

Abstract
In this study, we document that efficient interaction between arenavirus nucleoprotein (NP) and RNA-dependent RNA polymerase (L protein), the two trans-acting viral factors required for both virus RNA replication and gene transcription, requires the presence of virus-specific RNA sequences located within the untranslated 5′ and 3′ termini of the viral genome.
TEXT
Several arenaviruses, chiefly Lassa virus (LASV) and Junin virus (JUNV), cause hemorrhagic fever disease in humans and pose important public health concerns in regions where the viruses are endemic (1–6). Moreover, evidence indicates that the worldwide-distributed prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) is a neglected human pathogen of clinical relevance especially in cases of congenital and immunocompromised-individual infections (7–12). Arenaviruses are enveloped viruses with a bisegmented, negative-strand RNA genome. Each of the two RNA segments, S and L, uses an ambisense coding strategy to direct the synthesis of two viral polypeptides in opposite orientations, separated by a noncoding intergenic region (IGR) (13). The S segment encodes the nucleoprotein (NP) and the surface glycoprotein precursor (GPC), whereas the L segment encodes the matrix (Z protein) and the RNA-dependent RNA polymerase (L protein). We reported that the IGR of LCMV is necessary for transcription termination and production of infectious progeny, indicating that noncoding viral RNA sequences play different functions during the arenavirus life cycle (14). In this work, we further extended our studies on the functional roles played by noncoding arenaviral RNA sequences by examining the contribution of these sequences to the interaction between the nucleocapsid template (NP-RNA) and L polymerase required for formation of a functional virus ribonucleoprotein (vRNP) responsible for directing RNA replication and gene transcription of the arenavirus genome.
To examine whether virus-specific RNA sequences affected L-NP interaction, we used our LCMV minigenome (MG) system (15, 16). We transfected human embryonic kidney (HEK) 293T cells with NP- and L-expressing as well as T7 RNA polymerase (T7 pol)-expressing plasmids together with or without a plasmid that directed T7 pol-mediated intracellular synthesis of an LCMV MG RNA containing the chloramphenicol acetyltransferase (CAT) gene in the NP locus (pT7-MG) (15–18) and examined L-NP interaction by use of pulldown (PD) assays (Fig. 1). To facilitate PD assays and protein detection, we used tagged versions of NP and L that we had confirmed to be functionally active in the MG rescue assay (Fig. 1F and G), although FLAG-tagged L appeared to have some overall reduced activity in this assay. In the presence of MG RNA, L protein was readily detected by Western blotting (WB) in the complex PD by NP (Fig. 1B). In contrast, in the absence of MG RNA, the complex PD by NP showed dramatically reduced levels of L protein (Fig. 1B). Likewise, NP was efficiently pulled down with L protein in the presence, but not in the absence, of virus-specific RNA (Fig. 1C). These results indicated that the presence of virus-specific RNA sequences promotes L-NP interaction. As a control, we confirmed, consistent with previous findings (19), that NP-NP interaction did not depend on the presence of virus-specific RNA (Fig. 1D). We also found that expression of LASV MG RNA promoted LASV L-NP interaction (Fig. 1E), supporting the generality of this finding.
FIG 1.

