

ABSTRACT
The host-targeted antiviral drug UV-4B reduces viral replication and promotes survival in a mouse model of experimental dengue virus (DENV) infection. UV-4B is an iminosugar that inhibits the α-glucosidase family of enzymes and subsequently the folding of glycosylated proteins, both viral and host. Here, we utilized next-generation sequencing to investigate evolution of a flavivirus under selective pressure by a host-targeted antiviral in vivo. In viral populations recovered from UV-4B-treated mice, there was a significant increase in the number of single-nucleotide polymorphisms (SNPs) and the ratio of nonsynonymous to synonymous SNPs compared to findings in viral populations from vehicle-treated mice. The strongest evidence of positive selection was in the glycosylated membrane protein, thereby providing in vivo validation of the mechanism of action of an iminosugar. In addition, mutations in glycosylated proteins were present only in drug-treated mice after a single passage. However, the bulk of the other mutations were present in both populations, indicating nonspecific selective pressure. Together with the continued control of viremia by UV-4B, these findings are consistent with the previously predicted high genetic barrier to escape mutations in host-targeted antivirals.
IMPORTANCE Although hundreds of millions of people are infected with DENV every year, there is currently no approved vaccine or antiviral therapy. UV-4B has demonstrated antiviral activity against DENV and is expected to enter clinical trials soon. Therefore, it is important to understand the mechanisms of DENV resistance to UV-4B. Host-targeted antivirals are thought to have a higher genetic barrier to escape mutants than directly acting antivirals, yet there are very few published studies of viral evolution under host-targeted antivirals. No study to date has described flavivirus evolution in vivo under selective pressure by a host-based antiviral drug. We present the first in vivo study of the sequential progression of viral evolution under selective pressure by a host-targeted antiviral compound. This study bolsters support for the clinical development of UV-4B as an antiviral drug against DENV, and it provides a framework to compare how treatment with other host-targeted antiflaviviral drugs in humans and different animal models influence viral genetic diversity.
INTRODUCTION
Dengue virus (DENV) is the most common mosquito-borne human pathogen. DENV can cause illnesses ranging from self-limited mild disease to severe and potentially life-threatening dengue hemorrhagic fever/dengue shock syndrome. Approximately 400 million people are infected each year with DENV (1), generally in tropical and subtropical areas, with 20,000 infections resulting in death (2). The current standard of care consists of fluid replacement and other supportive care. There is presently no vaccine to prevent or antiviral to treat DENV infection. Recent phase IIb and III trials of an attenuated, tetrameric vaccine showed limited efficacy, especially in naive individuals and against certain DENV serotypes (3, 4). Chemotherapeutic agents effective against DENV are a pressing unmet medical need.
Replication of DENV relies on an RNA polymerase with poor proofreading capabilities. Due to this error-prone replication, many variants are formed, resulting in a quasispecies (5, 6). Viral fitness, the replication environment, and other factors determine which mutations become fixed in the viral population. Studies suggest that there is an optimal mutation rate that produces genetic diversity while limiting the accumulation of deleterious mutations (7–10).
Antiviral treatment typically results in the eradication of susceptible variants. This eradication, however, leaves replicative space for minor populations of resistant viruses to expand. This can result in the antiviral regimen failing (11, 12). The important role of minor subspecies in accelerated resistance has been shown in various viruses, including HIV (13) and hepatitis C virus (HCV) (12). In the case of HCV and influenza virus, drug-resistant strains arise due to single-nucleotide polymorphisms (SNPs) (14, 15), which are easily acquired due to the error rate of the RNA polymerase.
An antiviral that targets a common host pathway could subvert the issues of viral heterogeneity and the emergence of drug-resistant mutants. Host-targeted antiviral therapies are thought to have an elevated barrier to resistance because host factors are genetically more stable than viral factors. Additionally, in the case of α-glucosidase inhibitors, multiple viral proteins are affected, presumably making the route to an escape mutant more complex. Finally, targeting a host pathway that is commonly used by all replicating DENV serotypes, not to mention numerous other viruses, has the potential for broad application.
Dengue viral particles assemble in the endoplasmic reticulum where the glycosylation machinery of the host is required to modify envelope and membrane glycoproteins to form immature viral particles (16). Compounds that disrupt glycosylation can cause viral glycoproteins to misfold, ultimately resulting in reduced assembly, secretion, and infectivity of viral particles (17, 18). Iminosugars are monosaccharides that target the host enzymes α-glucosidase I and II (19, 20). It has been shown that iminosugars affect the folding of the prM protein of DENV and subsequently the formation of the prM-E complex (21). Recently, we tested the iminosugar UV-4B for efficacy in a mouse model of severe dengue-like disease (22). Treatment of 129/Sv mice deficient in type I and type II interferon (IFN) receptors (AG129) with UV-4B, even when it is administered as late as 48 h postinfection, reduces viral titers and cytokine levels and promotes survival.
