

Abstract
Glioblastoma multiforme (GBM) is the most lethal form of primary brain tumors, characterized by highly invasive and aggressive tumors that are resistant to all current therapeutic options. GBMs are highly heterogeneous in nature and contain a small but highly tumorigenic and self-renewing population of stem or initiating cells (Glioblastoma stem cells or GSCs). GSCs have been shown to contribute to tumor propagation and resistance to current therapeutic modalities. Recent studies of human GBMs have elucidated the genetic alterations common in these tumors, but much remains unknown about specific signaling pathways that regulate GSCs. Here we identify a distinct fraction of cells in a genetically engineered mouse model of EGFR-driven GBM that respond to anti-EGFR therapy by inducing high levels of c-MET expression. The MET positive cells displayed clonogenic potential and long-term self-renewal ability in vitro and are capable of differentiating into multiple lineages. The MET positive GBM cells are resistant to radiation and highly tumorigenic in vivo. Activation of MET signaling led to an increase in expression of the stemness transcriptional regulators Oct4, Nanog and Klf4. Pharmacological inhibition of MET activity in GSCs prevented the activation of Oct4, Nanog and Klf4 and potently abrogated stemness. Finally, the MET expressing cells were preferentially localized in perivascular regions of mouse tumors consistent with their function as GSCs. Together, our findings indicate that EGFR inhibition in GBM induces MET activation in GSCs, which is a functional requisite for GSCs activity and thus represents a promising therapeutic target.
Keywords: cancer stem cells, Glioblastoma multiforme, EGFR inhibition
Introduction
Glioblastoma Multiforme (GBM) is the most malignant form of primary brain tumors with a median survival of less than 15 months, a prognosis that has virtually not improved over the past five decades [1]. GBM tumors have a tendency to invade the brain parenchyma and are highly heterogeneous in nature, both at the molecular and cellular levels [2]. These salient features of GBM have prevented the development of an effective treatment for this cancer and as such, GBM treatment regimen are palliative rather than curative. The standard of care treatment for newly diagnosed GBM patients with adequate functional status includes debulking surgical resection, radiation and concurrent temozolomide, a DNA alkylating agent, followed by adjuvant temozolomide [3]. Although the bulk of the tumor can be removed and therapeutically targeted, evidence suggests that there exists a population of cells with stem-like features that can survive treatment and eventually repopulate the tumor [4].
Cancer stem-like cells or tumor-initiating cells are functionally defined as cells capable of self-renewal and highly enriched with tumorigenic potential [5–7]. Glioblastoma Stem Cells (GSCs) have been shown to display the capability for unlimited growth as multicellular spheres in defined medium, differentiate into multiple lineages and efficiently initiate tumors in immunodeficient mice [8, 9]. GSCs are also believed to play a leading role in therapeutic resistance and tumor recurrence. In contrast to bulk tumor cells, GSCs survive irradiation and chemotherapy treatment better and therefore are thought to contribute to therapeutic resistance and tumor recurrence [10–14].
Signaling by the MET receptor tyrosine kinase (RTK) regulates cell growth, survival and motility in many cancers including gliomas [15]. MET overexpression has been associated with poor prognosis and enhanced tumor invasiveness in GBM patients [16–18]. Large-scale genomic studies in GBMs confirmed frequent genomic amplification of MET [19–21] and studies on the genomic heterogeneity of GBMs at the single cell level revealed that a small fraction of GBM cells within a tumor contain focal amplification of c-MET that is independent of other RTKs [22, 23]. These studies show that a relatively small population of GBM cells is MET positive and recent work demonstrated that MET plays a central role in maintaining GSC populations in human GBMs, suggesting a link between MET signaling and GSCs [24–27]. The precise mechanism of how MET signaling confers GSC phenotypes, however, remains unclear.
In this study, we examine the physiological consequences of EGFR inhibition in a genetically engineered mouse model of GBM and demonstrate that treatment of EGFR-positive GBM with a TKI (gefitinib) result in the induction of c-MET expression in a subset of cells that have GSC characteristics. We further establish that MET signaling is a requisite for initiation and maintenance of the GSC features. Our results show the capacity for c-Met to support GSC phenotype that involves an endogenous dynamic mechanism analogous to cellular reprogramming.
Materials and Methods
EGFR Conditional Transgenic Mice
All mouse procedures were performed in accordance with Tufts University’s recommendations for the care and use of animals and were maintained and handled under protocols approved by the Institutional Animal Care and Use Committee. Cre/Lox-mediated conditional expression of the human wild type EGFR and conditional firefly luciferase transgenes was achieved as described before [28–30]. GBM tumor induction were achieved by stereotactic injections of adult transgenic animals (LSL-EGFR; Cdkn2a−/−; PTEN2lox) of a bicistronic TGFα-Cre recombinant virus described in detail elsewhere [28, 29]. Resulting GBM tumors express activated human EGFR protein and firefly luciferase and are null for p16INK4a/p19ARF (Cdkn2a−/−) and PTEN.