Efficient interaction of arenavirus NP and L requires the presence of virus-specific RNA. (A) Schematic diagrams of MG constructs. LCMV and LASV MGs were placed under the control of a modified T7 promoter (T7pΔ2G) (15), followed downstream by an additional C residue (16), the hepatitis delta ribozyme (drz), and T7 RNA polymerase terminator (T7T). The additional C residue was introduced to allow for the generation of the authentic virus 3′ end (genome polarity) following drz-mediated cleavage of unprocessed MG RNA produced intracellularly by the T7 RNA polymerase. The 3′ untranslated region (3′UTR), 5′UTR, and IGR highlighted in gray indicate cis-acting LCMV genomic sequences. Nr, residual NP ORF-derived sequence; Gr, residual GPC ORF-derived sequence; L1 to L3, linker sequences for cloning. (B, C) LCMV NP-L interaction. 293T cells were seeded (4.5 × 106 cells/10-cm plate) and cultured overnight prior to transfection with plasmids expressing either WT or Strep-tagged NP (2 μg), FLAG- or Strep-tagged L (8 μg), T7 polymerase (T7 pol; 2 μg), and LCMV MG (pT7-MG, previously described as pMG#7Δ2G [15]; 2 μg), using 1 mg/ml polyethyleneimine Max (PEI Max) (2 μl PEI Max/μg of DNA). “+” or “−” indicates the presence or absence, respectively, of plasmid in the transfection mixture. At 72 h posttransfection, cells were washed with phosphate-buffered saline (PBS) and lysed with 1 ml of cell lysis buffer (CLB; 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% NP-40, 1 mM EDTA) supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific). Cell lysates were clarified by centrifugation at 15,000 rpm at 4°C for 10 min and incubated for 2 h at 4°C with Strep-Tactin resin (Strep-Tactin Super flow Plus, Qiagen). Beads were washed three times (1 ml of CLB each wash) and the Strep-tagged protein complex was eluted using CLB containing 2.5 mM desthiobiotin. Proteins present in cleared cell lysates (Input) and eluates (PD: Strep) were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 4 to 20% gradient polyacrylamide gels and electroblotted onto polyvinylidene difluoride membranes (Immobilon transfer membranes, Millipore). To detect FLAG-, Strep-, and hemagglutinin (HA)-tagged proteins, membranes were incubated with anti-FLAG (rabbit polyclonal), anti-Strep (mouse monoclonal), and anti-HA (rabbit polyclonal) antibodies, respectively, followed by the corresponding secondary antibodies conjugated with horseradish peroxidase. SuperSignal West Pico, Dura, or Femto chemiluminescent substrate (Thermo Scientific) was used to produce chemiluminescent signals that were visualized using ImageQuant LAS 4000 (GE Healthcare Life Science). In some experiments, a mouse monoclonal antibody to FLAG (clone 2H8) was used for the detection of FLAG-tagged proteins. (D) Virus-specific RNA is not required for NP-NP interaction. 293T cells were seeded (9.0 × 105 cells/M6 well) and cultured overnight prior to transfection with pCAGGS-NP-HA (2 μg) and 2 μg of either pC-NP (WT NP) or pC-Strep-NP (Strep-NP) (18, 20). At 72 h posttransfection, pulldown assays were performed as described for panel B. (E) LASV L-NP interaction. 293T cells were seeded (6.6 × 106 cells/T75 flask) and cultured overnight prior to transfection with 3 μg of either pC-LASV-NP (WT LASV-NP) or pC-Strep-LASV-NP (Strep-LASV-N), 12 μg of pC-FLAG-LASV-L (FLAG-LASV-L), and 3 μg of pCAGGS-T7pol (T7 pol), together with 3 μg of pT7-LASV-MG (pT7-LASV-MG), using PEI Max. “+” or “−” indicates the presence or absence, respectively, of plasmid in the transfection mixture. At 72 h posttransfection, cleared cell lysates (Input) were prepared as described for panel B, and two-thirds of the cell lysate was subjected to Strep-Tactin-based PD performed as described for panel B. One-third of the cell lysates was incubated for 2 h at 4°C with protein A+G Sepharose beads (1:1 mixture) pretreated with anti-FLAG antibody (2H8). After three washes of the Sepharose beads with 1 ml of CLB, FLAG-tagged protein was detached from the Sepharose beads by boiling in SDS loading buffer (IP: anti-FLAG). (F) MG activity of tagged LCMV NP and L proteins. 