

ABSTRACT
Severe dengue virus (DENV)-associated diseases can occur in patients who have preexisting DENV antibodies (Abs) through antibody-dependent enhancement (ADE) of infection. It is well established that during ADE, DENV-antibody immune complexes (ICs) infect Fcγ receptor-bearing cells and increase the systemic viral burden that can be measured in the blood. For protection against infection with DENV serotypes 1 to 4, strongly neutralizing Abs must be elicited to overcome the effect of ADE. Clinical observations in infants who have maternal DENV Abs or recent phase II/III clinical trials with a leading tetravalent dengue vaccine suggested a lack of correlation between Ab neutralization and in vivo disease prevention. In addressing this gap in knowledge, we found that inoculation of ICs formed with serotype cross-reactive Abs that are more than 98% neutralized in vitro promotes high mortality in AG129 mice even though peak viremia was lower than that in direct virus infection. This suggests that the serum viremia level is not always correlated with disease severity. We further demonstrated that infection with the ICs resulted in increased vascular permeability, specifically in the small intestine, accompanied with increased tissue viral load and cytokine production, which can be suppressed by anti-tumor necrosis factor alpha (anti-TNF-α) Abs. Flow cytometric analysis identified increased infection in CD11bint CD11cint/hi CD103− antigen-presenting cells by IC inoculation, suggesting that these infected cells may be responsible for the increase in TNF-α production and vascular permeability in the small intestine that lead to mortality in mice. Our findings may have important implications for the development of dengue therapeutics.
IMPORTANCE We examined the relationship between the neutralizing level of Abs at the time of infection and subsequent disease progression in a mouse model in order to understand why patients who are shown to have a neutralizing quantity of Abs still allow sufficient DENV replication to induce severe dengue manifestations, which sometimes do not correlate with viremia level. Strikingly, we found that high mortality was induced in AG129 mice by the increase in TNF-α-induced vascular permeability accompanied by an increased viral load, specifically in the small intestine, even when the initial infection level is suppressed to less than 5% and the peak viremia level is not enhanced. This suggests that ADE overcomes the protective efficacy of Abs in a tissue-dependent manner that leads to severe small intestinal pathology. Our findings may serve to address the pathogenic role of Abs on severe dengue disease and also help to develop safe Ab-based therapeutic strategies.
INTRODUCTION
Dengue virus (DENV) infection with any of the 4 related viral serotypes (DENV1 to DENV4) causes a variety of clinical manifestations, ranging from self-limiting febrile illness, known as dengue fever (DF), to the life-threatening severe diseases, such as dengue hemorrhagic fever (DHF) or dengue shock syndrome (DSS), characterized by vascular leakage, thrombocytopenia, bleeding, and elevated levels of cytokines (1–3). Dengue is emerging as a global public health threat, with an estimated 400 million human infections and several hundred thousand cases of severe dengue occurring yearly (4). Severe dengue diseases are often associated with secondary heterotypic infections or primary infection in the case of infants born from DENV-immune mothers. Preexisting cross-reactive antibodies (Abs) or neutralizing Abs at suboptimal concentrations are hypothesized to enhance DENV infection through the phenomenon termed “antibody-dependent enhancement” (ADE) of infection (5–7). Understanding the detailed disease mechanisms underlying ADE is important in order to develop safe vaccines and other Ab therapeutic interventions for DENV diseases. The study of the pathogenic properties of specific Abs in humans is complicated by various factors: variable precision in grading the severity of dengue cases in different countries where it is endemic, rapid changes in the clinical presentation of patients, and difficulty in obtaining tissue specimens from patients to directly observe the pathology (8). Understanding dengue pathogenesis is therefore challenging, and animal models that recapitulate essential elements of major pathologies of human infection are needed to address the complications associated with severe dengue infections.
ADE is believed to occur through the formation of dengue virus-antibody immune complexes (ICs) that bind to Fcγ receptors on cells, such as monocytes/macrophages and dendritic cells, subsequently facilitating viral entry and replication; this in turn results in an increase in the number of infected cells and systemic viral burden (9, 10). Experimental evidence in support of ADE has been reported in both in vitro and in vivo studies. DENV infection is enhanced in Fcγ receptor-bearing cells in the presence of immune sera or DENV monoclonal Abs in vitro (11–13). It has been reported that nonhuman primates passively immunized with DENV Abs developed a higher level of viremia than that seen with virus-only infection. (12, 14). More recently, in the AG129 mouse model lacking alpha interferon (IFN-α), IFN-β, and IFN-γ receptors, it was demonstrated that by injecting Abs into mice prior to DENV infection, nonlethal illness becomes a fatal disease; it resembles human DHF/DSS accompanied by elevated levels of cytokines, vascular leakage, intestinal bleeding, and thrombocytopenia, as well as increased viremia levels (15–18). These results support the hypothesis of ADE that Abs contribute to enhancement of viral burden and disease severity. However, it remains unclear whether Abs merely increase the systemic level of virus, which is referred to as serum viremia. Although some clinical studies have shown a statistical correlation between high levels of viremia and an increased risk for DHF/DSS (19–21), a more recent clinical study of 75 Vietnamese infants with DHF/DSS has shown significant heterogeneity in viremia and that viremia level is not always associated with clinical severity (22). Since the presence of maternal DENV Abs is associated with developing DHF/DSS in infants (23), it is possible that Abs might have a potential to facilitate disease severity besides responses arising from increased viremia.
Recently, clinical trial results of the leading dengue vaccine candidate, a recombinant live, attenuated tetravalent CYD vaccine (ChimeriVax), have been reported (24, 25). The clinical outcome showed varied efficacy among DENV serotypes; the vaccination failed to protect against DENV2 in a phase 2b trial in Thailand (25) and provided only 35% protection against DENV2 in a phase 3 trial in five Asian countries (24) even though highly neutralizing levels of Ab titers were suggested to be triggered in patients (assessed by the 50% plaque reduction neutralization test [PRNT50]). Although the vaccine efficacy was elevated to 42.3% against DENV2 infection in a phase 3 trial in five Latin American countries (26), these reports, together with the above-mentioned experience of the Vietnamese infants in the 5- to 9-month age bracket, suggest that the presence of Abs at the neutralizing level still allows sufficient DENV replication to induce severe dengue manifestations in patients. This reflects the complexity of the role of Abs in achieving the correct balance between protective efficacy and ADE in vivo.
In this study, we inoculated DENV ICs displaying various levels of neutralization as measured by in vitro assays into AG129 mice and examined the relationships between neutralization level, viremia level, and disease severity. Consequently, we found that inoculation of ICs, which showed a >98% neutralization level in vitro, accompanied with infusion of massive free Abs, still caused systemic infection in mice. Quite surprisingly, the infection caused higher mortality, without enhancement of the peak viremia level, than the virus infection control (no Abs). Furthermore, we found that DENV infection in the presence of a neutralizing quantity of Abs specifically caused severe small intestinal pathology accompanied with increased tissue viral load, cytokine production, and vascular permeability, which later could be therapeutically suppressed by anti-tumor necrosis factor alpha (anti-TNF-α) Abs without causing any mortality.
MATERIALS AND METHODS
Cells, virus strain, and antibodies.
BHK-21 cells (baby hamster kidney fibroblast cells; ATCC) were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S) at 37°C in 5% CO2. C6/36, an Aedes albopictus cell line (ATCC), was maintained in RPMI 1640 medium with 10% FBS, 25 mM HEPES, and 1% P/S at 28°C in the absence of CO2. A mouse-adapted strain, DENV2 S221 (27), was a gift from Sujan Shresta (La Jolla Institute for Allergy and Immunology, California). The S221 strain was grown in C6/36 cells, and the supernatants were stored at −80°C after filtration through a 0.45-μm-pore-size membrane. Virus titer was determined by standard plaque assay on BHK-21 cells. 4G2 (mouse IgG2a, anti-E of all serotypes) and 3H5 (mouse IgG1, anti-E of DENV2) hybridomas were purchased from ATCC and grown in PFHM-II (Gibco) with 1% P/S at 37°C in 5% CO2. The bulk supernatant was collected and filtered through a 0.45-μm-pore-size membrane. 4G2 and 3H5 Abs were then purified through a protein G column by using AKTApurifier UPC 10 (GE Healthcare) according to the manufacturer's instructions. After dialysis against phosphate-buffered saline (PBS), the concentration of Ab was quantified using a NanoDrop spectrophotometer (Thermo Scientific). The humanized 4G2 Ab was constructed using a previously published protocol (28). Monoclonal 6B6C-1 Ab (mouse IgG2a, anti-E of all serotypes) was purchased from Merck Millipore (MAB8744). The mouse IgG2a isotype control Ab was purchased from BioLegend. After dialysis against PBS to remove sodium azide, the concentration of Ab was remeasured before use by using a NanoDrop spectrophotometer.
In vitro and in vivo IC infection.
Sv/129 mice deficient in type I and II interferon (IFN) receptors (AG129), purchased from B&K Universal (United Kingdom), were housed in the biosafety level 2 (BSL-2) animal facility at Duke-NUS, Singapore, and all animal experiments were approved by the Animal Care Committee at Singapore General Hospital/NUS. Six- to 10-week-old mice were used for all in vivo experiments. If not stated otherwise, ICs were preformed by mixing 2 × 104 PFU of S221 with various concentrations of mouse monoclonal Ab, 4G2, 3H5, or 6B6C-1 for 1 h on ice and inoculated intravenously (i.v.) in a total volume of 200 μl into mice. For the in vitro neutralization assay, 1 × 105 BHK-21 cells were incubated with ICs in a total volume of 200 μl for 1 h at 37°C. Cells were then washed once with culture medium, resuspended with 500 μl of culture medium, and incubated for an additional 24 h at 37°C. For the in vitro ADE assay, 1 × 105 THP-1 cells were incubated with ICs in a total volume of 200 μl for 2 h at 37°C. Cells were then washed once with culture medium, replaced with 500 μl of culture medium, and incubated for an additional 48 h at 37°C. The supernatants were subjected to standard plaque assay using BHK-21 cells to determine virus titers.