GBM Primary Cultures and Drug Treatment
Primary cultures of tumors were established as follows: tumors were excised and minced in 0.25% trypsin (wt/vol) 1mM EDTA and allowed to disaggregate for 15 minutes at 37°C. The resulting cell suspension was then strained through a 100 µm cell strainer (Falcon). The single suspension of cells was washed in PBS twice and plated on 0.2% gelatin coated tissue culture plates. Cells were fed every 24 hours with fresh media that consisted of DMEM supplemented with 10% heat inactivated fetal bovine serum and antibiotics. For drug treatment, stock solutions of drugs (Supporting Information Table 1) were added at the indicated final concentrations in serum deficient media and cells were incubated for the indicated period of time at 37°C in 5% CO2.
Flow Cytometry Sorting
Drug-treated and control vehicle-treated cells were trypsinized and collected by centrifugation. The cells were washed with PBS three times and incubated with an anti-MET antibody (1:50, #5631 Cell Signaling Technology) for 1 hour at room temperature. After 3 washes with PBS, cells were incubated in FITC-conjugated goat anti-mouse IgG (1:500, Millipore) or Alexa Fluor 633-conjugated goat anti-mouse secondary antibodies (1:500, Invitrogen)for 1 hour at room temperature and washed in PBS. The stained cells were measured on a Legacy MOFLOs and analyzed using Summit version 4.3 software (Beckman Coulter).
Stereotactic Allograft Injections
Stereotactic injections of adult animals (3 months of age and above) have been described in detail previously [29, 30]. Briefly, 1 × 104 or 1 × 102 of the sorted MET-positive and MET-negative luciferase positive GBM tumor cells were injected into the striatum in PBS and tumor growth was monitored by bioluminescence over time using the IVIS 200 imaging system (Xenogen).
Neurosphere Formation Assays
Neurospheres were cultured in serum-free DMEM/F12 medium by adding EGF (20 ng/ml), FGF (20 ng/ml), and B27 supplement (1 ×). In brief, 1 × 103 of the sorted MET-positive and MET-negative cells were plated out in triplicate and incubated for 5 days into 24-well cell culture plate. Spheres were counted and plotted as average ± S.D.
Immunofluorescence
Neurospheres were dissociated and plated on chamber slides (Falcon). The cells were washed with PBS three times and fixed with freshly made 4% (w/v) paraformaldehyde in PBS for 10 minutes at room temperature. After 3 washes with PBS, cells were permeabilized with PBS-T (PBS with Tween 20 (0.1% v/v)) for 15 minutes (except those stained with the anti O4 antibody). The fixed and permeabilized cells were then blocked with PBS-T plus 10% goat serum (Sigma) for 20 minutes at room temperature, followed by an incubation with the indicated antibodies overnight at 4°C in blocking solution. After extensive washes in PBS-T, cells were incubated in FITC-conjugated goat anti-mouse IgG (1:500, Millipore) and Cy3-conjugated goat anti-rabbit secondary antibodies (1:1000, Millipore) in blocking solution for 1 hour at room temperature and washed in PBS-T. Fluorescence-stained slides were mounted with a coverslip in Vectashield mounting medium with DAPI for staining of nuclear DNA reagent and examined by fluorescence microscopy. Paraffin sections of mouse EGFR;Cdkn2a−/−;PTEN−/− GBMs treated with the EGFR TKI erlotinib (150 mg/kg oral gavage daily for 3 days) were processed for immunofluorescence as follows: sections were deparaffinized and rehydrated in graded xylenes and alcohol solutions. Once rehydrated, endogenous peroxidase activity was inhibited by incubation for 10 minutes in 0.3% H2O2 in methanol followed by PBS washes. Sections were treated for antigen retrieval solution (Dako, S1699) using a microwave for 20 minutes at low power then cooled down for 30 minutes. After 3 washes with PBS, slides were incubated in blocking solution (5% (v/v) goat serum (Sigma) in PBS/0.3% Triton-X100 (v/v)) for 1 hour at room temperature. The sections were then processed for indirect immunofluorescence as follow: primary antibodies were incubated for 24 hour at 4°C. Secondary antibodies used were FITC-conjugated goat anti-mouse IgG (1:500, Millipore) and Cy3-conjugated goat anti-rabbit secondary antibodies (1:1000, Millipore) for IHC in blocking solution. Information on primary antibodies is provided in Supporting Information Table 1.
Quantitative RT-PCR
Total RNA from treated and mock-treated cells was extracted with the RNeasy Mini Kit (QIAGEN). cDNA was prepared with a Superscript® III First-strand Synthesis Supermix for qRT-PCR kit (Invitrogen) according to the manufacturer’s protocol and PCR was carried out with Taqman probes (TaqMan® Gene Expression Assays, Applied Biosystems) against the indicated target genes. TBP was used as an internal control. Analysis was performed with the comparative ct method with the comparator being RNA extracted from paired non-treated cells. Sources of probes are provided in Supporting Information Table 1.
Immunoblots
Western blots were performed as described previously [29]. Source for antibodies is provided in Supporting Information Table 1.