293T cells were seeded (4.0 × 105 cells/M12 well) and cultured overnight prior to transfection with pT7-MG (0.5 μg), pCAGGS-T7pol (0.5 μg), and NP-expressing plasmid (WT, pC-NP; HA, pC-NP-HA; Strep, pC-Strep-NP) (0.3 μg), together with L protein-expressing plasmids (WT, pC-L; FLAG, pC-FLAG-L; Strep, pC-Strep-L) (0.3 μg). Empty pCAGGS was used as a negative control [L(−)]. At 72 h posttransfection, CAT expression levels were measured using a CAT enzyme-linked immunosorbent assay (ELISA) kit (Roche). CAT expression levels of WT NP- and L-transfected cells were set to 100%. Data represent means ± standard deviations (SD) of results from triplicate samples. (G) MG activity of tagged LASV NP and L proteins. 293T cells were seeded (4.5 × 105 cells/M12 well) and cultured overnight prior to transfection with pT7-LASV-MG (0.5 μg), pCAGGS-T7pol (0.5 μg), and 0.3 μg of NP-expressing plasmid (WT, pC-LASV-NP; Strep, pC-Strep-LASV-NP), together with 0.6 μg of L protein-expressing plasmid (WT, pC-LASV-L; FLAG, pC-FLAG-LASV-L). pCAGGS-Empty, instead of L protein-expressing plasmid, was used as a negative control [L(−)]. At 72 h posttransfection, CAT expression levels were measured using a CAT ELISA kit (Roche). CAT expression levels of WT NP- and L-transfected cells were set to 100%. Data represent means ± SD of results from triplicate samples. The total amount of DNA represented in panels B to G was adjusted using empty pCAGGS. For panels B to D, values for input and PD samples correspond to 1% and 20%, respectively, of values for whole-cell lysates. For panel E, values for input, IP, and PD samples correspond to 0.5%, 6.7%, and 6.7%, respectively, of values for whole-cell lysates. DNA fragments containing full-length ORFs of NP and L protein of either LCMV or LASV tagged at their N termini with either FLAG or two Strep sequences connected by the GGGS3 linker sequence were subcloned into the expressing plasmid pCAGGS. pT7-LASV-MG was generated using the backbone of LCMV MG plasmid pT7-MG. LCMV-specific sequence was removed by digestion with AvrII and replaced with commercially synthesized, AvrII-flanked LASV (Josiah strain) S segment-specific sequence containing 5′ and 3′ UTRs, the IGR, and BstI and BsmBI restriction sites at the NP and GPC loci, respectively, for cloning. The CAT reporter gene was cloned in to substitute for the NP ORF.
The IGR was shown to play a critical role in the control of transcription termination by the arenavirus polymerase complex, but overall levels of RNA replication were similar in the presence and absence of the IGR (14), suggesting that the IGR does not contribute significantly to the L-NP interaction required for RNA synthesis. To further examine this issue, we compared the effects of different IGR sequences on L-NP interaction. Consistent with previous findings (14), the activity of the MG construct lacking the IGR (MGΔIGR) was decreased to about 20% of the wild-type (WT) MG level (Fig. 2B). Moreover, replacement of the LCMV IGR with the LASV or JUNV IGR resulted only in very modestly decreased levels of CAT protein expression, which correlated with levels of cMG (complementary polarity to MG) RNA and CAT mRNA being not significantly affected (Fig. 2C). The MG construct that we used contains the CAT open reading frame (ORF) in the NP locus and an empty GPC locus that resulted in CAT mRNA and cMG RNA species that differed only by about 180 nucleotides, undistinguishable under our Northern blot conditions. Consistent with the MG activity results, all MG RNAs supported with similar efficiency L-NP interaction (Fig. 2D), suggesting that the 3′ and 5′ termini were critical for the enhancement of L-NP interaction. These results also support the view that structural features, rather than specific sequences, within the IGR contribute to the regulation of arenavirus gene transcription.
FIG 2.

