

Abstract
BRCA1 and BRCA2 mutation carriers are predisposed to develop breast and ovarian cancers, but the reasons for this tissue specificity are unknown. Breast epithelial cells are known to contain elevated levels of oxidative DNA damage, triggered by hormonally driven growth and its effect on cell metabolism. BRCA1- or BRCA2-deficient cells were found to be more sensitive to oxidative stress, modeled by treatment with patho-physiologic concentrations of hydrogen peroxide. Hydrogen peroxide exposure leads to oxidative DNA damage induced DNA double strand breaks (DSB) in BRCA-deficient cells causing them to accumulate in S-phase. In addition, after hydrogen peroxide treatment, BRCA deficient cells showed impaired Rad51 foci which are dependent on an intact BRCA1-BRCA2 pathway. These DSB resulted in an increase in chromatid-type aberrations, which are characteristic for BRCA1 and BRCA2-deficient cells. The most common result of oxidative DNA damage induced processing of S-phase DSB is an interstitial chromatid deletion, but insertions and exchanges were also seen in BRCA deficient cells. Thus, BRCA1 and BRCA2 are essential for the repair of oxidative DNA damage repair intermediates that persist into S-phase and produce DSB. The implication is that oxidative stress plays a role in the etiology of hereditary breast cancer.
Keywords: Cancer, Oxidative Stress, Homologous Recombination, BRCA, Chromosome
1. Introduction
Oxidative stress is known to be important in the development of aging, degenerative diseases and carcinogenesis [1]. Breast epithelium is subjected to oxidative stress in association with the menstrual cycle [2]. Thus, hormonally driven proliferation in concert with cellular metabolism can trigger the formation of reactive oxygen species (ROS), which in turn can produce oxidative DNA damage. The accumulation of oxidative DNA damage can be a critical initiating event in carcinogenesis [3]. Mammary tissues are exposed to a significant level of oxidative stress, during hormone induced growth and metabolism [4–6].
Base excision repair (BER) is the main pathway to repair oxidized DNA lesions in all cells, conserved from E. coli to humans [7, 8]. BER genes are essential in mouse embryonic development, providing housekeeping function for endogenous metabolism that produces oxidative DNA damage. There are two pathways of BER in mammalian cells, short-patch and long-patch, which are characterized by the size of the re-synthesis patch that occurs after strand-incision. Short-patch BER requires XRCC1 and ligase III, together with polymerase β, whereas long-patch utilizes the same machinery as Okazaki fragment joining, with FEN1, ligase I and either the replicative (ε or δ) or the repair polymerase (β). Recent evidence has suggested that single-strand break repair in the nucleus is repaired much like an Okazaki fragment, whereas ligase III is used predominantly in the mitochondria [9]. The repair of oxidative DNA lesions or repair intermediates by BER may be restricted during active DNA replication, where access to the lesion in the region of the replicative polymerase complex is limited. The involvement of BRCA1 and BRCA2 in the direct repair of oxidative DNA damage is largely unknown, with limited reported evidence that they may play a role in removing oxidative DNA damage from plasmids [10]. The repair of an oxidative lesion in a replicating plasmid could be mediated by replication-linked recombination (post-replication repair), but this possibility was not raised.
DNA double-strand breaks (DSBs) may arise spontaneously during DNA replication or following exposure to ionizing radiation (IR), chemotherapeutic drugs or oxidative stress [11]. Homologous recombination (HR) is involved in the repair of DSBs, especially those arising from stalled replication forks [12]. Defective HR results in chromatid exchanges proceeding to genomic instability. Cells deficient in HR are sensitive to IR and chemotherapeutic drugs [13, 14], that affect both strands of DNA and work in the S/G2-phases of the cell cycle where HR is the preferential pathway of DSB repair [15]. HR can be initiated when a DSB (arising from DNA damage or blocked DNA replication) is processed to reveal a 3′ single-strand DNA (ssDNA) tail after resection of the 5′-end strand. The ssDNA is rapidly bound by the ssDNA-binding protein, Replication Protein-A (RPA), which is a required precursor to the formation of the Rad51 filament that mediates DNA strand invasion and exchange.
The breast cancer susceptibility gene BRCA1 encodes a tumor suppressor protein, mutations of which account for 2.5–5% of all breast cancers and ~40% of hereditary breast cancers [16]. BRCA1 is a multifunctional protein that plays a role in cell cycle checkpoints, apoptosis and DNA repair [17]. There is debate about which of these functions of BRCA1 are responsible for its role in maintaining genomic integrity and suppressing breast cancer. We favor its role in HR since it is the only known defect in BRCA2-deficient cells and because BRCA1 and BRCA2 mutation carriers have a similar human phenotype [18]. The primary role of BRCA2 is to maintain genomic integrity via HR by mediating the formation of Rad51 filaments on ssDNA, which in turn, promote strand exchange [19, 20]. BRCA2 contains eight BRC repeats that bind Rad51 [20] and a DNA binding domain that binds ssDNA [21]. BRCA2 facilitates displacement of RPA from ssDNA and allows the formation of a Rad51 filament [22–24]. Cells carrying homozygous null mutations in BRCA1 or BRCA2 genes exhibit lower clonogenic survival and higher rates of chromosomal aberrations and mutations in response to IR [23–25] in addition to a variety of genotoxic stresses. Here we show that BRCA1 and BRCA2 participate in the repair of oxidized DNA lesions that persist into S-phase where they produce DSB necessitating the use of HR.
2. Materials and Methods
2.1. Cell culture and media
Human breast cancer MCF-7 cells were purchased from American Type Culture Collection (Manassas, VA). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20mM Hepes, 10% bovine growth serum and antibiotics (100U/ml Penicillin/0.1mg/ml Streptomycin). Cells were cultured at 37°C in 5% CO2. For routine culture, cells were grown in phenol red containing medium. Since phenol red and bovine growth serum can produce ROS, for oxidative stress conditions, cells were grown in phenol red-free medium supplemented with 5% charcoal stripped fetal bovine serum (Sigma Aldrich).
2.2. Transfections and establishment of clonal cell lines
BRCA1 and BRCA2 small hairpin RNAs (shRNAs) were generated using a lentiviral pLKO.1-puromycin vector (Sigma Aldrich). The sequences used for the small hairpin RNA complementary to BRCA1 and BRCA2 were as follows:
BRCA1:
5′-CGGGCCCACCTAATTGTACTGAATCTCGAGATTCAGTACAATTAGGTGGGCTTTTTG-3′
BRCA2:
5′-CCGGTGAAGAATGCAGGTTTAATATCTCGAGATATTAAACCTGCATTCTTCATTTTTG-3′
For establishment of clonally-derived cell lines, MCF-7 cells were electroporated using nucleofector system (Amaxa) according to manufacturer’s directions. The transfected cells were seeded in 10-cm dishes containing complete DMEM media. After 72h, the plates were washed with PBS and media containing 3μg/ml puromycin was added. The individual puromycin-resistant cells were allowed to grow for another 4 weeks to form colonies. The colonies were picked by placing cloning cylinders around clearly separate colonies. Immunoblotting was used to select the BRCA1 and BRCA2 depleted clones. Clones were maintained in a media containing 3μg/ml puromycin. For transient transfection, we used 4μg of XRCC1, FEN1 siGenome SMART pool siRNA and Non-Targeting siGenome siRNA as a control (Thermo Scientific). Cells were electroporated using the Nucleofector system (Amaxa), and were harvested 48h post electroporation.
2.3. Immunofluorescence
Cells were seeded in an eight-chamber slide. After the indicated treatment, the slides were washed with PBS, fixed using 4% formaldehyde and 0.2% Triton X-100 in PBS, blocked and permeabilized using 0.2% Triton X-100 and 5% bovine growth serum in PBS for 1h at room temperature. Cells were incubated with primary antibodies overnight. The slides were washed with PBS and the bound antibodies were revealed by IgG Alexa fluor antibodies (Invitrogen). Nuclei were counterstained with DAPI, and slides were mounted for immunofluorescence.
