to be at odds with that observed during infection of CrPV-2 and CrPV-3 (Fig. 10A). It is likely that the minigenome reporter mRNAs are not regulated in the same manner as the viral genome; thus, further investigations are needed to elucidate the role of the duplication in CrPV 5′ UTR translation using the infectious clones.
In conclusion, we have constructed the first infectious clones of a dicistrovirus. The CrPV clones are infectious to both Drosophila S2 cells and adult flies, thus providing invaluable tools in dissecting the molecular mechanisms behind the Dicistroviridae life cycle, pathogenesis, and interactions with the host. Furthermore, the CrPV clones may provide a framework to develop other dicistrovirus infectious clones that have been difficult to obtain. Our in vitro system showed that CrPV-2 and CrPV-3 RNAs are capable of producing CrPV nonstructural and structural proteins and that processing of the polyproteins is dependent on ORF1 protein expression, indicating that the viral 3C protease is active in Sf-21 cell extracts (Fig. 4) (21, 22, 38). As with poliovirus, it may be possible to exploit this in vitro system to investigate CrPV-2 or CrPV-3 replication and assembly (65, 66). Finally, because CrPV infections have a wide host range, the infectious clones can be exploited for biological controls, such as management of insect pests and induction of innate immune responses or expression of antiviral proteins in insects and arthropods.
ACKNOWLEDGMENTS
We acknowledge Erik Tammpere, who performed experiments for the data shown in Fig. 3B. We are grateful for the help with Drosophila injections from John Church and Michael Gordon.
This work was supported by a Canadian Institute of Child Health (CIHR) operating grant to E.J. (MOP-81244) and Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants to L.J.F. and E.J. E.J. is a CIHR new investigator and a Michael Smith Foundation for Health Research (MSFHR) scholar. L.J.F. is the Canada Research Chair in Quantitative Proteomics. C.H.K. is supported by a UBC four-year Ph.D. fellowship.
REFERENCES
- 1.Bonning BC, Miller WA. 2010. Dicistroviruses. Annu Rev Entomol 55:129–150. doi: 10.1146/annurev-ento-112408-085457. [DOI] [PubMed] [Google Scholar]
- 2.Cox-Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA, Quan PL, Briese T, Hornig M, Geiser DM, Martinson V, vanEngelsdorp D, Kalkstein AL, Drysdale A, Hui J, Zhai J, Cui L, Hutchison SK, Simons JF, Egholm M, Pettis JS, Lipkin WI. 2007. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- 3.de Miranda JR, Cordoni G, Budge G. 2010. The Acute bee paralysis virus-Kashmir bee virus-Israeli acute paralysis virus complex. J Invertebr Pathol 103:S30–S47. doi: 10.1016/j.jip.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Gallai N, Salles J-M, Settele J, Vaissière BE. 2009. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol Econom 68:810–821. doi: 10.1016/j.ecolecon.2008.06.014. [DOI] [Google Scholar]
- 5.Bonami JR, Hasson KW, Mari J, Poulos BT, Lightner DV. 1997. Taura syndrome of marine penaeid shrimp: characterization of the viral agent. J Gen Virol 78:313–319. [DOI] [PubMed] [Google Scholar]
- 6.Lightner DV, Redman RM. 1998. Strategies for the control of viral diseases of shrimp in the Americas. Fish Pathol 33:165–180. doi: 10.3147/jsfp.33.165. [DOI] [Google Scholar]
- 7.Muscio OA, LaTorre JL, Scodeller EA. 1987. Small nonoccluded viruses from triatomine bug Triatoma infestans (Hemiptera: Reduviidae). J Invertebr Pathol 49:218–220. doi: 10.1016/0022-2011(87)90163-7. [DOI] [PubMed] [Google Scholar]
- 8.Reinganum C, O'Loughlin GT, Hogan TW. 1970. A nonoccluded virus of the field crickets Teleogryllus aceanicus and T. commodus (Orthoptera: Gryllidae). J Invertebr Pathol 16:214–220. doi: 10.1016/0022-2011(70)90062-5. [DOI] [Google Scholar]