Clonogenic Assays
FACS sorted MET-positive and MET-negative cells were quantified with a hemocytometer. In the radiation survival experiment, cells were then serially diluted to the appropriate concentrations and plated out in triplicate per data point into 25-cm2 cell culture flasks. The cells were then permitted to attach for 24 hours at 37°C. Stock SU11274 solutions were resuspended in DMSO (Sigma) and then stored at −20°C. Immediately before the experiments, stock solutions were diluted in DMSO to the listed concentrations. Cells were exposed to a vehicle (DMSO) or to SU11274 (10µM) concentration for 4 hours. The cells were then irradiated with graded doses of γ-rays using the JL Shepherd Mark I Irradiator (JL Shepherd and Associates), at a dose rate of 2.38 Gy/min, as determined by thermoluminescence dosimetry for the system employed. After an additional 68 hours of incubation in medium containing either the vehicle or the drug, the cells were rinsed with PBS, and drug-free medium was added. The cells were then maintained at 37°C for 8 days to allow for the formation of colonies and then stained with 0.5% crystal violet (Sigma) in absolute methanol. The colonies were counted visually with a cutoff value of 50 viable cells. The surviving fraction was then calculated as follows: mean number of colonies/(number of cells inoculated × plating efficiency), where plating efficiency is defined as the mean number of colonies/number of cells inoculated for untreated controls. Surviving fractions in the cells exposed to radiation plus SU11274 were normalized by dividing by the surviving fraction obtained for SU11274 alone. We then calculated the dose enhancement ratio (DER) as the dose (Gy) for the radiation plus vehicle cells divided by the dose (Gy) for radiation plus SU11274 (normalized for drug toxicity) at a surviving fraction of 0.1. Error bars were also calculated as S.D. via the pooling of the results of three independent experiments.
In the self-renewal assay, neurospheres were dissociated with Accutase (Innovative Cell Technologies, Inc.) according to the manufacturer’s instructions, single cells were then assessed for viability by trypan blue staining and cells were replated immediately in stem cell media as above. In the neurosphere formation assay, an Extreme Limited Dilution Assay protocol was followed [31]. Briefly, 5, 20, 50, 100 and 250 MET-positive or MET-negative cells were plated per well (X24) in 96 well plates and grown in neurosphere media for 10 days. Wells containing neurospheres (almost exclusively one neurosphere per well) were scored and the data generated computed using ELDA’s online algorithm (http://bioinf.wehi.edu.au/software/elda/).
Results
EGFR Inhibition Induces c-MET Expression in a Subset of Cells
We previously demonstrated in our EGFR-driven genetically engineered mouse model of GBM(EGFR;Cdkn2a−/−;PTEN−/−) that pharmacological EGFR inhibition with gefitinib in the context of Cdkn2a and PTEN nullizygous GBM tumors result in a G1 cell cycle arrest [29]. This arrest is accompanied with a drastic change in gene expression profile of which the MET proto-oncogene is one of the most robustly induced genes [29]. Furthermore, we showed that the increased MET mRNA levels resulted from a rapid (within 30 min) and sustained transcriptional activation of the c-MET gene [29].
To gain further insight into the physiological consequences of this induction, we first determined the extent of MET expression on a per cell basis. Using early passage primary cultures of our EGFR;Cdkn2a−/−;PTEN−/− mouse GBMs, we performed flow cytometry analysis on gefitinib-treated cells using a MET antibody that recognized the extracellular domain of MET to determine the extent of MET expression within the population. We show that in three primary cell cultures isolated from individual GBM tumors, approximately 20% of the cells express MET at levels detectable by flow cytometry (Fig. 1). Untreated cells do not express MET protein, confirming our previous qRT-PCR observations that non treated cells do not express MET mRNA [29] and this response is specific to EGFR inhibition. Treatment of our cells with the non-EGFR tyrosine kinase inhibitors saracatinib and imatinib did not result in the expression and activation of MET (Supporting Information Fig. S1). These results demonstrate that inhibition of EGFR activity results in the induction of MET expression only in a subset of primary GBM cultured cells.
Figure 1.

Flow cytometry analysis of c-MET expression in mouse GBM cells. (A): Representative plots of three independent primary cultures of mouse GBMs were treated with gefitinib (10 µM for 16 hours), incubated with an anti-MET receptor antibody and processed for flow cytometry. (B): Consistency of EGFR inhibition-induced MET expression. Average of percent MET positive cells from triplicate experiments ± standard deviation.
Cells With Induced c-MET Expression are Highly Clonogenic, can Self-Renew, Express Stem Cell Markers and are Multipotent
Recent work with human GBMs suggests that MET positive GBM cells have characteristics associated with GSC phenotypes [24–26]. The capacity to grow as neurosphere-like masses has been used extensively as an in vitro indicator of one of the components that define stemness in GSCs [11, 32–35]. To ascertain the stem/progenitor potential of our cells, we treated cells with gefitinib and MET positive and MET negative cells were isolated using FACS and subjected to neurosphere growth assays. MET positive cells readily formed neurospheres whereas MET negative were completely devoid of this capacity (Fig. 2A). Moreover, non-treated non-MET expressing cells were incapable of forming neurospheres under these same conditions (Fig. 2A). To demonstrate that the formation of the neurospheres is not the result of cellular aggregation but rather result from single cells developing into neurosphere masses, we labeled our cultures separately with mCherry and mGFP fluorescent proteins and repeated the neurosphere forming assays in a 1:1 admixture of both fluorescently labeled MET positive cells. The resulting neurospheres formed were uniformly either mCherry or mGFP positive and never mosaic for both fluorescent marker (Supporting Information Fig. S2), demonstrating that they originated from single cells.
Figure 2.