2.4. Immunoblotting
Whole-cell extracts were prepared by lysing cells in radioimmunoprecipitation buffer (150 mM NaCl, 50 mM Tris [pH 8.0], 5 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1.0% Nonidet P-40, 2.0 mM phenylmethylsulfonyl fluoride, 1.0 mM Na3VO4, Halt protease inhibitor cocktail [Pierce]). Protein concentration was quantified using the Bradford assay. 30μg of protein was loaded into 4–12 % Bis-Tris (FEN1, XRCC1, APE1, OGG1, γH2AX and ACTIN) and 50μg of protein was loaded into 3% to 8% Tris acetate (BRCA1, BRCA2, ACTIN) precast gels (Invitrogen). These gels were subjected to SDS-PAGE, transferred onto a polyvinylidene difluoride Immobilon P membrane (Millipore), and blocked with 5% milk – TBST (50 mM Tris [pH 7.5], 150 mM NaCl, 0.05% Tween 20) for 1 h at room temperature. Immunodetection was performed using the following antibodies: anti-BRCA1, anti BRCA1, anti-FEN1, anti-XRCC1, anti-γH2AX, anti-APE1, anti-OGG1 and anti-Actin. Goat anti-mouse IgG and goat anti-rabbit IgG conjugated to horseradish peroxidase were used for secondary antibodies. Bands were detected using ECL chemiluminescence detection methods (PerkinElmer) and exposure to X-ray film (Molecular Technologies).
2.5. 8-oxo-dG analysis
Cells were treated with 200 μM H2O2 for an hour and were fixed using 1% (w/v) paraformaldehyde (in PBS, pH 7.4 + 2.5mM EDTA) for 15 min on ice. Cells were then stored in 70% ethanol at −20°C until processed. Cells were rinsed by centrifugation (800 g, 5 mins) and oxidative DNA damage was detected using primary antibody against 8-oxo-dG (Trevigen) 1:250 in PBS containing 1% w/v BSA, 0.1% v/v Tween20 (BSA-T-PBS) for 1 hr at RT. Cells were then washed with BSA-T-PBS and stained with 5μg/ml of Goat anti-mouse IgG conjugated to Alexa488 (Life technologies) in BSA-T-PBS for 1 hr at RT (protected form light). Samples were immediately analyzed by flow cytometry (LSR II). To keep the basal levels of oxidative DNA damage low, these experiments were conducted at 3% Oxygen tension.
2.6 EdU and Propidium iodide dual staining cell cycle analysis
For the analysis of cell cycle progression, after H2O2 treatment, exponentially growing cells were treated with 10 μM EdU (Click-iT® EdU Flow Cytometry Assay Kits, Invitrogen, Catalogue # C10424). Cells were fixed and permeabilized according to the manufacture’s protocol. For propidium iodide staining, fixed EdU stained cells were incubated with 5-μg/ml propidium iodide (PI) (Sigma Aldrich) and 100 μg/ml RNAse (Fermentas) in PBS for 30 mins at room temperature. Cells were analyzed by flow cytometer (BD-LSR II)
2.7. Antibodies
The following antibodies and dilutions were used for immunofluorescence: 53BP1: 1:200 (Abcam), BRCA1: 1:300 (Santa Cruz Biotechnology), BRCA2: 1:50 (Santa Cruz biotechnology), PCNA: 1:100 (Santa Cruz Biotechnology), Rad51: 1:600 (Calbiochem). The following antibodies and dilutions were used for immunoblotting: Actin: 1:2000 (Millipore), BRCA1: 1:100 (Calbiochem), BRCA2: 1:50 (Calbiochem), FEN1: 1:500 (BS Biosciences), SMC1: 1:1000 (Bethyl laboratories), XRCC1: 1:500 (Abcam), APE1: 1:500 (Novus biologicals), OGG1:1:500 (Novus biologicals) and γH2AX: 1:1000 (Millipore).
2.8. Survival assays
All the cells lines (Control, BRCA1 and BRCA2 shRNA depleted MCF-7 cells, HCC137 and EUFA), were treated with different concentrations of H2O2 (as per the experiment requirement) in serum-free media for an hour at 37 °C. After 1h H2O2 treatment, cells were rinsed, harvested and plated at various cell densities in P60 dishes. Cells were allowed to grow in fresh media for 15 days. Visible colonies were identified with methanol fixation and Crystal Violet staining. The number of colonies per dish was counted and the surviving fractions were calculated as the ratio of the plating efficiencies of treated cells to untreated cells. The mean value ± SD for three independent experiments was determined.
For the MTS assay, the CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega) was used following the manufacturer’s instruction. Briefly, MCF-7 cells were seeded into 96-well plates at a density of 1×10^4 per well and 4h later a media containing various concentrations of H2O2 was added. The cells were incubated for 24 h at 37°C. The following day, fresh media was added with 10μl of the MTS reagent into each well and cells were incubated at 37°C for 2h. The absorbance was detected at 490 nm with a Microplate Reader (Infinite M-200, Tecan).
2.9. Chromosome aberrations
Metaphase chromosome spreads were prepared from exponentially growing MCF-7 cells using standard procedures [26]. Cells were treated with 40μM H2O2 for 1h and then fresh media was added. After 8h, cells were treated with colcemid (0.1μg/ml) for 3h, trypsinized, and pelleted. Following hypotonic swelling in 0.075 M potassium chloride for 10 min at 37°C, cells were fixed in three changes of 3:1 methanol/glacial acetic acid at room temperature for 10 min each. Cells were dropped onto humidified, clean glass slides, dried overnight, then stained in 0.08μg/ml 4,6-diamidino-2-phenylindole (DAPI) in 2x SSC, rinsed, air dried and mounted in antifade (Vectorshield, Vector Labs). Individual metaphases were captured and analyzed using a Nikon E800 epifluorescence microscope equipped with a digital imaging system (Applied Spectral Imaging, Inc., Vista, CA). A minimum of 25 DAPI-banded images from each sample was scored for the presence of structural chromosomal aberrations (chromatid breaks, triradials, quadriradials, & complex chromatid exchanges).
3.0. Statistical analysis
All data were presented as mean ± standard deviation of the mean (SE) of at least three experiments. Statistical calculations were performed within the GraphPad Prism data plotting software. Differences between individual groups were analyzed by paired t-test.
3. Results
3.1. Functional validation of Isogenic BRCA1- and BRCA2-deficient cells
One of the rate-limiting steps in examining the function of BRCA1 and BRCA2 is the availability of stable isogenic pairs of BRCA-proficient and deficient cells. BRCA1 and BRCA2 shRNA depleted MCF-7 cells were generated and confirmed by western blotting and immunofluorescence. By western blotting, clone #12 for shBRCA1 and clone #A for shBRCA2 showed a >90% depletion of the target protein (Fig 1A, 1B) and were used in the subsequent experiments. IR treated BRCA1-deficient or BRCA2-deficient clones showed reduced BRCA1 and BRCA2 foci formation, respectively, by immunofluorescence, compared to wild type cells (Fig 1C). Cell lines defective in BRCA1 or BRCA2 are reported to be sensitive to IR [26–29]. The BRCA1 or BRCA2-deficient cell clones were exposed to increasing doses of IR and their colony forming ability was reduced compared to wild-type MCF-7 cells (Fig 1D).
Figure 1. Depletion of BRCA1 and BRCA2 by shRNA in MCF-7 cells.