PRNT.
Fifty-percent plaque reduction neutralization test (PRNT50) values of mouse 4G2, 6B6C-1, and 3H5 were determined by using 100 PFU of S221. S221 (100 PFU/100 μl) was incubated on ice with 0 to 10 μg/100 μl of Ab for 1 h and then added on confluent BHK-21 cells. After 1 h of incubation at 37°C, cells were washed once with culture medium and proceeded to the standard plaque assay. PRNT50 values were calculated using GraphPad Prism software.
Quantification of viral load in serum by quantitative real-time RT-PCR.
Blood samples were collected by submandibular bleeding or from the postcaval vein after euthanasia of mice. Serum viral RNA was extracted using the QIAamp viral RNA minikit (Qiagen) according to the manufacturer's instructions. Real-time reverse transcription (RT)-PCR was carried out in a Bio-Rad real-time thermal cycler CFX96 by the use of the qScript one-step quantitative RT-PCR kit (Quanta) with primers (forward, 5′-CATATTGACGCTGGGAAAGA-3′; and reverse, 5′-AGAACCTGTTGATTCAAC-3′) and TaqMan probe (6-carboxyfluorescein [FAM]-5′-CTGTCTCCTCAGCATCATTCCAGGCA-3′-6-carboxytetramethylrhodamine [TAMRA]) targeting for the DENV2 distal 3′ noncoding region (29). Plasmid containing whole-genome sequences of DENV2 strain 3295 (GenBank accession EU081177.1) was used to make a standard curve for the quantification of the viral genome copy number.
Measurement of viral load and cytokine levels in mouse tissue.
Mice were euthanized by CO2 inhalation and perfused with PBS after blood collection from the postcaval vein. Tissues were collected and snap-frozen in liquid nitrogen after removal of all visible luminal content from the intestine. Frozen tissues were then homogenized by TissueLyser (Qiagen) in PBS, and the supernatants after centrifugation were used to measure tissue viral load and cytokine levels. For the viral load measurement, RNA was extracted from the supernatants using the TRIzol extraction method and subjected to real-time RT-PCR as described above. The values of viral genome numbers in tissues were normalized to 18S rRNA expression levels with mouse 18S primers (forward, 5′-CGGCTACCACATCCAAGGAA-3′; and reverse, 5′-GCTGGAATTACCGCGGCT-3′) and TaqMan probe (FAM-5′-TGCTGGCACCAGACTTGCCCTC-3′-TAMRA). The levels of cytokines (TNF-α, interleukin 1β [IL-1β], IL-6, monocyte chemoattractant protein 1 [MCP-1], IL-10, and IL-12) in serum and tissue homogenates were measured by using Ready-SET-Go! enzyme-linked immunosorbent assay (ELISA) kits (eBioscience) according to the manufacturer's instructions.
Anti-TNF-α antibody treatment.
Rat anti-mouse TNF-α Ab (clone MP6-XT22; eBioscience) was dialyzed against PBS to remove sodium azide and used for the neutralization of TNF-α in mice (18) with modification of the therapeutic regimen. A total of 200 μg of TNF-α Ab was administered intraperitoneally (i.p.) into mice on day 3 postinfection.
Measurement of vascular leakage levels in tissues.
Vascular leakage levels in tissues were measured by Evans blue staining assay as described previously (18, 30) with modifications. Mice were i.p. administered 0.4 ml of Evans blue solution (0.5% in PBS), and 2 h later, mice were sacrificed by CO2 inhalation and perfused with PBS. Tissues were collected into formamide after the removal of all visible luminal content from the intestine. After 24 h of extraction at 37°C, the amount of Evans blue in the extract was quantified by measuring absorbance at 620 nm using a microplate reader, Infinite M200 (Tecan).
Flow cytometry.
Small intestines were collected, opened longitudinally, and washed with PBS to remove the contents. Intestines were then cut into approximately 1.5-cm pieces, placed into 50-ml tubes containing 30 ml Hanks balanced salt solution (HBSS) medium (Gibco) supplemented with 5% FBS, 1% P/S, and 2 mM EDTA (Merck), and shaken at 200 rpm for 30 min at 37°C. After being washed with PBS, tissue pieces were minced by scissors and digested in 20 ml of HBSS medium containing 5% FBS, 1% P/S, and 1.5 mg/ml collagenase VIII (Sigma) at 200 rpm for 15 min at 37°C. The cell suspension was collected, passed through a 70-mm strainer (BD) by pressing with the plunger of a 1-ml syringe (BD), and washed twice with HBSS supplemented with 5% FBS and 1% P/S. Cells were stained with LIVE/DEAD viability reagent (LIVE/DEAD fixable near-IR dead cell stain kit; Invitrogen) according to the manufacturer's instructions and then stained with FcR block/anti-mouse CD16/32 (clone 93; BioLegend). Cells were further stained using FITC anti-mouse CD103 (clone M290; BD Pharmingen) or Alexa Fluor 488 anti-mouse CD31 (clone 390; BioLegend), PerCP anti-mouse I-A/I-E (clone M5/114.15.2; BioLegend), phycoerythrin (PE)-Cy7 anti-mouse CD11c (clone N418; BioLegend), antigen-presenting cell (APC) anti-mouse CD11b (clone M1/70; BioLegend), and Alexa Fluor 700 anti-mouse CD45 (clone 30-F11; BioLegend). Cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Pharmingen), and PBS supplemented with 1% FBS and 0.1% saponin was used for further washing and the Ab dilution process. Cells were stained with anti-NS3 Ab (clone 3F8) (31) or human IgG1 isotype control (Ancell), which were biotinylated using EZ-Link NHS-PEG4-biotin (Thermo Scientific) according to the manufacturer's instructions. After being washed, cells were stained with streptavidin-conjugated PE (BioLegend). Data were obtained using a BD Fortessa and analyzed with FlowJo software (Tree Star). The gate for NS3-positive cells was set at less than 0.1% of the isotype control staining.
DLS assay.
S221 was grown in C6/36 cells and purified as described previously (32). Briefly, virus particles in the supernatant of virus-infected cells were precipitated with 8% polyethylene glycol 8000 (PEG 8000) in NTE buffer (12 mM Tris-Cl [pH 8.0], 120 mM NaCl, 1 mM EDTA). The virus particles were resuspended and centrifuged through a 30% sucrose cushion and further purified by using 10% to 30% potassium tartrate gradient centrifugation. The virus band was collected, buffer exchanged to NTE buffer, and concentrated using an Amicon Ultra-4 centrifugal concentrator (Millipore). The purity of the virus preparation was assessed by a Coomassie blue-stained SDS-PAGE gel (data not shown). Virus titer and absolute genome copy numbers were determined by standard plaque assay and real-time RT-PCR separately as described above. The ratio of virus titer to genome number was determined as comparable to that of the virus stock used for our mouse experiments. ICs were formed by mixing 2 × 104 PFU of the virus with 0 to 200 μg of 4G2 Ab as described above and immediately subjected to dynamic light scattering (DLS) measurement as well as neutralization assay. For DLS size measurement, 20 μl of IC was loaded into a quartz cuvette for analysis by a Zetasizer Nano S machine (Malvern) at 37°C. Samples were measured without any filtration. Data were analyzed using Zetasizer Nano software version 6.01. The deconvolution of the measured correlation curve to size distribution by numbers or intensities was done by using a nonnegative least-squares algorithm. Each generated diameter report is an average from more than 10 readings.
Statistical analysis.
Significant differences between data groups were determined by a 2-tailed Student t test analysis. For mouse survival, statistical analysis was performed by the log-rank test using the GraphPad Prism software. P values less than 0.05 were considered significant.
RESULTS
Inoculation with highly neutralized ICs causes lethal diseases without enhancing the viremia level in mice.