MET positive GBM cells display features characteristic of GSCs. (A): Growth as neurospheres. Representative photomicrograph of untreated, gefitinib treated MET positive and MET negative cells isolated by FACS that were plated in neurosphere growth medium and grown for 5 days in triplicate. 1×103 MET positive and MET negative cells were seeded per well whereas 1×104 untreated cells were seeded. Scale bar=100 µm. (B): Quantitation of MET positive neurospheres formed in (A) and after dissociation to single cells and replating serially and grown in neurosphere growth media. (C): Multipotent capacity of MET positive neurosphere cells. Cells from freshly dissociated neurospheres were grown in differentiation media, fixed and stained by indirect immunofluorescence for markers of differentiated astrocytes (GFAP), neurons (Tuj 1, Neuron-specific class III beta-tubulin) and oligodendrocytes (O4). Scale bar=25 µm. (D): MET positive cells express the stem cell marker Prominin 1 (CD133). Quantitative RT-PCR analysis for known markers of GSC from RNA isolated from MET positive and negative cells. * p=0.0004. (E): MET positive cells are clonogenic. Limiting dilution neurosphere forming assays to establish the clonogenic potentials of MET-positive and MET-negative subpopulations from 3 GBM cell cultures. Cells were seeded into 96-well plates with defined densities (5–250 cells per well, 24 wells/condition) and resulting wells with neurospheres scored.
The capacity to self renew is also a salient feature of neuronal/progenitor and GSCs. Upon serial disaggregation and replating, MET positive neurospheres retained their ability to form neurospheres with the same efficiencies over time (Fig. 2B) and retained high MET expression over several passages (Supporting Information Fig. S3). In addition, the MET positive cells within the neurospheres are multipotent. Upon disaggregation and plating in differentiation medium, cells expressing markers of terminally differentiated astrocytes (GFAP), neurons (TUJ1) and oligodendrocytes (O4) were observed (Fig. 2C), indicating the multipotent characteristic of MET positive cells. Several markers of GSC including CD44, CD133, L1CAM and Nestin have been identified in patient-derived GSCs [36]. We ascertained the extent of expression of these markers in our MET positive and negative populations by quantitative RT-PCR. Figure 2D demonstrates that the marker Prominin 1 (CD133) is expressed over 10 fold more in MET positive cells than in MET negative cells whereas none of the other markers show differences in expression between the two populations (Fig. 2D). Finally, clonogenic growth is a feature associated with normal neural stem/progenitor cells (NSC) and GSCs. To determine the clonogenic potentials of MET-positive and MET-negative populations, we performed limiting dilution assays (LDA) using all three GBM cell cultures (Fig. 2E). FACS-sorted MET-positive and MET-negative cells were plated into 96-well plates at defined seeding densities and allowed to grow for 10 days in neurosphere media. Neurospheres grew from MET-positive cells but not from MET-negative cells (Fig. 2E). The estimated frequency of stem cells for the MET-positive populations are 1/27 (GBM-4) and 1/29 (GBM-4 and -5). Taken together, these data indicate that MET positive GBM cells are highly clonogenic with self-renewal and differentiation capacities and that non gefitinib treated cells have no stem potential.
MET Positive GSCs are Resistant to Ionizing Radiation
Studies have demonstrated that GSCs are more resistant to ionizing radiation (IR) than GBM cells [11, 14, 36]. We examined whether MET positive GSCs are intrinsically more radioresistant than MET negative cells. Resistance to IR was quantified by standard colony formation assays. MET positive and MET negative cells were irradiated in vitro with increasing doses of radiation and serial dilutions of cells plated and grown for 14 days. The resulting colonies were stained and counted and the data is reported as surviving fraction. MET positive cells were dramatically more resistant to ionizing radiation than MET negative cells (Fig. 3A).
Figure 3.

MET positive GSCs are radioresistant. (A): Radiation survival of MET positive and MET negative cells treated with SU11274 and mock treated was quantified by colony formation assays. The fraction of surviving colonies (y axis) was plotted against corresponding radiation dose (x axis). Bars, SD of triplicate experiments. * p=0.0013. (B): Growth as neurospheres is dependent on MET activity. Representative photomicrographs of MET negative (−) and positive (+) cells isolated by FACS and plated in neurosphere growth medium with and without the indicated MET inhibitors for 5 days. SU11274 (10 µM), Foretinib (1 µM), PHA66572 (500 nM) and Crizotinib (1 µM). Scale bar=100 µm.
Inhibition of MET Activity Abrogates Radioresistance and Clonogenicity of GSCs
To further ascertain the role of MET signaling in GSC radioresistance, we inhibited MET activity with the tool compound inhibitor SU11274 in MET positive and negative cells and assayed the sensitivity of these cells to IR by colony formation assays. Treatment of MET positive cells with SU11274 eliminated the radioresistance observed in GSCs and had no effect on MET negative cells (Fig. 3A). Next we ascertained the effects of MET inhibition on neurosphere formation using a panel of inhibitors. We first determined the optimal concentration for each inhibitor to obtain complete MET kinase inhibition by western blot analyses (Supporting Information Fig. S4). Then we showed that inhibition of MET kinase activity with four different inhibitors completely abrogated the ability of MET positive cells to grow as neurospheres (Fig. 3B). These results demonstrate that salient features of GSCs are driven by the kinase activity of MET and its signaling pathways.