MCF-7 cells were stably transfected with empty vector (pKLO.1) or with short hairpin RNA constructs against BRCA1 or BRCA2. The efficacy of the shRNA constructs in depleting BRCA1 (A) and BRCA2 (B) was determined by immunoblotting (actin served as loading control) and confirmed by immunofluorescence. (C) Confocal microscopic images of cell clones using immunofluorescent staining for BRCA1 and BRCA2. Wild type (empty vector) and BRCA1 or BRCA2 deficient MCF-7 cells were irradiated (10 Gy) and 4h later, the cells were fixed and processed for BRCA1 (red) or BRCA2 (green) immunofluorescence. The nuclei were identified by DAPI staining (blue). (D) Relative survival of BRCA1- or BRCA2-deficient cells after exposure to IR. Data are the mean and SE of the mean from three independent experiments. The survival of BRCA1- and BRCA2- deficient cells relative to wild type MCF-7 at 6 Gy was compared for statistical significance. (E) Formation of Rad51 foci (green) in response to DNA damage induced by IR. Nuclei were stained with DAPI. Representative nuclei are displayed from either untreated or irradiated cells. Quantification of the percentage of foci positive cells is shown (mean and SE of the mean from three independent experiments).
BRCA2 is required for IR-induced assembly of Rad51 complex in vivo [30]. Wild type and BRCA-deficient MCF-7 cells were exposed to 10 Gy IR and were subjected to fluorescence microscopy. Rad51 foci formation was impaired in both the shBRCA1 and shBRCA2 cells following IR treatment, whereas the wild type cells showed a normal increase in the number of nuclear Rad51 foci in response to IR (Fig 1E and 1F). Therefore, these results are consistent with previous studies and suggest that BRCA1 and BRCA2 are required for Rad51 foci formation in response to IR-induced DNA damage in MCF-7 cells.
3.2. BRCA-deficient cells are sensitive to oxidative stress by H2O2 treatment
To investigate whether BRCA1 or BRCA2 deficiency results in sensitivity to H2O2, the clonogenic survival of cells treated with various concentrations of H2O2 for 1h was measured. It has been shown that 500μM of H2O2 produced about as many DSBs as 2 Gy IR [32]. However, we chose to use doses of H2O2 in the range of 1 to 40μM, to be more representative of the local concentrations in the nucleus and a dose that produces relatively little cell death in normal cells. Both BRCA1 and BRCA2-deficient cells were more sensitive to H2O2 compared to wild-type cells (Fig 2A). At the highest utilized dose of 40μM H2O2, BRCA1 and BRCA2 deficient cells showed a 10 and a 100-fold increase in sensitivity relative to wild-type cells, respectively. Measuring survival by the MTS cell growth assay also found increased sensitivity of both BRCA1 and BRCA2-deficient cells to H2O2 treatment (Fig 2B), although the measured decrease in survival is reduced using a growth assay compared with a clonogenic assay. To further support our findings we also performed same experiments in BRCA1/BRCA2 deficient and proficient cell line pairs (HCC1937 with the empty vector pcDNA3 and HCC1937 complemented with a full length wtBRCA1 construct; EUFA423 and EUFA423 complemented with a full length wtBRCA2 construct). Consistent with the BRCA-depletion experiments, BRCA-deficient cell lines were sensitive to H2O2 treatment relative to complemented cells (Fig 2C and 2D). BRCA1- and BRCA2- deficient cells are sensitive to mitomycin C and IR, both of which generate oxidative damage and double-stranded DNA lesions [33, 34].
Figure 2. BRCA1- and BRCA2-deficient cells are sensitive to H2O2.

(A) Clonogenic survival of BRCA-deficient cells after exposure to H2O2. Data shown are the mean and SE of the mean from three independent experiments. The survival of BRCA1 and BRCA2 deficient cells relative to wild type MCF-7 at 40μM was compared for statistical significance. (B) MTS assay of BRCA-deficient cells after exposure to H2O2. (C) Clonogenic survival of BRCA1-deficient cell line – HCC1937 containing pcDNA3 empty vector and HCC137 complemented with a full-length wtBRCA1 construct after exposure to H2O2. Data shown are the mean and SE from three independent experiments. (D) Clonogenic survival of BRCA2-deficient cell line – EUFA and EUFA complemented with a full-length wtBRCA2 construct after exposure to H2O2. Data shown are the mean and SE from three independent experiments. (E) XRCC1-depletion in wild type and BRCA-deficient cells. Cells were harvested 48h after siXRCC1 or control siRNA (nt) nucleofection and cell lysates were immuno-blotted for XRCC1. Clonogenic survival of wild type and BRCA deficient cells nucleotransfected with siXRCC1 or control siRNA (nt) after exposure to H2O2 as in A. (F) The effect of FEN1-depletion as in E. (G) Representative immunoblots confirming XRCC1 depletion in BRCA1 and BRCA2 deficient cells. (H) Representative immunoblots confirming FEN1 depletion in BRCA1 and BRCA2 deficient cells. Actin was used as loading control. Cell lines were nucleotransfected with FEN1/XRRC1 siRNA and post 48 hrs from transfection, cells were harvested for immunoblotting.
We assessed whether components of the BER pathway were involved in the removal of oxidative DNA lesions under the same conditions. We knocked down XRCC1 in wild-type and in BRCA-deficient cells (Fig. 2G) and 48 hours later, treated cells with H2O2 as previously indicated. The survival of cells transfected with siRNA against XRCC1 was not significantly reduced relative to the non-targeting siRNA (nt) in MCF-7 cells with wild-type BRCA1 and BRCA2 proteins (Fig 2E). More importantly, the effect of XRCC1-depletion in BRCA-depleted cells also showed little enhancement of cell killing up to 40-μM H2O2 (Fig 2E). To investigate the possibility that the lack of effect of XRCC1-depletion on survival was due to repair by long-patch BER in these proliferating cells, we also depleted FEN1 in the same series of MCF-7 cells (Fig. 2H). We found independent cell killing as a result of depleting FEN1 by approximately 2-fold in all cell types, but no interactive effect of combined FEN1 and BRCA1 or BRCA2 depletion (Fig 2E). If both FEN1 and XRCC1 are depleted simultaneously, the results were no different to FEN1-depletion alone (data not shown).
The levels of 8-oxodeoxyguanosine (8-oxo-dG) was measured as a marker of H2O2-mediated DNA damage [35]. Cells were treated with H2O2, fixed, labeled with an anti-8-oxo-dG antibody and observed by flow cytometry. We treated cells with 200μM H2O2 for 1 h, which induces base lesions, sugar modifications and single-strand breaks (SSB). To keep the basal levels of oxidative DNA damage low, these experiments were conducted in 3% oxygen. Higher H2O2 concentrations and longer treatments can lead to DSB that might impede the measurement of 8-oxo-dG. After H2O2 treatment, there was an induction of oxidative DNA base modification as read by 8-oxo-dG lesions in comparison to the untreated cells in wild-type cells (Fig. 3A and 3B). FEN1 and XRCC1 depletion resulted in higher basal levels of 8-oxo-dG, which increased after H2O2 treatment to a higher level than wild-type cells (Fig. 3A and 3B). BRCA1-deficient cells have been reported to have higher levels of ROS compared to wt cells (Saha et al., 2009), which was confirmed in our studies, showing increased basal and induced levels of 8-oxo-dG in BRCA1 deficient cells compared to wt cells (Fig. 3A). Interestingly, FEN1-BRCA1 or XRCC1-BRCA1 co-depletion showed no differences in 8-oxo-dG levels before and after H2O2 treatment (Fig 3A), perhaps due to an adaptive response the co-depleted cells have lower levels of 8oxodG when compared to BRCA1/FEN1/XRCC1 single depleted cells (Fig 3A). BRCA2 deficient cells also show higher levels of 8-oxo-dG levels compared to wt cells (Fig 3B), although the effect is less marked than for BRCA1-deficiency. FEN1-BRCA2 or XRCC1-BRCA2 co-depleted cells have moderately higher levels of 8oxodG after H2O2 treatment (Fig 3B). However, FEN1-BRCA2 or XRCC1-BRCA2 co-depleted cells have lower levels of oxidative DNA damage compared to the BRCA2/FEN1/XRCC1 single depleted cells, again suggestive of an adaptive response (Fig 3B). In order to investigate whether this adaptive response is due to upregulation of other major BER proteins we analyzed the expression of APE1 and OGG1 and found no change in the expression of these BER proteins (Supplementary Figure 1). These results suggest that by co-depleting either BRCA1 or BRCA2 and components of the BER pathway, cells are pushed into a high oxidative stress environment and as a survival mechanism there is an adaptive response. We reason that other mechanisms such as enhanced antioxidant response or enhanced removal of oxidized dNTP pool might be at work in these co-depleted cells, which is the subject of future work. Recently evidence for such adaptive response against high levels of oxidative environment has been shown in cancer cells [31, 32]. However, the status of BER proteins or the adaptive response, does not affect the sensitivity of BRCA-deficient cells to hydrogen peroxide. The implication is that BRCA1 and BRCA2 participate in the DNA repair and survival after treatment with hydrogen peroxide. The critical DNA lesion from oxidative DNA damage in MCF-7 cells is independent of the long-and short patch BER pathway.