Previous ADE mouse model studies showed that preinjection of DENV Abs followed by next-day inoculation with a mouse-adapted DENV strain significantly increased viremia and mortality (16, 18). As this model clearly showed the effect of ADE in vivo, we used this mouse system for the study of DENV pathogenesis (33) and the evaluation of therapeutic candidates (34–36). In the present study, in order to address the relationship between the neutralizing activity of Abs and dengue disease severity, we take a reductionist approach to examine the conditions where neutralizing levels of Abs at the time of infection cause severe disease progression in mice. For this, we used DENV-Ab immune complexes (ICs) preformed in vitro for infection in mice. The ability of ICs to infect BHK-21 cells was used as a surrogate to score for neutralization, since these cells do not induce Fcγ receptor-mediated infection (37), and furthermore, we used THP-1 to determine the Ab concentrations required for ADE in vitro. ICs were preformed with 2 × 104 PFU of S221 (mouse-adapted DENV2 strain) with various amounts of 4G2 Ab (cross-reactive anti-DENV E Ab) (38) and inoculated into mice. As shown in Fig. 1A, virus inoculation without Abs (virus control) and with 0.01 μg Ab (0.01 μg IC) induced only 10% and 20% mouse mortality, respectively, while 0.1 to 100 μg IC inoculation induced more than 80% mortality (80% for 0.1 μg [P = 0.0043] and 100 μg [P = 0.0043] and 100% for 1 μg [P = 0.0002], 10 μg [P = 0.0002], and 20 μg [P = 0.0005]) by day 6 postinfection. Infection levels in BHK-21 cells showed 1% (for infection with 0.01 μg IC), 49% (for 0.1 μg), 91% (for 1 μg), 98% (for 10 μg), 98% (for 20 μg), and 99.7% (for 100 μg) (Fig. 1B), indicating that inoculation with ICs displaying more than 90% neutralized levels in vitro can cause high mortality in mice. Infection levels in THP-1 cells showed that 0.01 to 0.1 μg IC induced ADE more efficiently than 1 to 100 μg IC (Fig. 1B). Taken together, the BHK-21 data and the THP-1 infection data indicate that highly neutralized virus formed in vitro causes severe diseases in vivo. The viremia level measured by RT-PCR (virus copies/milliliter) showed that 0.01 to 10 μg IC induced enhancement of peak viremia regardless of infection levels at the early stage (days 1 and 2) (Fig. 1C), indicating the clear effect of ADE in the mice. Surprisingly, however, viremia for the 20 μg and 100 μg IC inoculation on day 1 was only 4.2% and 3.6% of that of the virus control, respectively; this indicates that more than 95% of initial infection was restricted by Abs, and the peak viremia level during the subsequent days monitored could not reach as high as that of the virus control (Fig. 1C). Notably, although both 0.01 μg and 0.1 μg IC inoculation induced enhancement of viremia from day 1 and showed similar kinetics throughout (Fig. 1C), 0.01 μg IC caused only 20% mortality, which is comparable to the virus control mortality rate of 10% (P = 0.6038), whereas 0.1 μg IC caused 80% mortality (Fig. 1A). Also, viremia of 20 μg IC and 100 μg IC inoculation was lower than that of 0.01 μg IC at all time points examined (Fig. 1C); however, these IC amounts induced higher mortality (100% for 20 μg IC and 80% for 100 μg IC) than 0.01 μg IC (20%) (Fig. 1A). The calculation of area under the curve (AUC) for viremia showed increased values for 0.01 to 10 μg IC inoculation compared with that of the virus control, as expected from the levels of peak viremia. However, in the case of 20 μg or 100 μg IC inoculation, the AUC was 2.05-fold or 2.54-fold lower than that of the virus control (Table 1). This indicates that the systemic infection level was suppressed in 20 μg and 100 μg IC-inoculated mice even though it caused higher mortality (Fig. 1A). We further tested the 500 μg IC inoculation; however, the mice showed suppressed viremia levels throughout and no mortality was recorded (data shown below). This suggests that the neutralization overcomes the disease progression at this Ab concentration in the case of 4G2. We also confirmed that inoculation with 100 μg Ab alone or 100 μg Ab with UV-inactivated virus (2 × 104 PFU) did not cause any disease symptoms (data not shown), indicating that the mortality observed for mice receiving highly neutralizing ICs is directly related to virus infection.
FIG 1.

Mouse survival rate and kinetics of viremia after inoculation with immune complexes (ICs) having different levels of neutralization. ICs were formed in vitro by mixing 2 × 104 PFU/100 μl of S221 with 0 to 100 μg/100 μl of 4G2 Ab. A total of 200 μl of IC was used for inoculation i.v. into mice and infection in BHK-21 cells or THP-1 cells. (A) Mouse survival rate was monitored until day 10 after inoculation with ICs containing 0 μg, 0.01 μg, 0.1 μg, 1 μg, 10 μg, 20 μg, and 100 μg of Ab. (B) A total of 1 × 105 BHK-21 cells were infected with ICs for 1 h followed by replacement with 500 μl of culture medium and incubation for an additional 24 h. A total of 1 × 105 THP-1 cells were infected with ICs for 2 h followed by replacement with 500 μl of culture medium and incubation for an additional 48 h. Virus titer in the supernatants was measured by standard plaque assay using BHK-21 cells. The percentages of virus titer were calculated based on virus control. Error bars indicate standard deviations from duplicate samples. (C) Blood samples were collected on days 1 to 5 (24, 48, 72, 90, and 114 h) postinfection, and mixed serum from each group was subjected to quantitative real-time RT-PCR to measure virus genome numbers. The results were obtained from two separate experiments, the graphs show the average results from two independent measurements, and error bars indicate standard deviations. The numbers of mice per group are 10 for virus control and 5 for other IC inoculations.
TABLE 1.
Summary of the AUC for viremia until day 5 postinfection
| IC inoculation or Ab preinjection | Virus titer (PFU) | 4G2 Ab amt (μg) | % mouse survival | AUCa |
|---|---|---|---|---|
| IC inoculation | 2 × 104 | 0 | 90 | 1.00 |
| 2 × 104 | 0.01 | 80 | 1.78 | |
| 2 × 104 | 0.1 | 20 | 1.89 | |
| 2 × 104 | 1 | 0 | 2.15 | |
| 2 × 104 | 10 | 0 | 1.58 | |
| 2 × 104 | 20 | 0 | 0.49 | |
| 2 × 104 | 100 | 20 | 0.39 | |
| 1 × 105 | 0 | 0 | 1.00 | |
| 1 × 105 | 20 | 0 | 0.51 | |
| 1 × 104 | 0 | 100 | 1.00 | |
| 1 × 104 | 20 | 33 | 0.31 | |
| 1 × 103 | 0 | 100 | 1.00 | |
| 1 × 103 | 20 | 83 | 0.34 | |
| Ab preinjection | 2 × 104 | 0 | 80 | 1.00 |
| 2 × 104 | 0.2 | 40 | 1.68 | |
| 2 × 104 | 2 | 0 | 2.29 | |
| 2 × 104 | 20 | 0 | 1.67 | |
| 2 × 104 | 100 | 20 | 0.36 | |
| 2 × 104 | 200 | 60 | 0.17 |
AUC values are calculated by the sum of the area of each day until day 5 postinfection by using Microsoft Excel software. Data are expressed as fold increases compared to virus control (0 μg Ab) in each experiment.
Next, ICs formed with different titers of S221 and 20 μg Ab were tested. Infection levels in BHK-21 cells showed 95.5%, 98.8%, and 99.0% reductions, to 1 × 105 PFU, 1 × 104 PFU, and 1 × 103 PFU of IC infection, respectively (data not shown). Mice that received 1 × 105 PFU of IC died earlier than mice that received virus alone, and inoculation with 1 × 104 PFU or 1 × 103 PFU of virus alone did not induce mortality, while 1 × 104 PFU or 1 × 103 PFU of IC caused 67% or 17% mortality, respectively (data not shown). In all cases, peak viremia levels (data not shown) and AUC values until day 5 (Table 1) of IC infection were lower than those of the virus control in spite of earlier or higher mortality. Taken together, these results imply that DENV infection, even when neutralizing quantities of Abs are present, still has a potential to cause severe diseases without any correlation with viremia level in mice.
Inoculation with highly neutralized ICs formed with cross-reactive Abs, but not with serotype-specific Abs, causes lethal diseases.
Severe dengue diseases are often caused by sequential infection with a different serotype but not with infection with the same serotype (39). Therefore, the formation of ICs with preexisting cross-reactive Abs in patients may play a critical role in disease severity (40). To address this, we repeated the immune complex studies with a different cross-reactive monoclonal Ab, 6B6C-1, which recognizes an epitope similar but not identical to that of 4G2 Ab in domain II (fusion loop) of the E protein (41, 42), and serotype-specific monoclonal Ab, 3H5, which recognizes domain III of the DENV2 E protein (43). PRNT50 values of 4G2, 6B6C-1, and 3H5 using 100 PFU of S221 were 3.65 μg, 0.93 μg, and 0.19 μg, respectively, indicating the different neutralizing activities between these 3 Abs (data not shown). For 6B6C-1 ICs, clear enhancement of peak viremia and high mortality were induced by 0.2 μg and 2 μg IC. However, viremia for 20 μg IC on day 1 was restricted to 4.1% of the virus control, and the peak viremia level was observed to be reproducibly lower than the virus control (Fig. 2C) despite higher mortality (Fig. 2A), similar to ICs formed with cross-reactive 4G2 Ab (Fig. 1). For serotype-specific 3H5 Ab, high mortality was not induced by IC inoculation at any Ab concentrations (20% for 0.2 μg IC and 0% for other amounts of IC) (Fig. 2D). Infection levels in BHK-21 cells showed 44%, 94%, 99.3%, and 99.8% reductions in infections with 0.02 μg, 0.2 μg, 2 μg, and 20 μg IC, respectively (Fig. 2E), thereby indicating a higher neutralizing activity of 3H5 than of 4G2 (Fig. 1B) or 6B6C-1 (Fig. 2B). The enhancement of the peak viremia level was not observed at any Ab concentration for 3H5 (Fig. 2F), although 0.02 μg and 0.2 μg IC showed the effect of ADE in THP-1 cells (Fig. 2E). These results may reinforce an important aspect that highly neutralizing serotype-specific Abs are protective against severe diseases in vivo regardless of neutralized levels of ICs, while the presence of cross-reactive Ab causes lethal diseases at a wide range of Ab concentrations.
FIG 2.

Mouse survival rate and kinetics of viremia after inoculation with ICs formed with cross-reactive Ab, 6B6C-1, and serotype-specific Ab, 3H5. ICs were formed by mixing 2 × 104 PFU/100 μl of S221 with 0 to 20 μg/100 μl of 6B6C-1 or 3H5 Ab. A total of 200 μl of IC was used for inoculation i.v. into mice and infection in BHK-21 and THP-1 cells. (A and D) The mouse survival rate was monitored until day 10 after inoculation with ICs containing 0 μg, 0.02 μg, 0.2 μg, 2 μg, and 20 μg of Ab. (B and E) A total of 1 × 105 BHK-21 cells and THP-1 cells were infected with ICs and cultured as described in Fig. 1. The virus titer in the supernatants measured by plaque assay was shown as the percentages calculated based on virus control. Error bars indicate standard deviations from duplicate samples. (C and F) Virus genome numbers in serum on days 1 to 5 were measured as described for Fig. 1. The graphs show the average results from two independent measurements, and error bars indicate standard deviations. The number of mice per group is 5.