- 9.Sabin LR, Hanna SL, Cherry S. 2010. Innate antiviral immunity in Drosophila. Curr Opin Immunol 22:4–9. doi: 10.1016/j.coi.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson JE, Pestova TV, Hellen CU, Sarnow P. 2000. Initiation of protein synthesis from the A site of the ribosome. Cell 102:511–520. doi: 10.1016/S0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- 11.Pfingsten JS, Costantino DA, Kieft JS. 2006. Structural basis for ribosome recruitment and manipulation by a viral IRES RNA. Science 314:1450–1454. doi: 10.1126/science.1133281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costantino DA, Pfingsten JS, Rambo RP, Kieft JS. 2008. tRNA-mRNA mimicry drives translation initiation from a viral IRES. Nat Struct Mol Biol 15:57–64. doi: 10.1038/nsmb1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jan E, Kinzy TG, Sarnow P. 2003. Divergent tRNA-like element supports initiation, elongation, and termination of protein biosynthesis. Proc Natl Acad Sci U S A 100:15410–15415. doi: 10.1073/pnas.2535183100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pestova TV, Hellen CU. 2003. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev 17:181–186. doi: 10.1101/gad.1040803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terenin IM, Dmitriev SE, Andreev DE, Royall E, Belsham GJ, Roberts LO, Shatsky IN. 2005. A cross-kingdom internal ribosome entry site reveals a simplified mode of internal ribosome entry. Mol Cell Biol 25:7879–7888. doi: 10.1128/MCB.25.17.7879-7888.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts LO, Groppelli E. 2009. An atypical IRES within the 5′ UTR of a dicistrovirus genome. Virus Res 139:157–165. doi: 10.1016/j.virusres.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 17.Jan E. 2006. Divergent IRES elements in invertebrates. Virus Res 119:16–28. doi: 10.1016/j.virusres.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Benjeddou M, Leat N, Allsopp M, Davison S. 2002. Development of infectious transcripts and genome manipulation of black queen-cell virus of honey bees. J Gen Virol 83:3139–3146. [DOI] [PubMed] [Google Scholar]
- 19.Boyapalle S, Beckett RJ, Pal N, Miller WA, Bonning BC. 2008. Infectious genomic RNA of Rhopalosiphum padi virus transcribed in vitro from a full-length cDNA clone. Virology 375:401–411. doi: 10.1016/j.virol.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 20.Pal N, Boyapalle S, Beckett RJ, Miller WA, Bonning BC. 2014. A baculovirus-expressed dicistrovirus that is infectious to aphids. J Virol 88:3610. doi: 10.1128/JVI.03821-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson JE, Powell MJ, Hoover SE, Sarnow P. 2000. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol Cell Biol 20:4990–4999. doi: 10.1128/MCB.20.14.4990-4999.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrey JL, Lee YY, Au HHT, Bushell M, Jan E. 2010. Host and viral translational mechanisms during cricket paralysis virus infection. J Virol 84:1124–1138. doi: 10.1128/JVI.02006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krishna NK, Marshall D, Schneemann A. 2003. Analysis of RNA packaging in wild-type and mosaic protein capsids of flock house virus using recombinant baculovirus vectors. Virology 305:10–24. doi: 10.1006/viro.2002.1740. [DOI] [PubMed] [Google Scholar]
- 24.Klock HE, Lesley SA. 2009. The polymerase incomplete primer extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol 498:91–103. doi: 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- 25.Wang QS, Au HH, Jan E. 2013. Methods for studying IRES-mediated translation of positive-strand RNA viruses. Methods 59:167–179. doi: 10.1016/j.ymeth.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Brasey A, Lopez-Lastra M, Ohlmann T, Beerens N, Berkhout B, Darlix JL, Sonenberg N. 2003. The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J Virol 77:3939–3949. doi: 10.1128/JVI.77.7.3939-3949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy G, Miron M, Khaleghpour K, Lasko P, Sonenberg N. 2004. The Drosophila poly(A) binding protein-interacting protein, dPaip2, is a novel effector of cell growth. Mol Cell Biol 24:1143–1154. doi: 10.1128/MCB.24.3.1143-1154.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lauring AS, Andino R. 2010. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woolaway KE, Lazaridis K, Belsham GJ, Carter MJ, Roberts LO. 2001. The 5′ untranslated region of Rhopalosiphum padi virus contains an internal ribosome entry site which functions efficiently in mammalian, plant, and insect translation systems. J Virol 75:10244–10249. doi: 10.1128/JVI.75.21.10244-10249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masoumi A, Hanzlik TN, Christian PD. 2003. Functionality of the 5′- and intergenic IRES elements of cricket paralysis virus in a range of insect cell lines, and its relationship with viral activities. Virus Res 94:113–120. doi: 10.1016/S0168-1702(03)00139-4. [DOI] [PubMed] [Google Scholar]
- 31.Shibuya N, Nakashima N. 2006. Characterization of the 5′ internal ribosome entry site of Plautia stali intestine virus. J Gen Virol 87:3679–3686. doi: 10.1099/vir.0.82193-0. [DOI] [PubMed] [Google Scholar]