Figure 3. A: Levels of 8-oxo-dG in BRCA1-depleted and FEN1-BRCA1 or XRCC1-BRCA1 co-depleted cells.<.
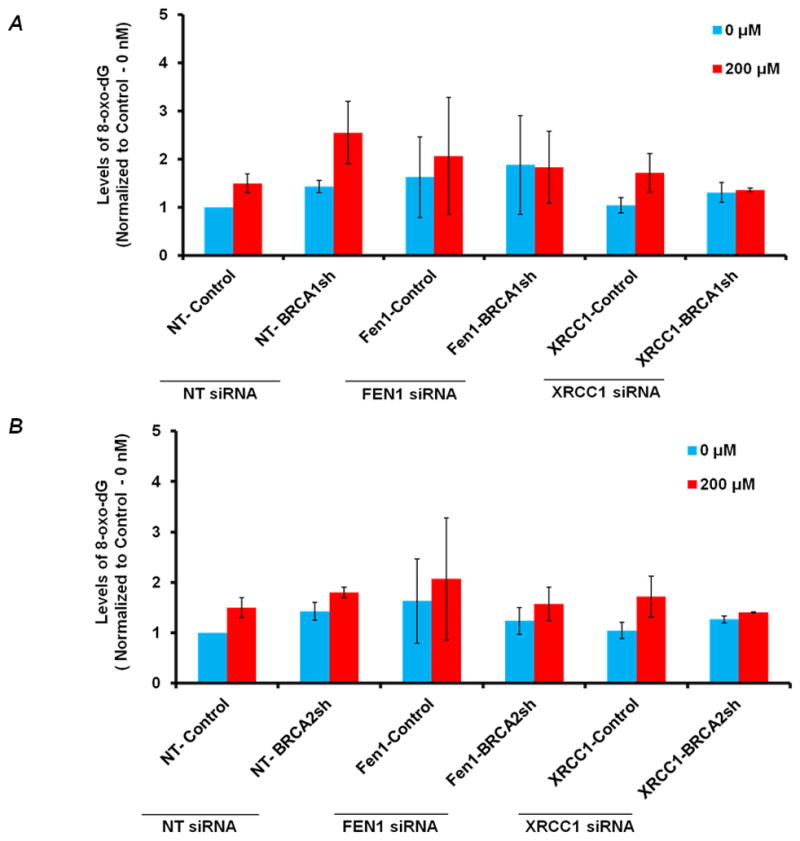
br>The relative levels of 8-oxo-dG (Mean Fluorescence Intensity, MFI –normalized to 0-nM of control cells) in BRCA1-depleted, FEN1-BRCA1 and XRCC1-BRCA1 co-depleted cells respectively before and after H2O2 treatment. Cells were nucleofected with FEN1 and XRCC1 siRNA respectively and 48 hrs after transfection treated with 200μM of H2O2 for 1 hr. Subsequently, harvested cells were subjected to 8-oxo-dG analysis by flow cytometry. 8-oxodG was determined by using primary antibody against 8-oxodG (Trevigen) and Alexa fluor 488-Rabbit-anti-Mouse secondary antibody IgG (H+L) (Molecular Probes®) using FACS (LSRII). Results are from three independent experiments and are expressed as the mean ± SE. To reduce the basal levels of oxidative DNA damage cells were incubated in 3% Oxygen incubator. B: Levels of 8-oxo-dG in BRCA2-depleted and FEN1-BRCA2 or XRCC1-BRCA2 co-depleted cells: Relative levels of 8-oxo-dG (Mean Fluorescence Intensity, MFI – normalized to 0-nM of control cells) in BRCA2-depleted, FEN1-BRCA2 and XRCC1-BRCA2 co-depleted cells respectively before and after H2O2 treatment as in A.
3.3. BRCA1 and BRCA2-deficient cells accumulate in S phase after oxidative stress
Proliferating cell nuclear antigen (PCNA) is a co-factor for the DNA replicative polymerases δ and ε, and is involved in DNA replication processivity. We used PCNA staining in chromatin bound foci as a marker of S-phase in MCF-7 cells [37]. PCNA is present throughout the cell cycle in proliferating cells, but only associates with chromatin in “replication centers” in S-phase [38]. Therefore, in cells where the nuclear envelope is broken down by gentle detergent treatment, only chromatin bound proteins remain readily visible. Similarly, 53BP1 staining was used as a marker of DNA DSBs, since other histone changes, such as γ-H2AX, can show foci independently of DSB formation. Cells were treated with H2O2 for various times of exposure [39], fixed and then stained for PCNA (green) and 53BP1 (red; Fig. 4A). At the zero time-point, approximately 30% of the wild type and the BRCA deficient cells were in S-phase and 53BP1 was barely detectable. After 4h of treatment with H2O2, the percentage of S-phase cells remained constant in the wild type sample (~30%), whereas the S-phase fraction accumulated significantly in the BRCA deficient cells (80%; Fig. 4B). After treatment with hydrogen peroxide, the number of cells with 53BP1 foci was similar at 2h in wild-type and BRCA-deficient cells (data not shown), becoming elevated at 4h in the BRCA-deficient cells (Fig. 4A). Thus, there was an accumulation of 53BP1 foci in S-phase cells in BRCA-deficient cells, which reflects the development of DSB in replicating cells, without the intact homologous recombination machinery.
Figure 4. BRCA-deficient cells accumulate in S phase due to oxidative stress-induced double-strand breaks.

(A) Cells were treated with 200μM H2O2 for 4h and the immunofluorescence staining pattern was imaged. Double-strand breaks are shown by 53BP1 (red); S-phase cells are shown by the presence of PCNA in nuclear foci (green). (B) Quantification of the percentage of PCNA foci-positive cells from A. Data shown are the mean and SE from three independent experiments. (C) Quantification of S phase arrested BRCA deficient cells. Cells were treated with 200μM H2O2 for 4h and flow-cytometric analysis of S phase arrested cells after exposure to H2O2 treatment (EdU negative – propidium iodide S phase DNA content) showing reduced incorporation of EdU were measured (additional data in Fig S3). Flow cytometric data is normalized to untreated control cells. Data shown are the mean and SE from three independent experiments. (D) Representative Immunoblots of γH2AX accumulation in BRCA1, BRCA2 deficient and control cells. Actin was used as loading control. Cell lines were treated with 200μM H2O2 for shown time points and were harvested for immunoblotting. (E) Formation of Rad51 foci (green) 4h after treatment with 200μM H2O2 for 1h.