Preexisting cross-reactive Abs at high levels of neutralizing activity cause lethal diseases in mice.
To address whether high lethality is reproducibly induced by preexisting Abs at a neutralizing quantity, mice were preinjected intravenously with 0 to 200 μg 4G2 Ab 1 h prior to infection and then inoculated with 2 × 104 PFU of S221. Virus infection control showed 20% mortality, while preinjection of 0.2 to 20 μg Ab induced 60% or 100% mortality (60% for 0.2 μg Ab [P = 0.2207] and 100% for 2 μg Ab [P = 0.0119] and 20 μg Ab [P = 0.008]) (Fig. 3A) accompanied by the enhancement of peak viremia levels (Fig. 3B). Although the survival rates for 100 μg and 200 μg Ab were not at a significant level, these concentrations of Abs still caused higher mortality (80% for 100 μg [P = 0.099] and 40% for 200 μg [P = 0.6071] Ab) than the virus control (20%) (Fig. 3A). Notably, viremia for 100 μg and 200 μg Ab preinjection on day 1 was limited to 5.9% and 4.5% of that of the virus control, respectively (Fig. 3B), indicating that the inoculated virus was highly neutralized through IC formation in vivo, and their peak viremia levels (Fig. 3B) and AUC values until day 5 (Table 1) were observed to be lower than those of the virus control. Inoculation with 200 μg of isotype control Ab (mouse IgG2a) did not affect mouse survival and viremia level (data not shown). These results confirm that even under conditions where preexisting circulating Abs at the time of infection result in more than 95% neutralization, there is still a potential to cause severe diseases in mice without increasing the viremia level, which is similar to inoculation with highly neutralized in vitro-formed ICs.
FIG 3.
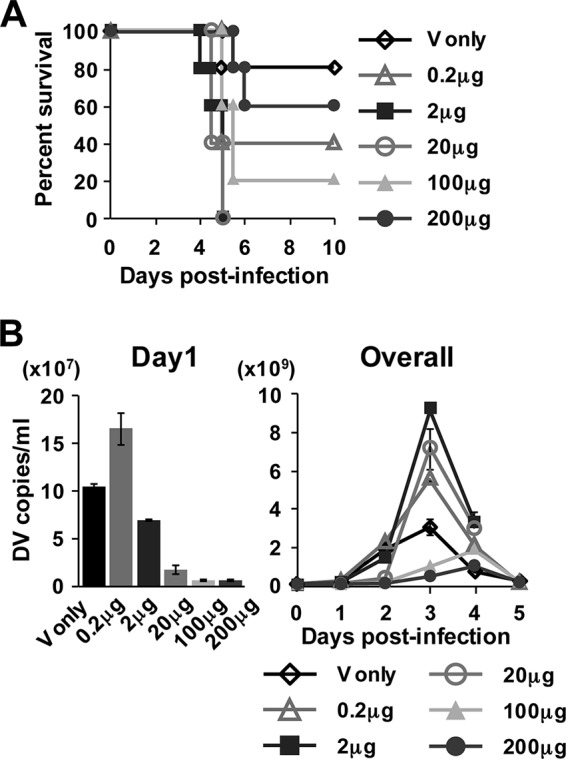
Survival rate and kinetics of viremia after infection of mice preinjected with various concentrations of 4G2 Ab. Mice were preinjected i.v. with 4G2 Ab at the concentrations of 0 to 200 μg (100 μl/mouse in PBS) 1 h prior to infection and then inoculated i.v. with 2 × 104 PFU of S221 (200 μl/mouse in PBS). (A) The mouse survival rate was monitored until day 10 after infection. (B) The virus genome numbers in serum on days 1 to 5 were measured as described for Fig. 1. The graphs show the average results from two independent measurements, and error bars indicate standard deviations. The number of mice per group is 5.
Highly neutralized IC inoculation is accompanied with the infusion of massive free Abs into mice.
In the experiments so far, we have not considered the physicochemical status of the Ab-virus mixture used in the in vivo experiments. In order to probe this, we resorted to measure the distribution and size of the ICs by dynamic light scattering (DLS). ICs were formed with 2 × 104 PFU of S221 with a range of 4G2 Ab amounts (0 to 200 μg), and their size distribution profiles were measured rapidly. The plot of the resulting size distribution data by numbers for ICs showed that their apparent particle sizes increased from 40.6 nm (virus only), a size close to the reported size measured by cryoelectron microscopy (cryo-EM) (44), to ∼60 nm when incubated with 0.02 μg or 0.2 μg of Ab (Fig. 4A). The plot by number normalized the data by taking into consideration the higher signal of the bigger molecules and readjusted the ratio of Ab to virus-Ab complex in solution. The signal for free Abs was not detectable in the IC mixture of 0.02 μg and 0.2 μg Ab, indicating that the majority of the Abs at this concentration are bound to the virus. In contrast, for ICs formed with 2 to 200 μg Ab, only the Ab molecules are detected (Fig. 4A), hence indicating that the free Ab/Ab-virus complex ratio in solution is very high. This suggests that the epitopes on the virus particles were almost saturated with Abs at the concentrations tested. Plotting the same results by intensity (Fig. 4B), which is biased toward the detection of larger molecules, showed the presence of virus-Ab complexes when incubated with 2 to 200 μg Ab even though the absolute numbers of these complexes are <1% of the total population of molecules in the samples. Although it is well established that the hydrodynamic radius of the ICs becomes larger with increased amounts of Abs, it is also possible that at these high concentrations of Abs, the solution becomes more viscous, resulting in less accurate size measurements. Alternatively, the virus-Ab complexes may have started to form bigger aggregates due to the cross-linking of particles by antibodies.
FIG 4.
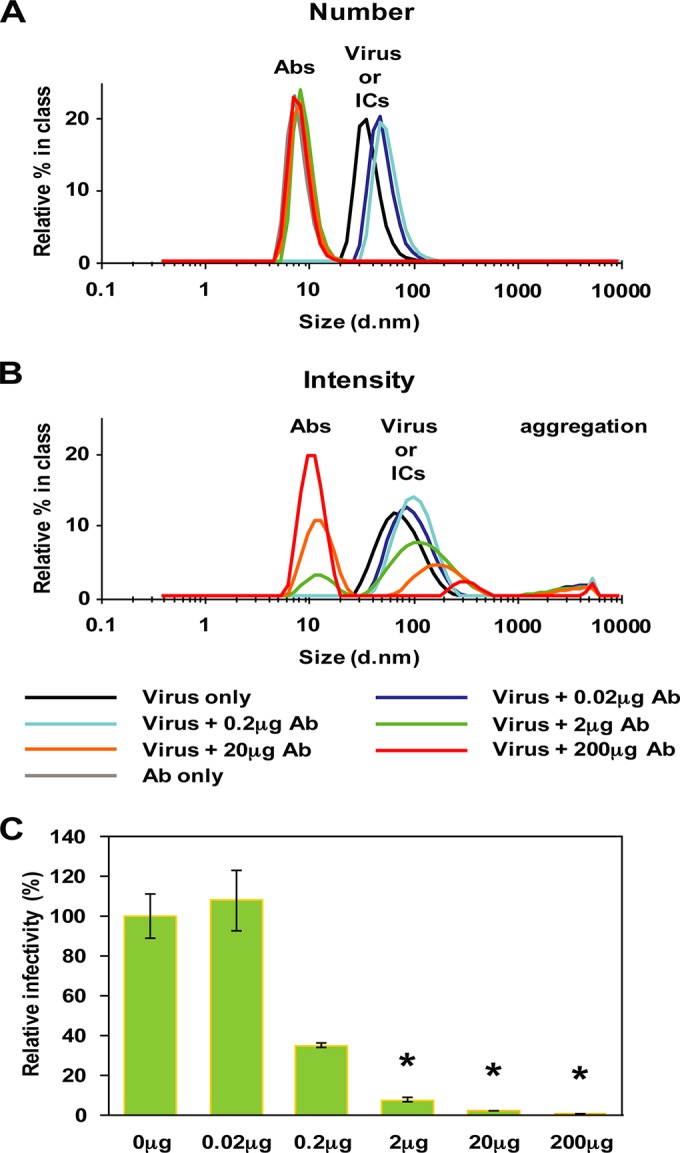
Size distribution of ICs formed with a series of Ab concentrations. ICs were formed by mixing 2 × 104 PFU of S221 with 0 to 200 μg of 4G2 Ab and subjected to analysis by dynamic light scattering (DLS), which detects the distribution of particle sizes and the ratio of molecules in solution. Size distributions of the major molecules in the solution were plotted by number (A) and by intensity (B). Abs and virus-Ab complexes have a diameter of ∼10 nm and ∼60 nm, respectively. Free Ab is detected at >2 μg IC, indicating that the ratio of free Ab to virus-Ab immune complex is very high. (C) The relative infectivity of the ICs in BHK-21 cells. Error bars indicate standard deviations from duplicate samples. An asterisk (*) shows the samples for which the ratio of Ab to virus-Ab complex is in far excess, as detected by DLS.
Neutralization tests of ICs in BHK-21 cells in the presence of 2 μg, 20 μg, and 200 μg Ab showed a reduction in infectivity of 93%, 98%, and 99.7%, respectively (Fig. 4C). This corresponded well with the large excess of Abs detected by DLS. Thus, inoculation of highly neutralized ICs is accompanied with the infusion of a massive quantity of free Abs in mice. These results, along with the observations in mice preinjected with Abs (Fig. 3), suggest that the presence of massive amounts of free Abs may contribute to further disease progression by forming infective ICs with newly produced virus in vivo.