The IGR is not required for efficient L-NP interaction. (A) Schematic diagrams of MG constructs. LCMV MG constructs containing either the LASV or the JUNV IGR or lacking the IGR were generated using PCR-based mutagenesis by replacing the IGR sequence of pT7-MG with LASV or JUNV IGR or deleting the IGR and also deleting residual NP and GPC ORF-derived sequences and a linker sequence (L1). L4 and L5, linker sequences for cloning. (B, C) Effect of the IGR on MG RNA synthesis and gene expression. 293T cells (4.5 × 105 cells/well; 12-well plate) were transfected with plasmids expressing NP (0.3 μg), L [L(+); 0.3 μg], and T7 RNA polymerase (0.5 μg) together with the indicated LCMV MG-expressing plasmid (0.5 μg) containing different IGR sequences or lacking the IGR (ΔIGR) (14). Empty pCAGGS plasmid was used instead of L-expressing plasmid [L(−)] as a negative control. At 72 h posttransfection, cells were collected and analyzed for CAT expression by ELISA and RNA synthesis by Northern blotting using a CAT antisense probe. 28S rRNA was detected by methylene blue staining. Representative Northern blot data from triplicate samples are shown. CAT and viral RNA expression levels in cells transfected with WT LCMV MG plasmid were set to 100%. Data represent means ± SD of results from triplicate samples. (D) Pulldown assay was performed as described for Fig. 1B. Values for input and PD samples correspond to 0.5% and 10%, respectively, of values for whole-cell lysates.
We next examined whether enhanced L-NP interaction could be observed by providing in trans an in vitro-transcribed RNA containing the virus-specific 3′ and 5′ termini of the MG RNA sequences in the absence of an active vRNP. For this, we prepared lysates of 293T cells transfected with NP- and L-expressing plasmids and added to them an in vitro-transcribed MG RNA (Fig. 3B). Subsequently, samples were subjected to PD assay as described in the legend to Fig. 1B. Consistent with our previous findings, L-NP interaction was enhanced in cells that supported vRNP-mediated viral RNA synthesis, whereas exogenously added in vitro-transcribed MG RNA failed to promote L-NP interaction (Fig. 3A), suggesting that the formation of an active RNP is required for support of L-NP interaction.
FIG 3.
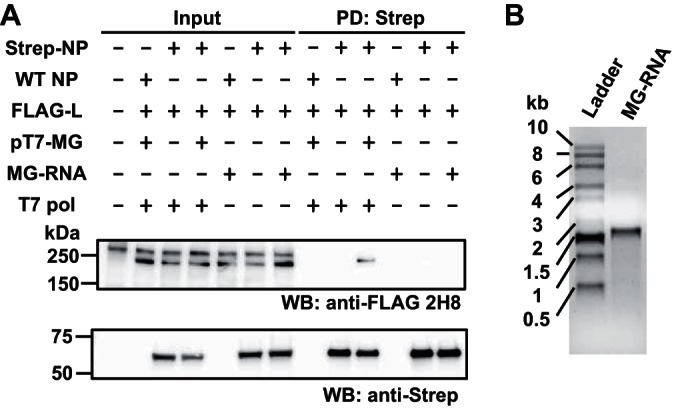
L-NP interaction is not supported by in vitro-transcribed MG RNA. (A) Effect of in vitro-transcribed virus-specific RNA on LCMV NP-L interaction. A DNA fragment containing the LCMV MG construct with enhanced green fluorescent protein (EGFP) and CAT genes in GP and NP loci, respectively, was amplified using a forward primer containing the T7 promoter sequence (5′-GAAATTAATACGACTCACTATAGGGCGCACCGG-3′), a reverse primer (5′-CGCACAGTGGATCCTAGGCATTTGATTGCG-3′), and pMG/S-CAT/GFP (14) as a template by PCR. Using this DNA fragment as a template, LCMV MG RNA in the genome polarity was transcribed using the T7 polymerase system (MAXIscript, Ambion) (MG-RNA). 293T cells (4.0 × 106 cells/10-cm dish) were cultured overnight and transfected with 3 μg of either pC-NP (WT NP) or pC-Strep-NP (Strep-NP) and 10 μg of pC-FLAG-L (FLAG-L), together with 3 μg of pCAGGS-T7pol (T7-pol) and 3 μg of pT7-MG using PEI Max. “+” or “−” indicates the presence or absence, respectively, of plasmid in the transfection mixture. Total amount of DNA was adjusted using empty pCAGGS. At 72 h posttransfection, cells were washed with PBS and lysed with 1 ml of CLB supplemented with a protease and phosphatase inhibitor cocktail. Cell lysates were clarified by centrifugation (15,000 rpm at 4°C for 10 min) and incubated with (+) or without (−) 500 ng of MG RNA at 4°C for 16 h, prior to PD with Strep-Tactin resin (Strep-Tactin Super flow Plus, Qiagen) at 4°C for 2 h. Beads were washed three times with 1 ml of CLB and the associated protein complex was eluted using CLB containing 2.5 mM desthiobiotin. Protein levels present in cleared cell lysate (Input) and eluate (PD: Strep) were determined by Western blotting. Values for input and PD-analyzed samples correspond to 0.5% and 10%, respectively, of values for whole-cell lysate. (B) The quality of RNA transcript (500 ng) used for panel A was examined by agarose gel electrophoresis.