The increase in S-phase population seen by immuno-fluorescence for PCNA foci was confirmed by EdU-PI-dual staining flow cytometry analysis (Fig. 4C). EdU incorporation was analyzed in cells stained with propidium iodide after and before H2O2 treatment. It has been shown that EdU (and similar nucleoside analogs such as BrdU) incorporation after H2O2 treatment is severely attenuated in S phase cells since these cells are arrested in S phase [33, 34]. Thus, in order to measure S phase arrested cells, S phase cells (assessed by propidium iodide staining) that showed reduced incorporation of EdU (EdU negative cells) were measured (Fig 4C). Similar studies were performed on FEN1-BRCA1, XRCC1-BRCA1, FEN1-BRCA2 and XRCC1-BRCA2 co-depleted cells (Fig S3) also revealing that S phase cells were increased in BRCA1 or BRCA2 depleted cells independent of the status of FEN1 or XRCC1. These observations suggest that the BRCA and BER pathways handle oxidative DNA damage independently. There is an adaptive response observed in co-depleted cells demonstrated by relatively low levels of 8-oxo-dG and no enhanced cell kill in co-depleted cells (Fig 3A–B, 2E–F). The interpretation is that oxidative DNA damage can produce DNA DSBs in S-phase and that BRCA-deficient cells have difficulty in processing the S-phase DSBs, which result in an accumulation of cells in S-phase and increased cell killing.
γ-H2AX acts as a scaffold for many proteins during the DNA damage response (DDR) and it governs the persistence of the DDR complex [34, 35]. ATM and Rad3 related (ATR) phosphorylates γH2AX during S phase in response to SSB induced replication fork stalling [36, 37]. ATR mediated γH2AX can also be observed during UV induced nucleotide excision repair, while processing single strand intermediates in G0, G1 and G2 phases [38–40]. Recently, it was reported that H2O2 induces a heterogeneous ATR induced γH2AX signal which is DSB independent [35]. In BRCA1 or BRCA2 deficient cells, there is a measurable increase in γH2AX signal at 30 minutes (Fig. 4D), but by 60 minutes there is no detectable difference between BRCA-deficient cells and wild-type. At later time-points, the BRCA-deficient cells have a reduced γH2AX signal, which may be due to an impairment of DNA damage signaling that is dependent on BRCA1 and BRCA2. The conclusion is that γH2AX in not significantly increased in BRCA-deficient cells after treatment with H2O2, suggesting that the generation of DSB is not different in BRCA-deficient cells, but there is an inability of the cells to mount the appropriate DNA repair response.
When BRCA-deficient MCF-7 cells were exposed to 200μM H2O2, they showed reduced Rad51 foci compared to wild-type cells (see Fig. 4E), which suggests that BRCA1 and BRCA2 are required for Rad51 foci formation in response to H2O2-induced DNA damage. The implication is that the DSB that are formed in S-phase cells cannot be processed by the HR machinery. These results suggest that HR is utilized in the repair of this subset of oxidative DNA damage, DSB produced in S-phase, which in turn is responsible for cytotoxicity.
3.4. Oxidative damage causes chromosomal instability in BRCA1- and BRCA2- deficient cells
One direct hypothesis of the increase and persistence of DSBs in S-phase is that illegitimate end-joining will occur as a consequence of the failure to process the DSBs in the normal time frame. We questioned whether BRCA-deficient cells could produce chromatid-type aberrations as a direct result of oxidative DNA damage, which would support the hypothesis. We analyzed metaphase chromosome spreads of wild type and BRCA-deficient cells after treatment with a low concentration of H2O2: 40μM. Compared with the untreated wild-type cells, the untreated BRCA1- and BRCA2- deficient cells exhibited a higher frequency of chromatid-type breaks, but did not quite reach statistical significance (Fig. 5A). However, H2O2 treatment induced a 2.9- and 5.8-fold increase in the number of chromatid-type aberrations/cell in BRCA1- and BRCA2-deficient cells respectively compared to wild type cells (Fig. 5A, 5B). In addition, the H2O2 led to dicentrics, triradials and quadriradials in the BRCA-deficient cells, which were rarely seen in wild-type cells. The number of cells with multiple chromatid breaks per cell was higher in the BRCA-deficient cells compared to wild type cells (Supplementary Figure 2). These results suggest the involvement of BRCA1 and BRCA2 in the repair of H2O2-induced DSBs arising in S phase by HR. The implication is that BRCA1- or BRCA2-deficiency results in significant genetic instability as a consequence of oxidative stress and its consequential DNA damage in S-phase.
Figure 5. H2O2 induces chromosome aberrations in BRCA1- and BRCA2-deficient cells.
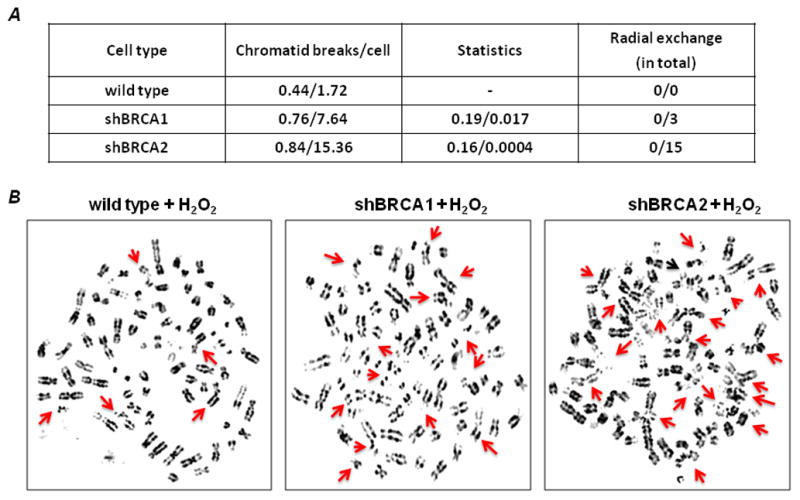
Untreated and H2O2-treated cells were subjected to cytogenetic analysis. (A) Chromatid breaks are shown as the average number of breaks per cell. Spontaneous aberrations were derived from the analysis of 50 metaphase spreads and are shown as the first result for each cell type; the second result is following treatment with H2O2. Statistical comparisons are shown for BRCA1- and BRCA2-deficient cells compared with wild type cells. Radial exchanges are shown in the fourth column. (B) Representative images of H2O2-treated metaphase spreads. Arrows indicate chromatid/chromosome breaks and radial exchanges in each cell type. (See additional details in Supplementary Figure 2).
4. Discussion
4.1. Oxidative Stress, DNA damage in breast epithelium, homologous recombination and breast cancer
BRCA1 and BRCA2 are tumor suppressor genes that participate in DNA repair by homologous recombination. Humans with mutations of either the BRCA1 or BRCA2 genes are at a higher risk of developing breast and ovarian cancer, but why is the cancer risk tissue-specific? Mammary tissue is exposed to an elevated level of oxidative stress because of hormonally regulated metabolism [6, 41]. As proliferation signals increase with the development of cancer, additional growth factor stimulation by estrogens and non-estrogenic pathways result in measurable oxidative stress and oxidative DNA damage. The results in this paper have clearly shown that cytotoxicity from hydrogen peroxide is not due to defects in BER, since the two pathways, BER and BRCA, when manipulated independently show that the BRCA-status dominates the effect on cell survival. When single-strand break repair intermediates are created and persist into S-phase, they can be converted into DSB, which we surmise is the cytotoxic lesion from hydrogen peroxide.