MET Expression Upon EGFR Inhibition is Induced in Perivascular Cells
Work from various groups indicates that neuronal stem cells and GSCs frequently reside within a perivascular niche [33, 37–40]. To determine the extent and location of the EGFR inhibitor-induced MET activation, we treated GBM-bearing mice with the EGFR inhibitor erlotinib p.o. daily for 72 hours and sacrificed the animals and processed their brains for histology. We then performed indirect immunofluorescence on paraffin sections using the endothelial cell marker CD31 to label the vasculature and with an anti MET antibody to label the cells that have acquired MET expression upon EGFR inhibitor treatment. Figure 4A demonstrates that EGFR inhibition in animals resulted in the appearance of MET positive cells that are located within perivascular areas and that the origin of these MET positive cells are from tumor cells since they are positive for human EGFR (Fig. 4B). In addition, MET receptors in these cells are activated as detected by the presence of the autophosphorylation site pTyr1234/1235 (Fig. 4B).
Figure 4.

MET positive cells are located within the perivascular space. GBM-bearing mice were treated by oral gavage with the EGFR kinase inhibitor erlotinib daily for 72 hours, sacrificed and their brains processed for indirect immunofluorescence. Representative photomicrographs of (A): control and erlotinib-treated tumor endothelial cells stained for CD31 (green) and MET positive cells (red) within GBM tumors. Insets: higher magnification photomicrographs. * blood vessels. Scale bars upper row=100µm& lower row=25 µm. (B): Perivascular MET receptors are activated. Colocalization of EGFR positive GBM tumor cells (green) and MET receptor expression (red) or phospho MET pTyr1234/1235 (red) surrounding blood vessels (*). Scale bars = 12.5µm.
MET Positive Cells are Highly Tumorigenic in an Orthotopic Transplantation Model
Tumorigenicity in vivo is a strong indicator of cancer stemness. To assess the relative enrichment in tumorigenicity for MET positive cells compared to MET negative cells, we carried out tumorigenic titration assays in vivo. We stereotactically injected 100 and 104 of either MET positive or MET negative cells (all are expressing a firefly luciferase reporter gene) into the brains of immunodeficient NcrNu/Nu mice and monitored tumor formation and growth over time using bioluminescence (Fig. 5A). Injection of 100 MET positive cells generated tumors more efficiently than their corresponding MET negative cells and as rapidly as injecting 104 MET negative cells (Fig 5B). Mice injected with 104 MET positive cells created tumors within the shortest amount of time whereas 100 MET negative cells showed no sign of tumor development 10 weeks after the injection (Fig. 5B).
Figure 5.
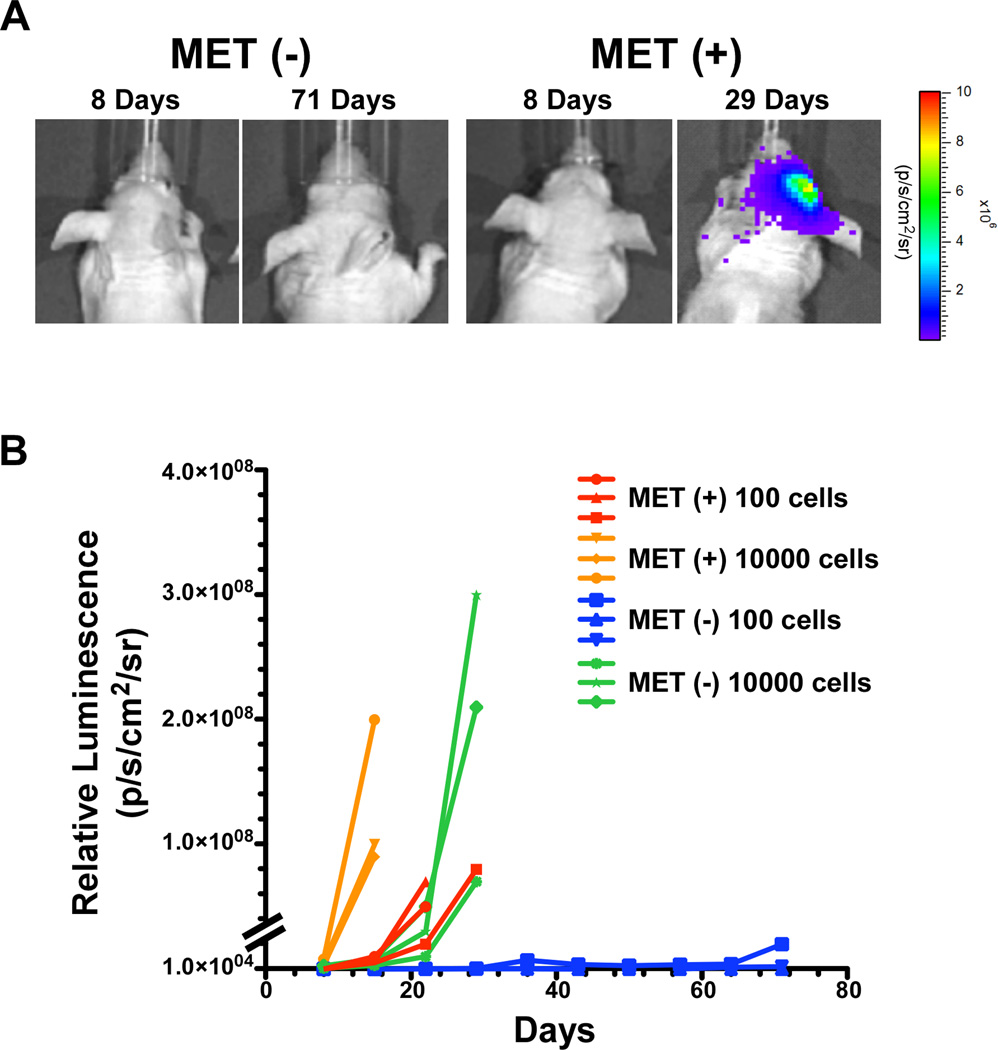
Tumorigenic potential of MET positive and negative cells. The indicated number of MET positive and MET negative cells expressing firefly luciferase were injected intracranially into immunodeficient mice and tumor growth monitored by bioluminescence imaging (BLI). (A): Representative photomicrograph of the BLI output from mice injected with 100 MET positive and 100 MET negative cells at the indicated time points. (B): Graphical representation of BLI values for mice injected with the indicated cells.