- 32.Khromykh AA, Westaway EG. 1994. Completion of Kunjin virus RNA sequence and recovery of an infectious RNA transcribed from stably cloned full-length cDNA. J Virol 68:4580–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryan RM, Morris RE, Rice WR, Ciraolo G, Whitsett JA. 1989. Binding and uptake of pulmonary surfactant protein (SP-A) by pulmonary type II epithelial cells. J Histochem Cytochem 37:429–440. doi: 10.1177/37.4.2926121. [DOI] [PubMed] [Google Scholar]
- 34.Sumiyoshi H, Hoke CH, Trent DW. 1992. Infectious Japanese encephalitis virus RNA can be synthesized from in vitro-ligated cDNA templates. J Virol 66:5425–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai CJ, Zhao BT, Hori H, Bray M. 1991. Infectious RNA transcribed from stably cloned full-length cDNA of dengue type 4 virus. Proc Natl Acad Sci U S A 88:5139–5143. doi: 10.1073/pnas.88.12.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyer JC, Haenni AL. 1994. Infectious transcripts and cDNA clones of RNA viruses. Virology 198:415–426. doi: 10.1006/viro.1994.1053. [DOI] [PubMed] [Google Scholar]
- 37.Li D, Aaskov J, Lott WB. 2011. Identification of a cryptic prokaryotic promoter within the cDNA encoding the 5′ end of dengue virus RNA genome. PLoS One 6:e18197. doi: 10.1371/journal.pone.0018197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore NF, Kearns A, Pullin JSK. 1980. Characterization of cricket paralysis virus-induced polypeptides in Drosophila cells. J Virol 33:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peyrefitte CN, Pastorino B, Bessaud M, Tolou HJ, Couissinier-Paris P. 2003. Evidence for in vitro falsely-primed cDNAs that prevent specific detection of virus negative strand RNAs in dengue-infected cells: improvement by tagged RT-PCR. J Virol Methods 113:19–28. doi: 10.1016/S0166-0934(03)00218-0. [DOI] [PubMed] [Google Scholar]
- 40.Plaskon NE, Adelman ZN, Myles KM. 2009. Accurate strand-specific quantification of viral RNA. PLoS One 4:e7468. doi: 10.1371/journal.pone.0007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gunji T, Kato N, Hijikata M, Hayashi K, Saitoh S, Shimotohno K. 1994. Specific detection of positive and negative stranded hepatitis C viral RNA using chemical RNA modification. Arch Virol 134:293–302. doi: 10.1007/BF01310568. [DOI] [PubMed] [Google Scholar]
- 42.Carillo-Tripp J, Bonning BC, Miller WA. 24 November 2014. Challenges associated with research on RNA viruses of insects. Curr Opin Insect Sci doi: 10.1016/j.cois.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 43.Flynt A, Liu N, Martin R, Lai EC. 2009. Dicing of viral replication intermediates during silencing of latent Drosophila viruses. Proc Natl Acad Sci U S A 106:5270–5275. doi: 10.1073/pnas.0813412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li WX, Ding SW. 2010. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci U S A 107:1606–1611. doi: 10.1073/pnas.0911353107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nayak A, Berry B, Tassetto M, Kunitomi M, Acevedo A, Deng C, Krutchinsky A, Gross J, Antoniewski C, Andino R. 2010. Cricket paralysis virus antagonizes Argonaute 2 to modulate antiviral defense in Drosophila. Nat Struct Mol Biol 17:547–554. doi: 10.1038/nsmb.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huszar T, Imler JL. 2008. Drosophila viruses and the study of antiviral host-defense. Adv Virus Res 72:227–265. doi: 10.1016/S0065-3527(08)00406-5. [DOI] [PubMed] [Google Scholar]
- 47.Costa A, Jan E, Sarnow P, Schneider D. 2009. The Imd pathway is involved in antiviral immune responses in Drosophila. PLoS One 4:e7436. doi: 10.1371/journal.pone.0007436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kemp C, Mueller S, Goto A, Barbier V, Paro S, Bonnay F, Dostert C, Troxler L, Hetru C, Meignin C, Pfeffer S, Hoffmann JA, Imler JL. 2013. Broad RNA interference-mediated antiviral immunity and virus-specific inducible responses in Drosophila. J Immunol 190:650–658. doi: 10.4049/jimmunol.1102486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Rij RP, Saleh MC, Berry B, Foo C, Houk A, Antoniewski C, Andino R. 2006. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev 20:2985–2995. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang XH. 2006. RNA Interference directs innate immunity against viruses in adult Drosophila. Science 312:452–454. doi: 10.1126/science.1125694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sasaki J, Nakashima N, Saito H, Noda H. 1998. An insect picorna-like virus, Plautia stali intestine virus, has genes of capsid proteins in the 3′ part of the genome. Virology 244:50–58. doi: 10.1006/viro.1998.9094. [DOI] [PubMed] [Google Scholar]