To protect against hydrogen peroxide resulting in DSB in S-phase, homologous recombination by the BRCA1/BRCA2 pathway is needed to repair the potentially clastogenic DNA lesions that arise. We have focused on the consequences of oxidative DNA damage and the requirement of HR for the subset of cytotoxic lesions that form in S-phase. Thus, although reactive oxygen species produces many types of DNA lesions, the vast majority of lesions are repaired by rapid and efficient DNA repair pathways of BER (and potentially nucleotide excision repair (NER) as a back-up to BER). We hypothesize that the relatively rare lesions that persist into S-phase such as the single-strand break could be converted into an S-phase associated DSB. Defects in any member of the BRCA1-BRCA2 pathway would show the same sensitivity to hydrogen peroxide, which is why we hypothesize that all members of this pathway will ultimately show links to hereditary breast and ovarian cancer.
Lymphoblasts from women with BRCA1 mutations show deficient processing of oxidative DNA lesions [42]. The authors also reported that oxidized DNA lesions inherently accumulated in the BRCA1-deficient breast cancer cell line, HCC1937 [43]. In addition, elevated background levels of oxidative DNA damage were also observed in sporadic breast tumors compared to control cells [6, 42, 44–46]. Therefore, breast cancers arising in the background of increased levels of oxidative DNA damage could be due to functional deficiency in the BRCA1-BRCA2 pathway of DNA repair, which has also been observed in a subset of sporadic cancers [47]. The recovery from oxidative DNA lesions by the BRCA1-BRCA2 pathway may explain why BRCA-deficient cells (and tumors) accumulate certain types of oxidative DNA lesions, resulting in genome instability and chromosome aberrations. The cells used in these studies had either depletion of >90% of the endogenous BRCA1 or BRCA2 protein by shRNA or were a known BRCA1 or BRCA2-deficient cell compared to a wild-type complemented cell. For BRCA1 or BRCA2 mutation carriers, where the heterozygous state is likely to produce an approximately 50% protein reduction based on gene dosage, we do not know if that alone is sufficient to detect some degree of sensitivity to oxidative DNA damage. There may be required second events, such as loss of the second BRCA allele or loss of a cell cycle checkpoint response, which remains to be determined. We know that loss of heterozygosity (LOH) for the BRCA-gene appears to occur early in carcinogenesis, based genotyping of cancers and the adjacent pre-malignant lesions [48], in which LOH has occurred in the pre-malignant tissues in BRCA-mutation carriers.
4.2. Increased sensitivity of BRCA1-BRCA2 deficient cells after H2O2 is independent of Base-Excision Repair
Mammary tissue is exposed to an elevated level of oxidative stress linked to normal metabolism in the face of estrogen driven proliferation. One of the consequences of oxidative stress can be the formation of oxidative DNA lesions, whose repair is reasonably well understood, the first line being base excision repair (BER) [49–51]. The major novel observation in this paper is that cellular sensitivity to oxidative stress and oxidative DNA damage is found in cells with a deficiency in BRCA1 or BRCA2, which is independent of BER. The implication is that oxidative stress is an etiological component of certain types of breast cancer. The hypothesis is that a subset of oxidative DNA lesions persists into S-phase, where they produce DSB that are more cytotoxic and are repaired via replication-linked homologous recombination rather than BER.
The reason for targeting the XRCC1 and FEN1 was to test the integrity of short or long patch BER with single gene depletion. The DNA glycosylases show significant overlapping function and redundancy, which would necessitate simultaneous depletion of the multiple target glycosylases. Depletion of the XRCC1 protein, which affects short-patch BER, had no selective cytotoxic effect on either BRCA-proficient or deficient cells in the presence or absence of oxidative stress. Despite the known interactions of XRCC1 with a number of other repair proteins, such as apurinic/apyrimidinic endonuclease 1 (APE1) and polynucleotide kinase (PNK) that participate in BER and single-strand break repair, XRCC1-deficient cells showed only minor sensitivity to H2O2 up to 40μM [52]. The lack of marked sensitivity to oxidative DNA damage in XRCC1-deficient cells is in contrast to their hypersensitivity to methylating agents, suggesting that oxidized and methylated DNA bases are processed by different repair pathways. Polymerase β is another participant in BER – in the gap filling step [53, 54]; however, polymerase β null fibroblasts also did not show hypersensitivity to H2O2 [55, 56]. Given that BER includes both short-patch and long-patch options, we investigated FEN1-depletion and did find sensitivity to H2O2, which was entirely independent of BRCA1 or BRCA2 manipulations. These findings suggest that long-patch BER may be the predominant mechanism of oxidative DNA damage repair in the nucleus, as reported by [57]. Our findings suggest that when oxidative DNA damage is not accessible to BER proteins, the residual DNA damage can result in fork collapse or daughter-strand gaps, both of which would require HR to mediate their repair [18]. Thus, the cellular sensitivity to H2O2 is determined by the oxidative lesions that persist into S-phase, which need HR proteins to complete the repair process.
4.3. Oxidative DNA damage results in an accumulation of BRCA-deficient cells in S-phase
The absence of BRCA1 or BRCA2 reduces Rad51 foci formation after oxidative stress and ionizing radiation, suggesting that the repair of oxidative DNA damage recruits homologous recombination proteins to the lesions that persist into S-phase and cause DNA double-strand breaks. Homologous recombination preferentially repairs DSB in the S-phase of cell cycle [15]. We used PCNA as a marker of S-phase cells together with 53BP1, a marker for DNA double-strand breaks (DSB) to support the idea that after 4h of persistent oxidative stress, DSB were increased in both BRCA1 and BRCA2-deficient cells, with a resultant accumulation of cells in S-phase. We showed that the number of cells with PCNA foci increase when BRCA1 or BRCA2 is depleted, suggesting that these cells are accumulating in S-phase. EdU-PI dual staining flow cytometry cell cycle analysis also confirmed the observations by immunofluorescence. The accumulation of BRCA-deficient cells in S-phase after oxidative stress is consistent with a previous study that shows sensitivity to ionizing radiation in S-phase of the cell cycle [58]. In addition, Rad51 is more abundant and forms foci preferentially in S-phase cells [59, 60], which likely reflects the importance of HR in replication fidelity [61, 62]. Non-homologous end-joining is not thought to play a role in the one-ended DSB that occurs with replication fork collapse nor in the repair of a daughter strand gap.
4.4. Cytotoxic Oxidative DNA damage is repaired by Homologous Recombination
The major role of the BRCA2 protein is DSB repair by HR. BRCA2 functions as a recruiter of Rad51 protein to the damaged site. The function of BRCA1 in DNA repair includes homologous recombination, but it also has multiple functions in the cellular response to DNA damage, including contributing to the recruitment of BRCA2. The main repair pathway of oxidized DNA lesions in all cells, from E. coli to human, is base excision repair (BER) [7, 8]. The understanding of breast carcinogenesis in the context of BRCA1- or BRCA2-deficiency suggests that both proteins play a role in the repair of oxidative DNA damage, but not via BER.
ROS can directly induce oxidized nucleotides that are subsequently converted to DSBs during replication and then repaired via HR [63, 64]. In yeast, spontaneous recombination is enhanced in mutants with defects in BER (and NER) pathways [65], indicating that unrepaired base damage can be processed entirely by the recombinational pathway. In this study, we show that BRCA1 or BRCA2-deficient cells are sensitive to H2O2 treatment by accumulating oxidative damage and DSBs in S-phase. Residual oxidative DNA damage shows a punctuate pattern of staining in the nucleus, with the signal more intense on the periphery, probably in heterochromatic DNA [66], which is replicated late in S-phase. However, the pattern of DSB seen in BRCA-deficient cells in S-phase is not restricted to the nuclear periphery, suggesting that a subset of oxidative DNA damage is contributing to the observed genetic instability.