To further confirm that L-NP interaction required for RNA synthesis is enhanced by virus-specific RNA sequences, we used the LCMV NP mutant with a D-to-A change at position 382 (D382A), which was shown to be dramatically affected in NP 3′-5′ exonuclease activity and ability to interfere with induction of type I interferon (IFN-I), without being significantly affected in NP activity in the LCMV MG rescue system (20). We predicted that although the D382A mutation had a great impact on a biologically important function of NP, this mutation should not affect L-NP interaction in the presence of virus-specific RNA. Both the WT and the D382A NPs were similarly efficient in their ability to PD L protein in the presence of virus-specific RNA (Fig. 4A). Consistent with our previous findings, D382A substitution did not significantly affect MG activity (Fig. 4B).
FIG 4.
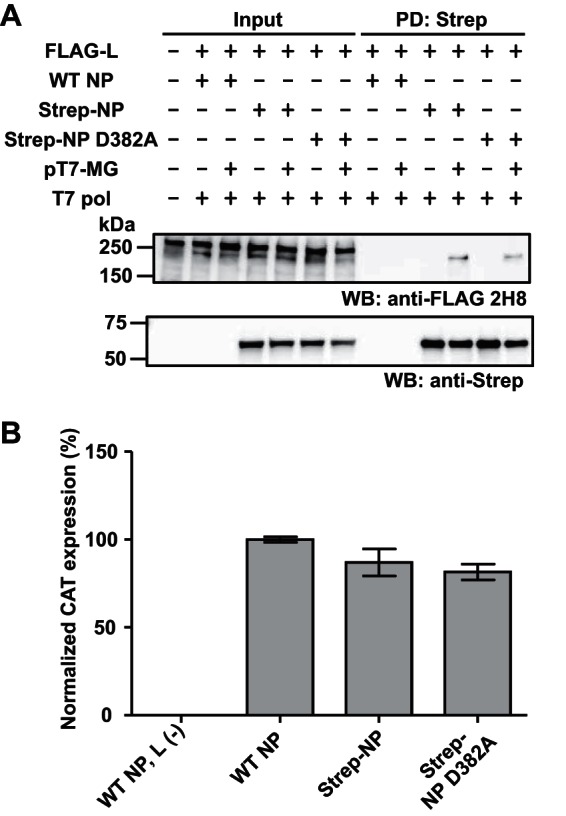
L-NP interaction is not affected by a mutation that abrogates the anti-IFN activity of NP. (A) Interaction between NP (D382A) and L in the presence of virus-specific RNA. To construct a plasmid expressing Strep-tagged NP D382A, a DNA fragment containing the D382A substitution of pC-NP D382A HA was subcloned into the corresponding region of Strep-tagged NP-expressing plasmid used for Fig. 1B by digestion with EcoNI and BbsI, followed by a ligation step (20). 293T cells (4.5 × 106 cells/10-cm dish) were cultured overnight prior to transfection with 8 μg of FLAG-L-expressing plasmid, 2 μg of untagged WT (WT NP) or Strep-tagged WT (Strep-NP) or D382A NP-expressing (Strep-NP D382A) plasmid, 2 μg of LCMV MG plasmid, and 2 μg of T7 pol-expressing plasmid, using PEI Max. “+” or “−” indicates the presence or absence, respectively, of plasmid in the transfection mixture. The total amount of DNA was adjusted using empty pCAGGS. At 72 h posttransfection, cells were lysed and PD was performed as described for Fig. 1B. Protein levels present in cleared cell lysate (Input) and eluate (PD: Strep) were examined by Western blotting. Values for input and PD-analyzed samples correspond to 0.5% and 10%, respectively, of values for whole-cell lysate. (B) Strep-tagged NP-D382A retained WT-like activity in the MG rescue assay. 293T cells (4.5 × 105 cells/well; 12-well plate) were cultured overnight and transfected with 0.5 μg of pT7-MG, 0.5 μg of pCAGGS-T7pol, and 0.3 μg of the indicated NP-expressing plasmid together with 0.3 μg of L protein-expressing plasmid. Empty pCAGGS was used instead of L protein-expressing plasmid as a negative control [L(−)]. At 72 h posttransfection, CAT expression levels were measured by a CAT ELISA kit (Roche). CAT expression levels of WT NP- and L-transfected cells were set to 100%. Data represent normalized means ± SD of results from triplicate samples.