DENV infection with high concentrations of Ab specifically enhances viral load and the production of TNF-α and IL-6 in the small intestine.
Increased cytokine production is one of the hallmarks of dengue pathogenesis and is thought to be a critical factor for DHF/DSS (45). Although viremia level was not enhanced by inoculation with highly neutralized ICs, it was conceivable that massive production of cytokines in IC-inoculated mice might affect their disease status. To investigate the detailed pathogenic events, tissues were harvested from the mice inoculated with virus alone (2 × 104 PFU) or ICs (2 × 104 PFU and 20 μg Ab) on days 1 to 5 postinfection. The homogenized tissues were then subjected to the measurement of viral load and cytokine levels. Figure 5A shows the overall kinetics of viral load in the various tissues measured by real-time RT-PCR. The enhancement of peak serum viremia was not observed in IC-inoculated mice, in which the AUC value until day 5 was 3.0-fold lower than that of the virus control (P = 0.0382). Corresponding with serum viremia level, the viral load in many tissues, such as spleen, large intestine, mesenteric lymph node (mLN), kidney, lung, and brain, was observed to be significantly lower in IC-inoculated mice during the early infection stages (days 1 to 3), and the peak viral load was not enhanced until a later stage (days 4 and 5) (Fig. 5A). However, we observed that the peak viral load was specifically enhanced by IC inoculation compared to the virus-only control (VC) and was 4.2-fold, 3.0-fold, and 540-fold higher on days 3, 4, and 5, respectively, in liver and 5.4-fold and 33.8-fold higher on days 4 and 5, respectively, in the small intestine, although the latter showed a lower viral load in IC-inoculated mice until day 3 (Fig. 5A). This implies that these tissues are more susceptible to infection than other tissues in the presence of Abs, as described in a previous study (18), and suggested that the effect of ADE overcomes the neutralizing activity of Abs in the tissues, although serum viremia level was not enhanced. Figure 5B shows the kinetics of the production of proinflammatory cytokines, such as TNF-α, IL-1β, IL-6, and monocyte chemoattractant protein-1 (MCP-1), levels of which are known to be enhanced in the serum of dengue patients (46–48). The cytokine profile showed a wide variation among the tissues and did not show significant enhancement by IC inoculation in serum as well as in many tissues, including spleen, liver, large intestine, mLN, kidney, lung, and brain (Fig. 5B). Notably, however, the levels of TNF-α and IL-6 in the small intestine were significantly enhanced by IC inoculation on days 4 and 5 postinfection (Fig. 5B), when these IC-inoculated mice started to display symptoms of severe diseases. Significant increases of IL-10 (anti-inflammatory) or IL-12 p70 (proinflammatory) were not observed in any tissues by IC inoculation (data not shown). So far in the tissues that we have examined, inoculation with ICs containing a highly neutralizing quantity of Abs specifically increased the production of TNF-α and IL-6 in the small intestine, accompanied by increased viral loads.
FIG 5.

Viral load and cytokine levels in various tissues after inoculation with virus alone or ICs formed with high concentration of Abs. Mice were inoculated with 2 × 104 PFU of S221 (VC) or ICs formed with 2 × 104 PFU of S221 and 20 μg 4G2 Ab (IC). Three mice were sacrificed on day 1 (24 h), day 2 (48 h), day 3 (72 h), day 4 (90 h), and day 5 (114 h) postinfection. Five mice were sacrificed before infection as a negative control (NC). Tissues were collected and homogenized in PBS, and the supernatants of the homogenates were used to measure viral load (A) and cytokine levels (B). (A) RNA was extracted from the homogenates and subjected to real-time RT-PCR. The values indicate the relative viral RNA genome numbers normalized to 18S rRNA. Samples below the detection limit of viral RNA, such as the negative control, were shown as the value of 0.1. (B) The levels of TNF-α, IL-1β, IL-6, and MCP-1 in the tissue homogenates were measured by ELISA. All graphs show the averages from 3 infected mice or 5 uninfected mice with standard deviations. A P value of less than 0.05 was considered significant (*, P < 0.05).
Viral load and the levels of TNF-α and IL-6 in the small intestine correlate with disease status in mice.
In order to address whether the small intestinal pathology is relevant to mouse disease status, tissues were harvested from mice inoculated with a lethal dose of 1 × 105 PFU of virus alone (V5) or with ICs formed with 2 × 104 PFU of virus and 20 μg Ab (IC) or nonlethally infected with 2 × 104 PFU of virus alone (V4) at 60 h (day 2.5) and 96 h (day 4) postinfection (Fig. 6A); they were then subjected to the measurement of viral load and cytokine levels. As shown in Fig. 6B, only the small intestine at 96 h postinfection showed significantly higher viral loads in both V5- and IC-inoculated mice than in V4-inoculated mice, with a 5.2-fold and 17.4-fold increase in V5 and IC, respectively. This indicates the correlation of viral load in the small intestine with disease status. Cytokine levels in tissues showed that the levels of TNF-α and IL-6 in the small intestine significantly increased in both V5 and IC compared with those of V4 at 96 h postinfection, although these levels in V5 at 60 h were significantly higher than in both V4 and IC (Fig. 6C). This suggests that inoculation with a high dose of virus alone results in high levels of cytokine production in the small intestine for an extended period of time, whereas inoculation with the ICs induces rapid increases in cytokine production at the later stage, corresponding with the increase in viral load in the small intestine. The levels of cytokine in other tissues, such as spleen, liver, large intestine (Fig. 6C), mLN, kidney, and lung (data not shown), did not show any correlation with disease severity at either 60 h or 96 h postinfection. Thus, only the levels of TNF-α and IL-6 in the small intestine appear to correlate with mouse disease status.
FIG 6.

Comparison of viral load and cytokine levels in tissues between lethally and nonlethally infected mice. Eighteen mice were infected with 1 × 105 PFU of S221 (V5) or 2 × 104 PFU of S221 (V4), and 21 mice were infected with ICs formed with 2 × 104 PFU of S221 and 20 μg 4G2 Ab (IC). Five or six mice from each group were sacrificed at 60 h (2.5 days) or at 96 h (4 days) postinfection, respectively, and tissues were used for the measurement of viral load and cytokine production. (A) The remaining mice were monitored for their survival rate until day 10 postinfection. The numbers of mice per group are 7 for the V5 and V4 groups and 10 for the IC group. (B) The viral load in tissues was measured as described for Fig. 4. (C) The levels of TNF-α, IL-1β, IL-6, and MCP-1 in spleen, liver, small intestine, and large intestine are shown. Five uninfected mice were used as a negative control (NC). The graphs show the averages from 5 mice (60 h) or 6 mice (96 h) with standard deviations. A P value of less than 0.05 was considered significant (*, P < 0.05).
ICs increase infection of CD11bint CD11cint/hi CD103− APCs in the small intestine.
Our results showed that increased levels of infection and cytokine production in the small intestine correlate with disease severity in mice. To identify the infected cells increased by IC infection, flow cytometric analysis was conducted with various cell markers and anti-DENV NS3 Ab. A previous study of the ADE mouse model showed that the infected cells in the small intestine are mainly major histocompatibility complex (MHC) class II+ CD103− CD11b+ F4/80+ macrophages for both Ab-dependent and Ab-independent infection (18), although the phenotype difference of target cells is still indistinct. We found that, after being gated on APCs (CD45+ MHC class II+ cells), NS3+ cells were 4.1-fold more abundant in IC-inoculated mice (1.69 ± 0.26%, n = 4) than in the virus infection control mice (0.41 ± 0.30%, n = 4) (P = 0.00063) on day 4 postinfection (Fig. 7). By analyzing for CD11b, CD11c, and CD103 expression, which can divide intestinal APCs into subsets of macrophages or dendritic cells (49), we found that the NS3+ population consists mainly of CD11bint CD11cint/hi cells (Fig. 7). CD103+ APCs were found to be negative for NS3 staining (data not shown); therefore, the infected cell population is suggested to be a subset of lamina propria macrophages (49, 50) with no difference between IC inoculation and virus control, which is consistent with a previous report (18). NS3 detection was negligible in the CD45− or CD31+ endothelial cell population (data not shown). Therefore, increased infection in CD11bint CD11cint/hi CD103− APCs by IC inoculation may be responsible for the increase of TNF-α and IL-6 production in the small intestine.
FIG 7.

Flow cytometric analysis for the detection of DENV-infected cells in the small intestine. Mice were infected with 2 × 104 PFU of S221 (VC) or ICs formed with 2 × 104 PFU of S221 and 20 μg 4G2 Ab (IC). Mice were sacrificed on day 4 (90 h) postinfection, and cells were isolated from the small intestines after digestion with 2 mM EDTA and 15 mg/ml collagenase solution. Flow cytometry (FCM) plots display cells pregated on CD45+ and MHC class II+ antigen-presenting cells (APCs) and then analyzed for NS3 or CD11b and CD11c expression. NS3+ cells were further analyzed for CD11b and CD11c expression and overlaid with the parent APC population. The FCM profile is representative of 1 in a group of 4 mice.
Anti-TNF-α Ab treatment protects mice from lethal IC infection by suppressing vascular permeability in the small intestine.