4.5. Hydrogen peroxide is clastogenic producing chromatid aberrations in BRCA-deficient cells
Chromatid breaks and exchanges can be induced by ionizing radiation in the S and G2 phases of the cell cycle, when the chromosome has replicated into 2 chromatids. Depletion of the BRCA1 or BRCA2 protein by RNA interference resulted in an increase in spontaneous chromatid aberrations. The replication-linked DSB produced by hydrogen peroxide are repaired by homologous recombination. The key observation in this paper is that hydrogen peroxide can induce clastogenic events, which is not the expected finding for sparse oxidative DNA damage. However, when cells move into S phase, there is conversion of single-strand breaks into DSB, the clastogenic lesion. When there is a deficiency of BRCA1 or BRCA2, a marked increase in chromatid aberrations is observed, as in this paper. In the absence of HR proteins, illegitimate end-joining is the only option for repairing the DSB, which results in the chromatid aberrations. The precise mechanism of illegitimate end-joining is not understood, as it could involve RAD52, alternative end-joining or classical non-homologous end-joining.
Breast cancers arising in BRCA mutation carriers occur as a consequence of the genetic instability created by the illegitimate end-joining of DSB in S phase [67]. The production of a radial chromosome or even a chromatid break is likely lethal to one or both daughter cells. However, illegitimate end-joining could produce interstitial deletions or insertions, or in rare cases a chromosomal translocation in one of the daughter cells without producing lethality. This is exactly the phenotype seen in human breast cancers arising in BRCA1 or BRCA2 mutation carriers. We have shown that BRCA1 or BRCA2 deficient cells are susceptible to DSB induced by oxidative stress and modeled by hydrogen peroxide. We propose that oxidative stress in hormonally-driven breast or ovarian epithelium can promote genetic instability in cells with a loss of function of the BRCA1-BRCA2 pathway of homologous recombination and hence promote carcinogenesis. Although there must be additional genetic (and perhaps epigenetic) alterations to produce a breast cancer, the vulnerability of breast epithelium to oxidative stress is likely to be a major driving force of breast cancer development.
Supplementary Material
(A) Representative Immunoblots of OGG1 and APE1 proteins in FEN1-BRCA-co-depleted and control cells. Actin was used as loading control. Cells were nucleofected with FEN1 siRNA and 48 hrs post transfection treated with 200μM of H2O2 for 1 hr and were harvested for immunoblotting. (B) Representative Immunoblots of OGG1 and APE1 proteins in XRCC1-BRCA-co-depleted and control cells treated as in A.
Untreated and H2O2-treated cells were analyzed cytogenetically. Number of chromatid-type breaks per cell is increased in BRCA-deficient cells.
BRCA deficient and control cells were nucleofected with FEN1 and XRCC1 siRNA respectively and 48 hrs post transfection treated with 200μM of H2O2 for 4 hrs. Subsequently, cells were subjected to dual staining (EdU + PI) to analyze S phase stuck cells using flow-cytometry. Graph represents relative folds of S phase population that are not able to uptake EdU (EdU negative) because they are stuck in S phase but are positive for PI staining. Data is normalized to untreated control cells. Data shown are the mean and SE from three independent experiments.
Highlights.
BRCA-deficient cells are sensitive to H2O2 induced oxidative DNA damage
Oxidative DNA damage results in the accumulation of DNA double-strand breaks during S-phase
Increased chromosomal breaks are triggered by oxidative stress in BRCA-deficient cells
Genetic instability in BRCA-deficient cells is increased in tissues subjected to oxidative stress
Acknowledgments
We would like to acknowledge the MSKCC Molecular Cytogenetics Core. This work was supported by PHS grants CA107640 and CA169306, plus Susan G. Komen for the Cure.
Footnotes
Conflict of Interest Statement
Simon Powell have no financial conflicts of interest with any of the work in this submitted paper.
Simon Powell have a single position of employment (Memorial Sloan-Kettering) and no other sources of income.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McCord JM. Superoxide dismutase in aging and disease: an overview. Methods Enzymol. 2002;349:331–41. doi: 10.1016/s0076-6879(02)49348-2. [DOI] [PubMed] [Google Scholar]
- 2.Brown NS, Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 2001;3(5):323–7. doi: 10.1186/bcr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hursting SD, et al. Mechanism-based cancer prevention approaches: targets, examples, and the use of transgenic mice. J Natl Cancer Inst. 1999;91(3):215–25. doi: 10.1093/jnci/91.3.215. [DOI] [PubMed] [Google Scholar]
- 4.Lavigne JA, et al. The effects of catechol-O-methyltransferase inhibition on estrogen metabolite and oxidative DNA damage levels in estradiol-treated MCF-7 cells. Cancer Res. 2001;61(20):7488–94. [PubMed] [Google Scholar]
- 5.Hamada J, et al. Increased oxidative DNA damage in mammary tumor cells by continuous epidermal growth factor stimulation. J Natl Cancer Inst. 2001;93(3):214–9. doi: 10.1093/jnci/93.3.214. [DOI] [PubMed] [Google Scholar]
- 6.Malins DC, et al. The etiology of breast cancer. Characteristic alteration in hydroxyl radical-induced DNA base lesions during oncogenesis with potential for evaluating incidence risk. Cancer. 1993;71(10):3036–43. doi: 10.1002/1097-0142(19930515)71:10<3036::aid-cncr2820711025>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 7.Aiub CA, et al. Participation of BER and NER pathways in the repair of DNA lesions induced at low N-nitrosodiethylamine concentrations. Toxicol Lett. 2004;154(1–2):133–42. doi: 10.1016/j.toxlet.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Evans MD, Saparbaev M, Cooke MS. DNA repair and the origins of urinary oxidized 2′-deoxyribonucleosides. Mutagenesis. 2010;25(5):433–42. doi: 10.1093/mutage/geq031. [DOI] [PubMed] [Google Scholar]
- 9.Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002;21(58):8981–93. doi: 10.1038/sj.onc.1206176. [DOI] [PubMed] [Google Scholar]
- 10.Le Page F, et al. BRCA1 and BRCA2 are necessary for the transcription-coupled repair of the oxidative 8-oxoguanine lesion in human cells. Cancer Res. 2000;60(19):5548–52. [PubMed] [Google Scholar]
- 11.O’Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7(1):45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- 12.Marians KJ. Mechanisms of replication fork restart in Escherichia coli. Philos Trans R Soc Lond B Biol Sci. 2004;359(1441):71–7. doi: 10.1098/rstb.2003.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Treszezamsky AD, et al. BRCA1- and BRCA2-deficient cells are sensitive to etoposide-induced DNA double-strand breaks via topoisomerase II. Cancer Res. 2007;67(15):7078–81. doi: 10.1158/0008-5472.CAN-07-0601. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005;3(10):531–9. doi: 10.1158/1541-7786.MCR-05-0192. [DOI] [PubMed] [Google Scholar]
- 15.Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004;32(12):3683–8. doi: 10.1093/nar/gkh703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miki Y, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 17.Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22(37):5784–91. doi: 10.1038/sj.onc.1206678. [DOI] [PubMed] [Google Scholar]
- 18.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12(1):68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 20.Wong AK, et al. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997;272(51):31941–4. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- 21.Bochkarev A, Bochkareva E. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr Opin Struct Biol. 2004;14(1):36–42. doi: 10.1016/j.sbi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467(7316):678–83. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharan SK, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386(6627):804–10. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 24.Shen SX, et al. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17(24):3115–24. doi: 10.1038/sj.onc.1202243. [DOI] [PubMed] [Google Scholar]
- 25.Tutt AN, et al. Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation. EMBO Rep. 2002;3(3):255–60. doi: 10.1093/embo-reports/kvf037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abbott DW, Freeman ML, Holt JT. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst. 1998;90(13):978–85. doi: 10.1093/jnci/90.13.978. [DOI] [PubMed] [Google Scholar]