Our results have shown that virus-specific RNA promotes arenavirus L-NP interaction. Intriguingly, a previous study reported coimmunoprecipitation (co-IP) of L-NP in the absence of arenavirus-specific RNA with the use of a vaccinia virus system (MVA-T7pol)-based expression system for L and NP (21). These apparently conflicting findings can be explained considering that the MVA-T7pol-based expression system results in hyperphysiological levels of L and NP that may overcome the requirement for virus-specific RNA. Expression levels of L protein are very low in infected cells, and direct L-NP interaction may also be of low affinity. Therefore, it is plausible that efficient vRNP-directed viral RNA synthesis is promoted by an enhanced interaction of L with the bona fide virus nucleocapsid template where NP tightly interacts with the viral RNA. On the other hand, a low affinity of the direct L-NP interaction might facilitate travel of the L protein through NP-encapsidated viral genomic and antigenomic RNA species during the biosynthetic processes of RNA replication and gene transcription.
ACKNOWLEDGMENTS
This work was supported by NIH grants RO1 AI047140 and RO1 AI077719 to J.C.D.L.T. M.I. was supported by the Japan Society for the Promotion of Science, the Daiichi Sankyo Foundation of Life Science, and the KANAE Foundation for the Promotion of Medical Science.
Footnotes
This article is contribution 29031 from The Scripps Research Institute.
REFERENCES
- 1.Enria DA, Briggiler AM, Sanchez Z. 2008. Treatment of Argentine hemorrhagic fever. Antiviral Res 78:132–139. doi: 10.1016/j.antiviral.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geisbert TW, Jahrling PB. 2004. Exotic emerging viral diseases: progress and challenges. Nat Med 10:S110–S121. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- 3.Khan SH, Goba A, Chu M, Roth C, Healing T, Marx A, Fair J, Guttieri MC, Ferro P, Imes T, Monagin C, Garry RF, Bausch DG, Mano River Union Lassa Fever Network. 2008. New opportunities for field research on the pathogenesis and treatment of Lassa fever. Antiviral Res 78:103–115. doi: 10.1016/j.antiviral.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 4.McCormick JB, Fisher-Hock SP. 2002. Lassa fever, vol 262 Springer-Verlag, Berlin, Germany. [Google Scholar]
- 5.Peters CJ. 2002. Human infection with arenaviruses in the Americans, vol 262 Springer-Verlag, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 6.Weissenbacher MC, Laguens RP, Coto CE. 1987. Argentine hemorrhagic fever. Curr Top Microbiol Immunol 134:79–116. [DOI] [PubMed] [Google Scholar]
- 7.Barton LL, Mets MB. 1999. Lymphocytic choriomeningitis virus: pediatric pathogen and fetal teratogen. Pediatr Infect Dis J 18:540–541. doi: 10.1097/00006454-199906000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Barton LL, Mets MB. 2001. Congenital lymphocytic choriomeningitis virus infection: decade of rediscovery. Clin Infect Dis 33:370–374. doi: 10.1086/321897. [DOI] [PubMed] [Google Scholar]
- 9.Barton LL, Mets MB, Beauchamp CL. 2002. Lymphocytic choriomeningitis virus: emerging fetal teratogen. Am J Obstet Gynecol 187:1715–1716. doi: 10.1067/mob.2002.126297. [DOI] [PubMed] [Google Scholar]