In previous studies, increased serum levels of TNF-α and IL-6 were observed in an ADE mouse model (16, 18), and treatment with anti-TNF-α Ab was shown to be effective against disease progression in lethal mouse models (18, 51), while anti-IL-6 Ab had no effect on mouse survival (18). Therefore, in order to address whether increased production of TNF-α in the small intestine is responsible for disease severity, anti-TNF-α Ab (200 μg/mouse, i.p.) was tested in IC-inoculated mice on day 3 postinfection, when the level of TNF-α was shown to increase drastically in the small intestine (Fig. 5B). Mice did not show any disease symptoms at the time of treatment on day 3 postinfection. At 24 h posttreatment, however, untreated mice exhibited severe disease symptoms, while TNF-α Ab-treated mice did not show severe clinical signs, such as reduced mobility or extensive ruffled fur on day 4. Mouse survival data demonstrated that treatment with TNF-α Ab completely protected IC-inoculated mice from lethal infection (Fig. 8A). Furthermore, TNF-α Ab treatment did not affect the serum viremia level as well as viral load in the small intestine (Fig. 8B), indicating that TNF-α is responsible for disease progression in the mice. Notably, the small intestine was observed to be swollen by an accumulation of fluid in IC-inoculated mice, leading to vascular leakage, and quite remarkably this fluid accumulation was suppressed by TNF-α Ab treatment (Fig. 8C). Evans blue staining assay demonstrated that massive vascular leakage was induced by IC inoculation only in the small intestine, and the leakage level was suppressed by TNF-α Ab treatment (Fig. 8D). This suggests that massive production of TNF-α in the small intestine is the leading cause of severe vascular leakage and ultimately death in the mice.
FIG 8.

The effect of anti-TNF-α antibody on disease progression in IC-inoculated mice. Mice were inoculated with ICs formed with 2 × 104 PFU of S221 and 20 μg 4G2 Ab. A total of 200 μg of anti-TNF-α Ab was administered i.p. into IC-inoculated mice on day 3 (72 h) postinfection. (A) The survival rates of untreated and treated mice were monitored until day10 postinfection. The number of mice per group is 5. (B) Blood was collected from untreated and treated mice at the time of treatment (day 3) and 24 h after treatment (day 4). Serum viral load was measured by real-time RT-PCR. The small intestine was harvested from untreated and treated mice at 24 h after treatment (day 4), and the viral load was measured as described for Fig. 5. The graph shows the averages from 5 mice (viremia) or 4 mice (small intestine) with standard deviations. (C) The pictures of the small intestine of an uninfected negative control (NC), an untreated IC-inoculated mouse, and a TNF-α Ab-treated IC-inoculated mouse on day 4 (24 h after treatment) postinfection are shown. Arrows indicate that the small intestine is swelling in the untreated IC-inoculated mouse. (D) Vascular leakage levels in tissues were measured by Evans blue staining assay. On day 4 (24 h after TNF-α Ab treatment) postinfection, mice were injected i.p. with 0.4 ml of 0.5% Evans blue solution, and tissues were collected into formamide after perfusion with PBS. The amount of Evans blue in the extract was quantified spectrophotometrically. Data are expressed as the fold increases in optical density at 620 nm per gram of tissue weight compared to the NC. The pictures shown are intestines prior to removal of luminal content. The graphs show the averages from 3 mice with standard deviations. A P value of less than 0.05 was considered significant (*, P < 0.05).
DISCUSSION
The contribution of DENV Abs in causing severe dengue disease in humans is not fully understood. DHF/DSS are characterized by plasma leakage, hemorrhage, and thrombocytopenia, and landmark studies have shown that the AG129 mouse model can recapitulate some of the aspects of human diseases (16, 18) and is therefore useful to investigate the mechanisms underlying dengue disease pathology caused by ADE infection. In this study, by using the AG129 mouse lethal model, we demonstrated that (i) inoculation with ICs containing a highly neutralizing quantity of cross-reactive Abs causes lethal infection even though the peak viremia level is lower than that in Ab-independent nonlethal infection, (ii) DENV infection with a highly neutralizing quantity of Abs specifically enhances the production of TNF-α and IL-6 and vascular permeability in the small intestine, accompanied by increasing the population of infected CD11bint CD11cint/hi CD103− APCs, and (iii) anti-TNF-α Ab treatment protects IC-inoculated mice from lethal infection by suppressing vascular permeability in the small intestine without significant changes in the viral load.
In general, ADE is thought to be a risk factor for severe diseases by increasing the number of infected cells and systemic viral burden (52). Although several in vivo experiments support this hypothesis (12, 14, 16, 18), it is still unclear whether Abs merely enhance systemic viral burden. Several clinical studies have shown a statistical correlation between viral burden and disease severity (19–21); however, a clinical study in Vietnam reported that viremia level was not always associated with clinical severity in infants with DHF/DSS (22). Notably, this report showed that infants who have substantial neutralizing maternal DENV Abs (calculated range of the PRNT50 titer of 138 to 580) can still develop severe diseases. Collectively, our data in the mouse model show that inoculation with highly neutralized ICs containing a massive excess of free Abs in the mixture can cause lethal infection regardless of viremia levels and appear to be relevant to observations made for some dengue patients.
The liver is implicated in DENV infections in humans and mouse models (18, 53, 54). In support of this, a recent report of the ADE mouse model suggested that Abs initially enhance infection in liver sinusoidal endothelial cells (LSECs), resulting in an increased systemic level of virus (viremia) and subsequent disease severity (18). Our results confirm that ADE in the liver precedes that in the small intestine in IC-inoculated mice; however, the enhancement of the serum viremia level was not induced in the mice. This indicates that increased viral load in the liver may not always be associated with serum viremia, although the possibility that infection in the liver may be the upstream event that is required for infection in the small intestine cannot be excluded. In addition, it is notable that there was no increase in inflammatory cytokine production in the liver of either the Ab-mediated or non-Ab-mediated infection. In contrast, our results demonstrated that the levels of TNF-α and IL-6 were specifically enhanced in the small intestine, accompanied by the increased tissue viral load and vascular leakage in IC-inoculated mice. Remarkably, a single dose of anti-TNF-α Ab on day 3 postinfection completely protected the mice from lethal infection, suggesting that TNF-α-induced vascular leakage in the small intestine is the leading cause of severe disease in this mouse model.
One of the interpretative limitations of our observations in the mouse model is its relevance to humans. The lack of IFN receptors in AG129 mice and the use of a mouse-adapted virus may alter the production of cytokines and the pathogenesis profile. Nevertheless, the similarity of an elevated serum cytokine profile, such as for TNF-α, IL-1β, IL-6, and MCP-1, in mice and human patients (46–48) supports the use of this mouse model for the study of host responses against dengue virus infection and its relation to disease status. It is worth noting that when patients are classified as having DHF/DSS, virus is no longer detectable in blood (55, 56), and therefore the host responses may play a critical role in disease progression. Nonetheless, the collective profile of host responses, such as tissue specificity of cytokine production in humans, remains unclear because of the difficulty of obtaining biopsy samples (57). Vascular leakage is a prominent feature of DHF/DSS in humans, which is characterized by increased hemoconcentration or fluid effusion in chest or abdominal cavities that is detectable by radiology or sonography (58–61). Also, the notable pathology that causes mortality in the IC-inoculated mice is the fluid accumulation in the small intestine as a consequence of TNF-α-induced vascular leakage. Even though no direct observations of fluid accumulation in the small intestine has been reported in human cases, it is well known that diarrhea is a common symptom found in dengue patients (62, 63). Our results showed that only limited cell types (CD11bint CD11cint/hi CD103−) among various subsets of APCs in the small intestine are positive for NS3, and, quite interestingly, there is no phenotype difference of the infected cells between the presence and the absence of Abs, although significantly higher infection is induced by IC inoculation. Therefore, it is conceivable that increased infection in the CD11bint CD11cint/hi CD103− APCs may have a major role in severe dengue pathology by inducing massive vascular leakage in the small intestine. Exactly how this event occurs requires further studies in order to understand the mechanism(s) by which the presence of Abs increases infection in a specific cell population regardless of viremia level in mice.
Recent reports of the ChimeriVax vaccination clinical trials, which were expected to induce serotype-specific Abs against all 4 serotypes in the recipients, showed only 30.2% efficacy in a phase 2b study in Thailand (25) and 56.5% efficacy in five Asian countries and 64.7% efficacy in five Latin American countries in a phase 3 study (24, 26). Notably, the vaccination provided lower efficacy against DENV2 (no protection in phase 2b and 35% or 42.3% protection in phase 3) even though highly neutralizing levels of Abs against this serotype were detected in the recipients when assessed by PRNT50 assay (mean titer, >300) (24, 25). This suggests that DENV infection even in the presence of a highly neutralizing quantity of Abs appears to retain the potential to induce severe dengue disease manifestations in humans, which is exactly what is observed in our mouse study. Our DLS data indicate that inoculation with ICs (2 to 100 μg Ab) is accompanied with massive free Abs. It is conceivable that the free Abs may be continuously consumed by forming ICs with newly produced virus, resulting in an enhancing status at a later stage. This is a plausible reason why a highly suppressed level of viral load on days 1 and 2 could be enhanced on days 3 to 5, especially in liver and the small intestine, although the peak viremia level is still lower than that in the virus-only control. The formation of ICs in a human subject with the repertoire of polyclonal antibodies, differing affinities, and concentrations will be challenging to model precisely (64). By correlating the DLS data on the size distribution of ICs and the quantum of neutralized virus used to infect AG129 mice, we have carefully reproduced a phenomenon that has been seen in the clinical setting with infants (22). We also showed that inoculation with ICs formed with serotype-specific Ab, 3H5 (anti-domain III of E), did not cause high mortality at any concentrations, while ICs formed with cross-reactive Abs, 4G2 or 6B6C-1 (anti-domain II of E), induced lethal diseases even at the highly neutralizing concentrations, which showed more than 95% suppression of viremia at the early stage of infection. The difference in neutralizing activities, epitopes, or IgG subclasses of the Abs might influence this phenomenon. Since many cross-reactive anti-domain III and premembrane (prM) Abs are also produced in patients (40, 65), further detailed studies to address the role of cross-reactive Abs in IC infection and disease status may provide critical information for vaccine strategies.