Kaplan-Meier survival analysis further showed significant survival differences between animals receiving these subpopulations (Fig. 6A) reflecting the growth parameters displayed by the bioluminescence results. All the mice injected with 104 MET positive cells succumbed from their tumors within 20 days post injection. Mice injected with 100 MET positive or 104 MET negative cells all died within 30 and 35 days post injection respectively (Fig. 6A) and mice injected with 100 MET negative cells did not develop tumors within the course of the study (Fig. 6A–B). In addition, we found that the end-stage tumors derived from the MET positive cells injections retained MET receptor expression (Supporting Information Fig. S5). Interestingly, histopathological analysis of the resulting tumors revealed major differences between the tumors originating from MET positive and MET negative cells. The tumors that arose from the injections of 100 MET positive cells display histological features that are consistent with GBMs whereas the tumors that arose from the injections of 104 MET negative cells have no GBM characteristics and are classified as sarcomas (Fig. 6B–C).
Figure 6.

Survival of mice injected with MET positive and negative cells. (A): Kaplan–Meier survival plot of mice injected with the indicated MET positive and MET negative cells. Log-rank (Mantel-Cox) tests of survival between 100 MET (+) and 100 MET (−) cells p=0.022; 100 MET (+) and 1000 MET (+) p=0.029; 100 MET (+) and 1000 MET (−) p=0.022. (B–C): Representative microphotographs of H&E stained coronal sections of brains injected with MET positive and MET negative cells–derived allograft tumors. The black squares in (B) are areas of high magnification shown in (C). Scale bars= 2 mm (B) and 50 µm (C).
MET Signaling Regulates Stem Cells Markers
It has been reported that MET regulates the stem-like reprogramming transcription factors Sox2, Klf4, c-Myc, Oct4, and Nanog in human GSCs [25]. Here we measured the levels of transcript for these genes by quantitative RT-PCR in MET positive and MET negative cells in the presence and absence of MET kinase inhibition with four different MET inhibitors. We found that MET positive cells expressed high levels of Oct4, Nanog and Klf4 when compared to MET negative cells. We also demonstrate that the expression of these reprogramming factors returned to basal levels in response to MET inhibition (Fig. 7) strongly suggesting that MET kinase activity and signaling pathways are turning on the expression of these reprogramming transcription factors.
Figure 7.

MET induces stem cell reprogramming transcription factors. Total RNA was isolated from MET positive and MET negative cells treated with the MET inhibitors (SU11274 (10 µM), Foretinib (1 µm), PHA665752 (500 nM) and crizotinib (1 µM)) and the relative expression of the indicated transcription factors determined by quantitative RT-PCR. Mean from triplicate ± standard deviation. * p=0.0005, ** p=0.0013 and # p<0.0001.
Discussion
Different aspects of GSCs remain controversial because of unresolved questions with respect to their frequency, the identity of the cell surface markers by which they can be identified and isolated, and the nature of the cell(s) from which they originate [41, 42]. Using a reproducible genetically engineered mouse model of EGFR-driven Cdkn2a−/−;PTEN−/− GBM, we previously demonstrated that EGFR inhibition in GBM tumors leads to the rapid transcriptional activation of the c-met gene and ensuing expression of an active MET receptor both in vivo and in vitro [29]. In this study, we identified a distinct fraction of cells expressing high levels of MET in response to EGFR inhibitor treatment and showed that this subpopulation has key characteristics of GSCs.
The transcriptional induction of the c-met gene in a subset of cancer cells as a result of EGFR inhibition is novel. This observation led us to ask whether any non-GSC cells has the potential to become a GSC upon the acquisition of MET activity/signaling or are GSCs that are already present within the population respond to EGFR inhibition by turning on MET expression? The lack of neurosphere formation that we observed in the untreated cell population suggests that active GSCs are not present in the population. We cannot exclude however the presence of quiescent (G0) GSCs within our untreated cultures since quiescent stem cells do not give rise to neurospheres [43, 44]. To further address this issue, future directions should include cell cycle profiles of our cell population to determine if cells within a given cycle are acquiring MET expression and signalling upon EGFR inhibition.