- 27.Foray N, et al. Gamma-rays-induced death of human cells carrying mutations of BRCA1 or BRCA2. Oncogene. 1999;18(51):7334–42. doi: 10.1038/sj.onc.1203165. [DOI] [PubMed] [Google Scholar]
- 28.Morimatsu M, Donoho G, Hasty P. Cells deleted for Brca2 COOH terminus exhibit hypersensitivity to gamma-radiation and premature senescence. Cancer Res. 1998;58(15):3441–7. [PubMed] [Google Scholar]
- 29.Scully R, et al. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4(6):1093–9. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- 30.Yuan SS, et al. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59(15):3547–51. [PubMed] [Google Scholar]
- 31.Gorrini C, et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci U S A. 2014;111(12):4472–7. doi: 10.1073/pnas.1324136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gad H, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508(7495):215–21. doi: 10.1038/nature13181. [DOI] [PubMed] [Google Scholar]
- 33.Barnouin K, et al. H2O2 induces a transient multi-phase cell cycle arrest in mouse fibroblasts through modulating cyclin D and p21Cip1 expression. J Biol Chem. 2002;277(16):13761–70. doi: 10.1074/jbc.M111123200. [DOI] [PubMed] [Google Scholar]
- 34.Celeste A, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5(7):675–9. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 35.Katsube T, et al. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J Biochem. 2014 doi: 10.1093/jb/mvu021. [DOI] [PubMed] [Google Scholar]
- 36.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward IM, Minn K, Chen J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem. 2004;279(11):9677–80. doi: 10.1074/jbc.C300554200. [DOI] [PubMed] [Google Scholar]
- 38.Hanasoge S, Ljungman M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis. 2007;28(11):2298–304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- 39.Marti TM, et al. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103(26):9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsumoto M, et al. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J Cell Sci. 2007;120(Pt 6):1104–12. doi: 10.1242/jcs.03391. [DOI] [PubMed] [Google Scholar]
- 41.Sipe HJ, Jr, et al. The metabolism of 17 beta-estradiol by lactoperoxidase: a possible source of oxidative stress in breast cancer. Carcinogenesis. 1994;15(11):2637–43. doi: 10.1093/carcin/15.11.2637. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez H, et al. Lymphoblasts of women with BRCA1 mutations are deficient in cellular repair of 8,5′-Cyclopurine-2′-deoxynucleosides and 8-hydroxy-2′-deoxyguanosine. Biochemistry. 2007;46(9):2488–96. doi: 10.1021/bi062022p. [DOI] [PubMed] [Google Scholar]
- 43.Georgakilas AG, et al. BRCA1 involvement in toxicological responses and human cancer etiology. Toxicol Lett. 2009;188(2):77–83. doi: 10.1016/j.toxlet.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Li D, et al. Oxidative DNA damage and 8-hydroxy-2-deoxyguanosine DNA glycosylase/apurinic lyase in human breast cancer. Mol Carcinog. 2001;31(4):214–23. doi: 10.1002/mc.1056. [DOI] [PubMed] [Google Scholar]
- 45.Malins DC, Haimanot R. Major alterations in the nucleotide structure of DNA in cancer of the female breast. Cancer Res. 1991;51(19):5430–2. [PubMed] [Google Scholar]
- 46.Malins DC, Polissar NL, Gunselman SJ. Progression of human breast cancers to the metastatic state is linked to hydroxyl radical-induced DNA damage. Proc Natl Acad Sci U S A. 1996;93(6):2557–63. doi: 10.1073/pnas.93.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willers H, et al. Utility of DNA repair protein foci for the detection of putative BRCA1 pathway defects in breast cancer biopsies. Mol Cancer Res. 2009;7(8):1304–9. doi: 10.1158/1541-7786.MCR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber F, et al. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am J Hum Genet. 2006;78(6):961–72. doi: 10.1086/504090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bae I, et al. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004;64(21):7893–909. doi: 10.1158/0008-5472.CAN-04-1119. [DOI] [PubMed] [Google Scholar]
- 50.Nitta M, et al. Targeting EGFR induced oxidative stress by PARP1 inhibition in glioblastoma therapy. PLoS One. 2010;5(5):e10767. doi: 10.1371/journal.pone.0010767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saha T, et al. Transcriptional regulation of the base excision repair pathway by BRCA1. J Biol Chem. 2010;285(25):19092–105. doi: 10.1074/jbc.M110.104430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dianova II, et al. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004;32(8):2550–5. doi: 10.1093/nar/gkh567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dianov G, et al. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J Biol Chem. 1998;273(50):33811–6. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- 54.Fortini P, et al. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J Biol Chem. 1999;274(21):15230–6. doi: 10.1074/jbc.274.21.15230. [DOI] [PubMed] [Google Scholar]
- 55.Fortini P, et al. DNA polymerase beta is required for efficient DNA strand break repair induced by methyl methanesulfonate but not by hydrogen peroxide. Nucleic Acids Res. 2000;28(16):3040–6. doi: 10.1093/nar/28.16.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sobol RW, et al. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379(6561):183–6. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 57.Simsek D, et al. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. 2011;471(7337):245–8. doi: 10.1038/nature09794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rothkamm K, et al. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23(16):5706–15. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flygare J, Benson F, Hellgren D. Expression of the human RAD51 gene during the cell cycle in primary human peripheral blood lymphocytes. Biochim Biophys Acta. 1996;1312(3):231–6. doi: 10.1016/0167-4889(96)00040-7. [DOI] [PubMed] [Google Scholar]
- 60.Tashiro S, et al. Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J Cell Biol. 2000;150(2):283–91. doi: 10.1083/jcb.150.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lundin C, et al. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol. 2002;22(16):5869–78. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sonoda E, et al. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. Embo J. 1998;17(2):598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brennan RJ, Schiestl RH. Free radicals generated in yeast by the Salmonella test-negative carcinogens benzene, urethane, thiourea and auramine O. Mutat Res. 1998;403(1–2):65–73. doi: 10.1016/s0027-5107(98)00050-5. [DOI] [PubMed] [Google Scholar]
- 64.Haber JE. DNA recombination: the replication connection. Trends Biochem Sci. 1999;24(7):271–5. doi: 10.1016/s0968-0004(99)01413-9. [DOI] [PubMed] [Google Scholar]
- 65.Swanson RL, et al. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19(4):2929–35. doi: 10.1128/mcb.19.4.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Souza-Pinto NC, et al. The recombination protein RAD52 cooperates with the excision repair protein OGG1 for the repair of oxidative lesions in mammalian cells. Mol Cell Biol. 2009;29(16):4441–54. doi: 10.1128/MCB.00265-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bunting SF, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative Immunoblots of OGG1 and APE1 proteins in FEN1-BRCA-co-depleted and control cells. Actin was used as loading control. Cells were nucleofected with FEN1 siRNA and 48 hrs post transfection treated with 200μM of H2O2 for 1 hr and were harvested for immunoblotting. (B) Representative Immunoblots of OGG1 and APE1 proteins in XRCC1-BRCA-co-depleted and control cells treated as in A.
Untreated and H2O2-treated cells were analyzed cytogenetically. Number of chromatid-type breaks per cell is increased in BRCA-deficient cells.
BRCA deficient and control cells were nucleofected with FEN1 and XRCC1 siRNA respectively and 48 hrs post transfection treated with 200μM of H2O2 for 4 hrs. Subsequently, cells were subjected to dual staining (EdU + PI) to analyze S phase stuck cells using flow-cytometry. Graph represents relative folds of S phase population that are not able to uptake EdU (EdU negative) because they are stuck in S phase but are positive for PI staining. Data is normalized to untreated control cells. Data shown are the mean and SE from three independent experiments.