In summary, our results showed that the presence of cross-reactive Abs, which can suppress more than 95% of the initial infection level, induces lethal DENV infection regardless of serum viremia level, by causing rapid severe small intestinal pathology accompanied by increased tissue viral load, TNF-α production, and vascular permeability in mice. This finding may have an important implication for vaccine development, serve to address the role of DENV Abs in exacerbating diseases in dengue patients, and also help in the development of novel therapeutic agents, such as therapeutic antibodies or immunomodulatory drugs.
ACKNOWLEDGMENTS
We thank Sujan Shresta for providing the DENV2 S221 strain and Alan Barrett for suggesting the use of 6B6C-1 to confirm our observations with 4G2 immune complexes. We thank Eng Eong Ooi and Taara Nadathur Madhavan for the comments on our manuscript. We also thank Kevin King, Ashley L. St. John, Nasirah B. S. Hamee, and Abhay P. S. Rathore for technical advice and support.
This work is supported by the Duke-NUS Signature Research Program (funded by the Agency for Science, Technology and Research, Singapore, and the Ministry of Health, Singapore) and in part by the National Medical Research Council, Singapore (www.nmrc.gov.sg), under grant NMRC/1315/2011.
REFERENCES
- 1.Gubler DJ. 2006. Dengue/dengue haemorrhagic fever: history and current status. Novartis Found Symp 277:3–16. doi: 10.1002/0470058005.ch2. [DOI] [PubMed] [Google Scholar]
- 2.Halstead SB. 2007. Dengue. Lancet 370:1644–1652. doi: 10.1016/S0140-6736(07)61687-0. [DOI] [PubMed] [Google Scholar]
- 3.Simmons CP, Farrar JJ, Nguyen van Vinh C, Wills B. 2012. Dengue. N Engl J Med 366:1423–1432. doi: 10.1056/NEJMra1110265. [DOI] [PubMed] [Google Scholar]
- 4.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fink J, Gu F, Vasudevan SG. 2006. Role of T cells, cytokines and antibody in dengue fever and dengue haemorrhagic fever. Rev Med Virol 16:263–275. doi: 10.1002/rmv.507. [DOI] [PubMed] [Google Scholar]
- 6.Guzman MG, Alvarez M, Halstead SB. 2013. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: an historical perspective and role of antibody-dependent enhancement of infection. Arch Virol 158:1445–1459. doi: 10.1007/s00705-013-1645-3. [DOI] [PubMed] [Google Scholar]
- 7.Halstead SB. 2003. Neutralization and antibody-dependent enhancement of dengue viruses. Adv Virus Res 60:421–467. doi: 10.1016/S0065-3527(03)60011-4. [DOI] [PubMed] [Google Scholar]
- 8.Whitehorn J, Farrar J. 2010. Dengue. Br Med Bull 95:161–173. doi: 10.1093/bmb/ldq019. [DOI] [PubMed] [Google Scholar]
- 9.Halstead SB. 1981. The Alexander D. Langmuir lecture. The pathogenesis of dengue molecular epidemiology in infectious disease. Am J Epidemiol 114:632–648. [DOI] [PubMed] [Google Scholar]
- 10.Halstead SB. 1988. Pathogenesis of dengue: challenges to molecular biology. Science 239:476–481. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- 11.Boonnak K, Slike BM, Burgess TH, Mason RM, Wu SJ, Sun P, Porter K, Rudiman IF, Yuwono D, Puthavathana P, Marovich MA. 2008. Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J Virol 82:3939–3951. doi: 10.1128/JVI.02484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goncalvez AP, Engle RE, St Claire M, Purcell RH, Lai CJ. 2007. Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc Natl Acad Sci U S A 104:9422–9427. doi: 10.1073/pnas.0703498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moi ML, Takasaki T, Saijo M, Kurane I. 2013. Determination of antibody concentration as the main parameter in a dengue virus antibody-dependent enhancement assay using FcγR-expressing BHK cells. Arch Virol 159:103–116. doi: 10.1007/s00705-013-1787-3. [DOI] [PubMed] [Google Scholar]
- 14.Halstead SB. 1979. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J Infect Dis 140:527–533. doi: 10.1093/infdis/140.4.527. [DOI] [PubMed] [Google Scholar]
- 15.Ng JK, Zhang SL, Tan HC, Yan B, Maria Martinez Gomez J, Tan WY, Lam JH, Tan GK, Ooi EE, Alonso S. 2014. First experimental in vivo model of enhanced dengue disease severity through maternally acquired heterotypic dengue antibodies. PLoS Pathog 10:e1004031. doi: 10.1371/journal.ppat.1004031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balsitis SJ, Williams KL, Lachica R, Flores D, Kyle JL, Mehlhop E, Johnson S, Diamond MS, Beatty PR, Harris E. 2010. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog 6:e1000790. doi: 10.1371/journal.ppat.1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierson TC. 2010. Modeling antibody-enhanced dengue virus infection and disease in mice: protection or pathogenesis? Cell Host Microbe 7:85–86. doi: 10.1016/j.chom.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Zellweger RM, Prestwood TR, Shresta S. 2010. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe 7:128–139. doi: 10.1016/j.chom.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Libraty DH, Endy TP, Houng HS, Green S, Kalayanarooj S, Suntayakorn S, Chansiriwongs W, Vaughn DW, Nisalak A, Ennis FA, Rothman AL. 2002. Differing influences of virus burden and immune activation on disease severity in secondary dengue-3 virus infections. J Infect Dis 185:1213–1221. doi: 10.1086/340365. [DOI] [PubMed] [Google Scholar]
- 20.Libraty DH, Young PR, Pickering D, Endy TP, Kalayanarooj S, Green S, Vaughn DW, Nisalak A, Ennis FA, Rothman AL. 2002. High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J Infect Dis 186:1165–1168. doi: 10.1086/343813. [DOI] [PubMed] [Google Scholar]
- 21.Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. 2000. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis 181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- 22.Simmons CP, Chau TN, Thuy TT, Tuan NM, Hoang DM, Thien NT, Lien le, Quy BNT, Hieu NT, Hien TT, McElnea C, Young P, Whitehead S, Hung NT, Farrar J. 2007. Maternal antibody and viral factors in the pathogenesis of dengue virus in infants. J Infect Dis 196:416–424. doi: 10.1086/519170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kliks SC, Nimmanitya S, Nisalak A, Burke DS. 1988. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am J Trop Med Hyg 38:411–419. [DOI] [PubMed] [Google Scholar]
- 24.Capeding MR, Tran NH, Hadinegoro SR, Ismail HI, Chotpitayasunondh T, Chua MN, Luong CQ, Rusmil K, Wirawan DN, Nallusamy R, Pitisuttithum P, Thisyakorn U, Yoon IK, van der Vliet D, Langevin E, Laot T, Hutagalung Y, Frago C, Boaz M, Wartel TA, Tornieporth NG, Saville M, Bouckenooghe A, the CYD14 Study Group. 2014. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet 384:1358–1365. doi: 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- 25.Sabchareon A, Wallace D, Sirivichayakul C, Limkittikul K, Chanthavanich P, Suvannadabba S, Jiwariyavej V, Dulyachai W, Pengsaa K, Wartel TA, Moureau A, Saville M, Bouckenooghe A, Viviani S, Tornieporth NG, Lang J. 2012. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. Lancet 380:1559–1567. doi: 10.1016/S0140-6736(12)61428-7. [DOI] [PubMed] [Google Scholar]
- 26.Villar L, Dayan GH, Arredondo-Garcia JL, Rivera DM, Cunha R, Deseda C, Reynales H, Costa MS, Morales-Ramirez JO, Carrasquilla G, Rey LC, Dietze R, Luz K, Rivas E, Miranda Montoya MC, Cortes Supelano M, Zambrano B, Langevin E, Boaz M, Tornieporth N, Saville M, Noriega F, CYD15 Study Group. 2015. Efficacy of a tetravalent dengue vaccine in children in Latin America. N Engl J Med 372:113–123. doi: 10.1056/NEJMoa1411037. [DOI] [PubMed] [Google Scholar]
- 27.Yauch LE, Zellweger RM, Kotturi MF, Qutubuddin A, Sidney J, Peters B, Prestwood TR, Sette A, Shresta S. 2009. A protective role for dengue virus-specific CD8+ T cells. J Immunol 182:4865–4873. doi: 10.4049/jimmunol.0801974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanson BJ, Boon AC, Lim AP, Webb A, Ooi EE, Webby RJ. 2006. Passive immunoprophylaxis and therapy with humanized monoclonal antibody specific for influenza A H5 hemagglutinin in mice. Respir Res 7:126. doi: 10.1186/1465-9921-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houng HH, Hritz D, Kanesa-thasan N. 2000. Quantitative detection of dengue 2 virus using fluorogenic RT-PCR based on 3′-noncoding sequence. J Virol Methods 86:1–11. doi: 10.1016/S0166-0934(99)00166-4. [DOI] [PubMed] [Google Scholar]