The identification of cell surface molecules that are preferentially expressed in GSCs and functionally associated with the maintenance of GSCs is not a trivial task. For example, several molecules including CD133 [45, 46], CD44 [47] and L1CAM [48] have been identified as cell surface markers of GSCs and used for their isolation. It remains unclear however, whether these markers have specific functions intrinsic to GSC phenotypes [49, 50]. Here we demonstrate that MET positive cells express CD133 and that MET activity is a necessary component of GSC physiology, supporting that MET is not only a cell surface marker of GSCs but also a key regulator of GSC phenotypes.
Our present work on MET expression in GSCs furthers that of others [24–26] in demonstrating that MET activation reprograms cells to acquire stem-like properties, thus reinforcing the importance of MET in GSC biology. GSCs share critical characteristics with normal neural stem cells and embryonic stem (ES) cells. Accordingly, transcription factors that are fundamental in regulating normal stem cells are also required for the maintenance of a GSC phenotype. The stem cell transcription factors Sox2, Oct4, Nanog, c-Myc, Olig2, Klf4 and the stem cell chromatin regulator Bmi1 have been shown to be critical for maintaining the self-renewal, proliferation, survival, and multi-lineage differentiation potential of GSCs [14, 25, 36, 51]. Sox2, Oct4 and Nanog are core components of a stem cell transcription factor network. Not only are they playing essential roles in maintaining the stemness of ES cells and somatic stem cells [52–55], they are also critical factors for cellular reprogramming and when expressed ectopically, for the generation of inducible pluripotent stem cells (iPS) [56, 57]. Our results demonstrate that MET positive cells differentially expressed Nanog, Oct4 and Klf4 in the GSC subpopulation and that MET activity turn on and/or maintain expression of these factors. We show that pharmacological inhibition of MET activity is sufficient to abrogate this response and conclude that expression of Nanog, Oct4 and Klf4 are under the control of MET signaling pathways. Our data suggest that expression of Oct4, Klf4 and Nanog induce an active reprogramming of MET expressing cells into GSCs. The capacity for MET signaling to induce and/or maintain GBM cells toward a GSC phenotype could result from an active reprogramming of differentiated GBM cells into GSCs. Indeed, our findings that the expression of the reprogramming factors Oct4, Nanog and Klf4 are rapidly induced within 24 hours of MET activation support a molecular mechanism similar to cellular reprogramming.
One of the key defining features of cancer stem cells is their high tumorigenic potential when grown orthotopically in vivo, which is experimentally measured by serial dilution implantation experiments. We show that 100 MET positive cells are not only capable of forming tumors within a short period of time but also result in tumors with histopathological features of GBMs. Interestingly, 100 MET negative cells did not result in tumor growth and tumors arising from the implantation of 104 MET negative cells do not show characteristics of GBMs but rather are classified as high grade sarcomas. These results suggest that MET negative cells are poorly tumorigenic and require a high threshold number of cells to produce a tumor under in vivo conditions, perhaps reflecting the necessity of sufficient levels of paracrine factors for the growth of these cells.
There has been several reports demonstrating that GSCs are not scattered randomly throughout GBMs but rather tend to reside close to blood vessels. Similar to normal neural stem cells (NSCs), which reside in perivascular locations called vascular niches that are primed to provide essential pro-survival and maintenance cues [58–60], GSCs also reside in perivascular niches [11, 24, 33, 36, 37, 40, 48]. Collectively, these studies strongly suggest that the perivascular niche may provide a microenvironment that enables and promotes the maintenance of GSCs. In light of our results and those of others on MET expression and activation in perivascular GSCs, it is tempting to speculate that brain endothelial cells are a source of the MET ligand hepatocyte growth factor (HGF). It has been reported that endothelial cells in liver and lung tissues express and secrete HGF [18, 61]. Our results show that the induction of MET expression in vivo occurs at a define location surrounding vasculature, consistent with the perivascular niche location of GSCs.
In conclusion, we demonstrate that MET is an enrichment marker for GSCs and a functional requisite for cancer stem cell phenotype in GBM. Our data provide further evidence for the implication of MET signaling in the induction and maintenance of GSCs and offer a model system for future studies on cancer stem cell biology and unveil MET as a promising therapeutic target.
Supplementary Material
Acknowledgments
The authors would like to thank Drs. Vicky Appleman and Katy Wong for critical review of the manuscript and Dr. William Miele for technical assistance.
Grant Support. This research was supported (in part) by NIH grant NCI U01 CA141556 (A.C.), by American Cancer Society Research Scholar Award 117409 (A.C.).