- 52.Mari J, Poulos BT, Lightner DV, Bonami JR. 2002. Shrimp Taura syndrome virus: genomic characterization and similarity with members of the genus cricket paralysis-like viruses. J Gen Virol 83:915–926. [DOI] [PubMed] [Google Scholar]
- 53.Reddy KE, Yoo M-S, Kim Y-H, Kim N-H, Jung H-N, Thao LTB, Ramya M, Doan HTT, Nguyen LTK, Jung S-C, Kang S-W. 2014. Analysis of the RdRp, intergenic and structural polyprotein regions, and the complete genome sequence of Kashmir bee virus from infected honeybees (Apis mellifera) in Korea. Virus Genes 49:137–144. doi: 10.1007/s11262-014-1074-8. [DOI] [PubMed] [Google Scholar]
- 54.Reddy KE, Noh JH, Kim YH, Yoo MS, Doan HT, Ramya M, Jung SC, Quyen DV, Kang SW. 2013. Analysis of the nonstructural and structural polyprotein regions, and complete genome sequences of Israel acute paralysis viruses identified from honeybees (Apis mellifera) in Korea. Virology 444:211–217. doi: 10.1016/j.virol.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 55.Moon JS, Domier LL, McCoppin NK, D'Arcy CJ, Jin H. 1998. Nucleotide sequence analysis shows that Rhopalosiphum padi virus is a member of a novel group of insect-infecting RNA viruses. Virology 243:54–65. doi: 10.1006/viro.1998.9043. [DOI] [PubMed] [Google Scholar]
- 56.Leat N, Ball B, Govan V, Davison S. 2000. Analysis of the complete genome sequence of black queen-cell virus, a picorna-like virus of honey bees. J Gen Virol 81:2111–2119. [DOI] [PubMed] [Google Scholar]
- 57.Simon-Loriere E, Holmes EC. 2011. Why do RNA viruses recombine? Nat Rev Microbiol 9:617–626. doi: 10.1038/nrmicro2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel N, Dykeman EC, Coutts RH, Lomonossoff GP, Rowlands DJ, Phillips SE, Ranson N, Twarock R, Tuma R, Stockley PG. 2015. Revealing the density of encoded functions in a viral RNA. Proc Natl Acad Sci U S A 112:2227–2232. doi: 10.1073/pnas.1420812112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pickl-Herk A, Luque D, Vives-Adrian L, Querol-Audi J, Garriga D, Trus BL, Verdaguer N, Blaas D, Caston JR. 2013. Uncoating of common cold virus is preceded by RNA switching as determined by X-ray and cryo-EM analyses of the subviral A-particle. Proc Natl Acad Sci U S A 110:20063–20068. doi: 10.1073/pnas.1312128110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harutyunyan S, Kumar M, Sedivy A, Subirats X, Kowalski H, Kohler G, Blaas D. 2013. Viral uncoating is directional: exit of the genomic RNA in a common cold virus starts with the poly-(A) tail at the 3′-end. PLoS Pathog 9:e1003270. doi: 10.1371/journal.ppat.1003270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manrubia SC, Escarmís C, Domingo E, Lázaro E. 2005. High mutation rates, bottlenecks, and robustness of RNA viral quasispecies. Gene 347:273–282. doi: 10.1016/j.gene.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 62.Flanegan JB, Petterson RF, Ambros V, Hewlett NJ, Baltimore D. 1977. Covalent linkage of a protein to a defined nucleotide sequence at the 5′-terminus of virion and replicative intermediate RNAs of poliovirus. Proc Natl Acad Sci U S A 74:961–965. doi: 10.1073/pnas.74.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Racaniello VR, Baltimore D. 1981. Cloned poliovirus complementary DNA is infectious in mammalian cells. Science 214:916–919. doi: 10.1126/science.6272391. [DOI] [PubMed] [Google Scholar]
- 64.Groppelli E, Belsham GJ, Roberts LO. 2007. Identification of minimal sequences of the Rhopalosiphum padi virus 5′ untranslated region required for internal initiation of protein synthesis in mammalian, plant and insect translation systems. J Gen Virol 88:1583–1588. doi: 10.1099/vir.0.82682-0. [DOI] [PubMed] [Google Scholar]
- 65.Molla A, Paul AV, Wimmer E. 1991. Cell-free, de novo synthesis of poliovirus. Science 254:1647–1651. doi: 10.1126/science.1661029. [DOI] [PubMed] [Google Scholar]
- 66.Barton DJ, Black EP, Flanegan JB. 1995. Complete replication of poliovirus in vitro: preinitiation RNA replication complexes require soluble cellular factors for the synthesis of VPg-linked RNA. J Virol 69:5516–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]