- 30.Tan GK, Ng JK, Trasti SL, Schul W, Yip G, Alonso S. 2010. A non mouse-adapted dengue virus strain as a new model of severe dengue infection in AG129 mice. PLoS Negl Trop Dis 4:e672. doi: 10.1371/journal.pntd.0000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreland NJ, Tay MY, Lim E, Paradkar PN, Doan DN, Yau YH, Geifman Shochat S, Vasudevan SG. 2010. High affinity human antibody fragments to dengue virus non-structural protein 3. PLoS Negl Trop Dis 4:e881. doi: 10.1371/journal.pntd.0000881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725. doi: 10.1016/S0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe S, Tan KH, Rathore AP, Rozen-Gagnon K, Shuai W, Ruedl C, Vasudevan SG. 2012. The magnitude of dengue virus NS1 protein secretion is strain dependent and does not correlate with severe pathologies in the mouse infection model. J Virol 86:5508–5514. doi: 10.1128/JVI.07081-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fraser JE, Watanabe S, Wang C, Chan WK, Maher B, Lopez-Denman A, Hick C, Wagstaff KM, Mackenzie JM, Sexton PM, Vasudevan SG, Jans DA. 2014. A nuclear transport inhibitor that modulates the unfolded protein response and provides in vivo protection against lethal dengue virus infection. J Infect Dis 210:1780–1791. doi: 10.1093/infdis/jiu319. [DOI] [PubMed] [Google Scholar]
- 35.Rathore AP, Paradkar PN, Watanabe S, Tan KH, Sung C, Connolly JE, Low J, Ooi EE, Vasudevan SG. 2011. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model. Antiviral Res 92:453–460. doi: 10.1016/j.antiviral.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe S, Rathore AP, Sung C, Lu F, Khoo YM, Connolly J, Low J, Ooi EE, Lee HS, Vasudevan SG. 2012. Dose- and schedule-dependent protective efficacy of celgosivir in a lethal mouse model for dengue virus infection informs dosing regimen for a proof of concept clinical trial. Antiviral Res 96:32–35. doi: 10.1016/j.antiviral.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Moi ML, Lim CK, Kotaki A, Takasaki T, Kurane I. 2011. Detection of higher levels of dengue viremia using FcγR-expressing BHK-21 cells than FcγR-negative cells in secondary infection but not in primary infection. J Infect Dis 203:1405–1414. doi: 10.1093/infdis/jir053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henchal EA, McCown JM, Burke DS, Seguin MC, Brandt WE. 1985. Epitopic analysis of antigenic determinants on the surface of dengue-2 virions using monoclonal antibodies. Am J Trop Med Hyg 34:162–169. [DOI] [PubMed] [Google Scholar]
- 39.Rothman AL. 2004. Dengue: defining protective versus pathologic immunity. J Clin Invest 113:946–951. doi: 10.1172/JCI200421512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dejnirattisai W, Jumnainsong A, Onsirisakul N, Fitton P, Vasanawathana S, Limpitikul W, Puttikhunt C, Edwards C, Duangchinda T, Supasa S, Chawansuntati K, Malasit P, Mongkolsapaya J, Screaton G. 2010. Cross-reacting antibodies enhance dengue virus infection in humans. Science 328:745–748. doi: 10.1126/science.1185181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crill WD, Chang GJ. 2004. Localization and characterization of flavivirus envelope glycoprotein cross-reactive epitopes. J Virol 78:13975–13986. doi: 10.1128/JVI.78.24.13975-13986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roehrig JT, Bolin RA, Kelly RG. 1998. Monoclonal antibody mapping of the envelope glycoprotein of the dengue 2 virus, Jamaica. Virology 246:317–328. doi: 10.1006/viro.1998.9200. [DOI] [PubMed] [Google Scholar]
- 43.Gromowski GD, Barrett AD. 2007. Characterization of an antigenic site that contains a dominant, type-specific neutralization determinant on the envelope protein domain III (ED3) of dengue 2 virus. Virology 366:349–360. doi: 10.1016/j.virol.2007.05.042. [DOI] [PubMed] [Google Scholar]
- 44.Kostyuchenko VA, Chew PL, Ng TS, Lok SM. 2013. Near-atomic resolution cryo-electron microscopic structure of dengue serotype 4 virus. J Virol 88:477–482. doi: 10.1128/JVI.02641-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martina BE, Koraka P, Osterhaus AD. 2009. Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev 22:564–581. doi: 10.1128/CMR.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bozza FA, Cruz OG, Zagne SM, Azeredo EL, Nogueira RM, Assis EF, Bozza PT, Kubelka CF. 2008. Multiplex cytokine profile from dengue patients: MIP-1beta and IFN-gamma as predictive factors for severity. BMC Infect Dis 8:86. doi: 10.1186/1471-2334-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee YR, Liu MT, Lei HY, Liu CC, Wu JM, Tung YC, Lin YS, Yeh TM, Chen SH, Liu HS. 2006. MCP-1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J Gen Virol 87:3623–3630. doi: 10.1099/vir.0.82093-0. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen TH, Lei HY, Nguyen TL, Lin YS, Huang KJ, Le BL, Lin CF, Yeh TM, Do QH, Vu TQ, Chen LC, Huang JH, Lam TM, Liu CC, Halstead SB. 2004. Dengue hemorrhagic fever in infants: a study of clinical and cytokine profiles. J Infect Dis 189:221–232. doi: 10.1086/380762. [DOI] [PubMed] [Google Scholar]
- 49.Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, Karp CL, Pulendran B. 2011. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol 187:733–747. doi: 10.4049/jimmunol.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, Sato S, Tsujimura T, Yamamoto M, Yokota Y, Kiyono H, Miyasaka M, Ishii KJ, Akira S. 2008. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol 9:769–776. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 51.Shresta S, Sharar KL, Prigozhin DM, Beatty PR, Harris E. 2006. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J Virol 80:10208–10217. doi: 10.1128/JVI.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guzman MG, Vazquez S. 2010. The complexity of antibody-dependent enhancement of dengue virus infection. Viruses 2:2649–2662. doi: 10.3390/v2122649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paes MV, Pinhao AT, Barreto DF, Costa SM, Oliveira MP, Nogueira AC, Takiya CM, Farias-Filho JC, Schatzmayr HG, Alves AM, Barth OM. 2005. Liver injury and viremia in mice infected with dengue-2 virus. Virology 338:236–246. doi: 10.1016/j.virol.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 54.Seneviratne SL, Malavige GN, de Silva HJ. 2006. Pathogenesis of liver involvement during dengue viral infections. Trans R Soc Trop Med Hyg 100:608–614. doi: 10.1016/j.trstmh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Nimmannitya S, Halstead SB, Cohen SN, Margiotta MR. 1969. Dengue and Chikungunya virus infection in man in Thailand, 1962-1964. I. Observations on hospitalized patients with hemorrhagic fever. Am J Trop Med Hyg 18:954–971. [DOI] [PubMed] [Google Scholar]
- 56.Nisalak A, Halstead SB, Singharaj P, Udomsakdi S, Nye SW, Vinijchaikul K. 1970. Observations related to pathogenesis of dengue hemorrhagic fever. 3. Virologic studies of fatal disease. Yale J Biol Med 42:293–310. [PMC free article] [PubMed] [Google Scholar]
- 57.Jessie K, Fong MY, Devi S, Lam SK, Wong KT. 2004. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J Infect Dis 189:1411–1418. doi: 10.1086/383043. [DOI] [PubMed] [Google Scholar]
- 58.Balasubramanian S, Janakiraman L, Kumar SS, Muralinath S, Shivbalan S. 2006. A reappraisal of the criteria to diagnose plasma leakage in dengue hemorrhagic fever. Indian Pediatr 43:334–339. [PubMed] [Google Scholar]
- 59.Srikiatkhachorn A, Krautrachue A, Ratanaprakarn W, Wongtapradit L, Nithipanya N, Kalayanarooj S, Nisalak A, Thomas SJ, Gibbons RV, Mammen MP Jr, Libraty DH, Ennis FA, Rothman AL, Green S. 2007. Natural history of plasma leakage in dengue hemorrhagic fever: a serial ultrasonographic study. Pediatr Infect Dis J 26:283–290. doi: 10.1097/01.inf.0000258612.26743.10. [DOI] [PubMed] [Google Scholar]
- 60.Thulkar S, Sharma S, Srivastava DN, Sharma SK, Berry M, Pandey RM. 2000. Sonographic findings in grade III dengue hemorrhagic fever in adults. J Clin Ultrasound 28:34–37. doi:. [DOI] [PubMed] [Google Scholar]
- 61.Wang CC, Wu CC, Liu JW, Lin AS, Liu SF, Chung YH, Su MC, Lee IK, Lin MC. 2007. Chest radiographic presentation in patients with dengue hemorrhagic fever. Am J Trop Med Hyg 77:291–296. [PubMed] [Google Scholar]
- 62.Low JG, Sung C, Wijaya L, Wei Y, Rathore AP, Watanabe S, Tan BH, Toh L, Chua LT, Hou Y, Chow A, Howe S, Chan WK, Tan KH, Chung JS, Cherng BP, Lye DC, Tambayah PA, Ng LC, Connolly J, Hibberd ML, Leo YS, Cheung YB, Ooi EE, Vasudevan SG. 2014. Efficacy and safety of celgosivir in patients with dengue fever (CELADEN): a phase 1b, randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet Infect Dis 14:706–715. doi: 10.1016/S1473-3099(14)70730-3. [DOI] [PubMed] [Google Scholar]
- 63.Low JG, Ong A, Tan LK, Chaterji S, Chow A, Lim WY, Lee KW, Chua R, Chua CR, Tan SW, Cheung YB, Hibberd ML, Vasudevan SG, Ng LC, Leo YS, Ooi EE. 2011. The early clinical features of dengue in adults: challenges for early clinical diagnosis. PLoS Negl Trop Dis 5:e1191. doi: 10.1371/journal.pntd.0001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pierson TC, Diamond MS. 2008. Molecular mechanisms of antibody-mediated neutralisation of flavivirus infection. Expert Rev Mol Med 10:e12. doi: 10.1017/S1462399408000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NT, Sukupolvi-Petty S, Navarro-Sanchez E, Young PR, de Silva AM, Rey FA, Varani L, Whitehead SS, Diamond MS, Harris E, Lanzavecchia A, Sallusto F. 2010. The human immune response to dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8:271–283. doi: 10.1016/j.chom.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]