Footnotes
Author Contributions: Hyun Jung Jun: Conception and design, Collection and assembly of data, Data analysis and interpretation and Manuscript writing. Roderick T. Bronson: Data analysis and interpretation. Al Charest: Conception and design, Financial support, Administrative support, Provision of study material, Collection and assembly of data, Data analysis and interpretation and Manuscript writing.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 2.Dunn GP, Rinne ML, Wykosky J, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee EQ, Nayak L, Wen PY, et al. Treatment Options in Newly Diagnosed Glioblastoma. Curr Treat Options Neurol. 2013 doi: 10.1007/s11940-013-0226-9. [DOI] [PubMed] [Google Scholar]
- 4.Nduom EK, Hadjipanayis CG, Van Meir EG. Glioblastoma cancer stem-like cells: implications for pathogenesis and treatment. Cancer J. 2012;18:100–106. doi: 10.1097/PPO.0b013e3182452e0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 7.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 8.Dirks PB. Cancer: stem cells and brain tumours. Nature. 2006;444:687–688. doi: 10.1038/444687a. [DOI] [PubMed] [Google Scholar]
- 9.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 10.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 12.Salmaggi A, Boiardi A, Gelati M, et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia. 2006;54:850–860. doi: 10.1002/glia.20414. [DOI] [PubMed] [Google Scholar]
- 13.Kang MK, Kang SK. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007;16:837–847. doi: 10.1089/scd.2007.0006. [DOI] [PubMed] [Google Scholar]
- 14.Facchino S, Abdouh M, Chatoo W, et al. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci. 2010;30:10096–10111. doi: 10.1523/JNEUROSCI.1634-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 16.Kong DS, Song SY, Kim DH, et al. Prognostic significance of c-Met expression in glioblastomas. Cancer. 2009;115:140–148. doi: 10.1002/cncr.23972. [DOI] [PubMed] [Google Scholar]
- 17.Nabeshima K, Shimao Y, Sato S, et al. Expression of c-Met correlates with grade of malignancy in human astrocytic tumours: an immunohistochemical study. Histopathology. 1997;31:436–443. doi: 10.1046/j.1365-2559.1997.3010889.x. [DOI] [PubMed] [Google Scholar]
- 18.Moriyama T, Kataoka H, Koono M, et al. Expression of hepatocyte growth factor/scatter factor and its receptor c-Met in brain tumors: evidence for a role in progression of astrocytic tumors (Review) Int J Mol Med. 1999;3:531–536. doi: 10.3892/ijmm.3.5.531. [DOI] [PubMed] [Google Scholar]
- 19.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 22.Snuderl M, Fazlollahi L, Le LP, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20:810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Szerlip NJ, Pedraza A, Chakravarty D, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A. 2012;109:3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joo KM, Jin J, Kim E, et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012;72:3828–3838. doi: 10.1158/0008-5472.CAN-11-3760. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Li A, Glas M, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U S A. 2011;108:9951–9956. doi: 10.1073/pnas.1016912108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Bacco F, Casanova E, Medico E, et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012;72:4537–4550. doi: 10.1158/0008-5472.CAN-11-3490. [DOI] [PubMed] [Google Scholar]
- 27.Kim KH, Seol HJ, Kim EH, et al. Wnt/beta-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 2013;15:161–171. doi: 10.1093/neuonc/nos299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acquaviva J, Jun HJ, Lessard J, et al. Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation. Cancer Res. 2011;71:7198–7206. doi: 10.1158/0008-5472.CAN-11-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jun HJ, Acquaviva J, Chi D, et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene. 2012;31:3039–3050. doi: 10.1038/onc.2011.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu H, Acquaviva J, Ramachandran P, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A. 2009;106:2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Eyler CE, Wu Q, Yan K, et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011;146:53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Read TA, Fogarty MP, Markant SL, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–147. doi: 10.1016/j.ccr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Son MJ, Woolard K, Nam DH, et al. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–452. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Z, Cheng L, Guryanova OA, et al. Cancer stem cells in glioblastoma--molecular signaling and therapeutic targeting. Protein Cell. 2010;1:638–655. doi: 10.1007/s13238-010-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 38.Shen Q, Goderie SK, Jin L, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 39.Ramirez-Castillejo C, Sanchez-Sanchez F, Andreu-Agullo C, et al. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat Neurosci. 2006;9:331–339. doi: 10.1038/nn1657. [DOI] [PubMed] [Google Scholar]
- 40.Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141–152. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149:36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pastrana E, Silva-Vargas V, Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;8:486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reynolds BA, Rietze RL. Neural stem cells and neurospheres--re-evaluating the relationship. Nat Methods. 2005;2:333–336. doi: 10.1038/nmeth758. [DOI] [PubMed] [Google Scholar]
- 45.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 46.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 47.Anido J, Saez-Borderias A, Gonzalez-Junca A, et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 48.Bao S, Wu Q, Li Z, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bidlingmaier S, Zhu X, Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J Mol Med (Berl) 2008;86:1025–1032. doi: 10.1007/s00109-008-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nguyen LV, Vanner R, Dirks P, et al. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- 51.Zbinden M, Duquet A, Lorente-Trigos A, et al. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29:2659–2674. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellis P, Fagan BM, Magness ST, et al. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci. 2004;26:148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- 53.Loh YH, Wu Q, Chew JL, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 54.Tay Y, Zhang J, Thomson AM, et al. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–1128. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- 55.Fong H, Hohenstein KA, Donovan PJ. Regulation of self-renewal and pluripotency by Sox2 in human embryonic stem cells. Stem Cells. 2008;26:1931–1938. doi: 10.1634/stemcells.2007-1002. [DOI] [PubMed] [Google Scholar]
- 56.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 57.Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 58.Shen Q, Wang Y, Kokovay E, et al. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell. 2008;3:289–300. doi: 10.1016/j.stem.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tavazoie M, Van der Veken L, Silva-Vargas V, et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–288. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mirzadeh Z, Merkle FT, Soriano-Navarro M, et al. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. 2008;3:265–278. doi: 10.1016/j.stem.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morisako T, Takahashi K, Kishi K, et al. Production of hepatocyte growth factor from human lung microvascular endothelial cells induced by interleukin-1beta. Exp Lung Res. 2001;27:675–688. doi: 10.1080/019021401317138478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.