- 10.Fischer SA, Graham MB, Kuehnert MJ, Kotton CN, Srinivasan A, Marty FM, Comer JA, Guarner J, Paddock CD, DeMeo DL, Shieh WJ, Erickson BR, Bandy U, DeMaria A Jr, Davis JP, Delmonico FL, Pavlin B, Likos A, Vincent MJ, Sealy TK, Goldsmith CS, Jernigan DB, Rollin PE, Packard MM, Patel M, Rowland C, Helfand RF, Nichol ST, Fishman JA, Ksiazek T, Zaki SR, LCMV in Transplant Recipients Investigation Team. 2006. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N Engl J Med 354:2235–2249. doi: 10.1056/NEJMoa053240. [DOI] [PubMed] [Google Scholar]
- 11.Jahrling PB, Peters CJ. 1992. Lymphocytic choriomeningitis virus. A neglected pathogen of man. Arch Pathol Lab Med 116:486–488. [PubMed] [Google Scholar]
- 12.Mets MB, Barton LL, Khan AS, Ksiazek TG. 2000. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am J Ophthalmol 130:209–215. doi: 10.1016/S0002-9394(00)00570-5. [DOI] [PubMed] [Google Scholar]
- 13.Buchmeier MJ, Peters CJ, de la Torre JC. 2007. Arenaviridae: the viruses and their replication, p 1791–1851. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 14.Pinschewer DD, Perez M, de la Torre JC. 2005. Dual role of the lymphocytic choriomeningitis virus intergenic region in transcription termination and virus propagation. J Virol 79:4519–4526. doi: 10.1128/JVI.79.7.4519-4526.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez M, Craven RC, de la Torre JC. 2003. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc Natl Acad Sci U S A 100:12978–12983. doi: 10.1073/pnas.2133782100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez M, de la Torre JC. 2003. Characterization of the genomic promoter of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 77:1184–1194. doi: 10.1128/JVI.77.2.1184-1194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC. 2000. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J Virol 74:3470–3477. doi: 10.1128/JVI.74.8.3470-3477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KJ, Perez M, Pinschewer DD, de la Torre JC. 2002. Identification of the lymphocytic choriomeningitis virus (LCMV) proteins required to rescue LCMV RNA analogs into LCMV-like particles. J Virol 76:6393–6397. doi: 10.1128/JVI.76.12.6393-6397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortiz-Riaño E, Cheng BY, de la Torre JC, Martinez-Sobrido L. 2012. Self-association of lymphocytic choriomeningitis virus nucleoprotein is mediated by its N-terminal region and is not required for its anti-interferon function. J Virol 86:3307–3317. doi: 10.1128/JVI.05503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Sobrido L, Emonet S, Giannakas P, Cubitt B, Garcia-Sastre A, de la Torre JC. 2009. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol 83:11330–11340. doi: 10.1128/JVI.00763-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerber R, Rieger T, Busch C, Flatz L, Pinschewer DD, Kummerer BM, Gunther S. 2011. Cross-species analysis of the replication complex of Old World arenaviruses reveals two nucleoprotein sites involved in L protein function. J Virol 85:12518–12528. doi: 10.1128/JVI.05091-11. [DOI] [PMC free article] [PubMed] [Google Scholar]