Next-generation sequencing (NGS) methods have greatly expanded our ability to characterize minor variants in viral populations and to monitor how these variants respond to antiviral agents (23–25). Deep-sequencing methods have been used to detect resistance mutations to virus-based antivirals during treatment for influenza virus (26), HIV (27), hepatitis B virus (28), and hepatitis C virus (29). In vivo studies of viral quasispecies evolving under selective pressure by antivirals have been completed in human subjects but typically only after antiviral failure. Most also focus on a few previously identified resistance mutations to virus-targeting antivirals (23, 30–32).
Here, we present the first in vivo study of flaviviral evolution under selective pressure by a host-targeted antiviral compound. We sequenced viral populations from treated or untreated mice at the peak of viremia (22). In the UV-4B treated mice, we found that there was an overall increase in SNPs and an increase in the ratio of nonsynonymous to synonymous SNPs. These findings suggest that there is increased selective pressure on DENV in the presence of UV-4B. Increased specific selective pressure appeared to be preferentially located in glycosylated viral proteins. However, the bulk of mutations were present in both treated and untreated populations, indicating some level of nonspecific selective pressure. Additionally, viremia remained controlled by the presence of UV-4B, with no evidence of viral escape even after several passages in vivo.
MATERIALS AND METHODS
Viruses.
Generation and preparation of DENV2 strain S221 was described previously (33, 34). Viral stocks were grown in C6/36 cells and concentrated by ultracentrifugation (35). The number of genomic equivalents (GE) was determined using real-time reverse transcription-PCR (RT-PCR) (35, 36).
Mouse experiments.
STAT1−/−/STAT2−/− (STAT1−/−/2−/−)129/Sv mice were bred and housed under specific-pathogen-free conditions at the La Jolla Institute (LJI). Mice were treated orally three times daily (TID; at 8-h intervals) with UV-4B (at 100 mg/kg of the active product UV-4) dissolved in water or with water alone (vehicle) for 72 h, starting at −1 h relative to infection. Doses were corrected for the 11.4% higher molecular weight (MW) of the test article, UV-4B (UV-4, MW 319.44; UV-4B is a UV-4 HCl salt, MW 355.90). After 30 min, 1 × 1010 (passage 1 and single-passage experiment) or 1 × 108 (passages 2 to 5 of multiple-passage experiment) GE of the S221 challenge virus resuspended in phosphate-buffered saline (PBS) with 5% fetal calf serum (FCS) was administered via intravenous (i.v.) tail vein injection. On day 3 of each passage, mice were euthanized, and blood was collected by cardiac puncture. Serum was separated, and a small aliquot was used for RNA isolation for the purpose of determining viral titer and sequencing. The next passage group of three mice was infected with serum diluted to 1 × 108 GE/dose (passages 2 to 5 of the multiple-passage experiment only). The animal work described here was conducted under animal protocols approved by the LIAI Institutional Animal Care and Use Committee.
Viral RNA harvest and quantification.
Immediately following isoflurane overdose, blood was collected from mice by cardiac puncture and separated using serum collection tubes. Viral RNA was isolated from serum using Qiagen Viral RNA isolation kits. To maximize RNA yield, the final product was eluted twice with 40 μl of buffer, yielding a total of 80 μl. DENV2 quantitative RT-PCR (qRT-PCR) was performed on serum samples pooled from three mice (multiple-passage experiment) or from each of the 10 mice (single-passage experiment), and the viral titer in GE per ml of serum was determined (35, 36).
Sequencing and analysis.
RNA library preparations, sequencing reactions, and initial bioinformatics analysis were conducted at Genewiz, Inc. (South Plainfield, NJ, USA). RNA samples were quantified using a NanoDrop 2000 (ThermoFisher Scientific, Waltham, MA, USA), and RNA integrity was checked with a 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Illumina TruSeq RNA library preparation, clustering, and sequencing reagents were used throughout the process according to the manufacturer's recommendations (Illumina, San Diego, CA, USA). The samples were clustered on one lane of a flow cell and sequenced using a 1-by-50 single-read (SR) configuration. Image analysis and base calling were conducted by the HiSeq Control software (HCS) on a HiSeq 2500 instrument. One mismatch was allowed for index sequence identification. To determine how the virus population changed with each passage, the sequence of each passage was compared to a reference sequence of the virus generated from the input virus used to infect the first set of mice (i.e., passage 1).
For assembly of the reference sequence (input virus grown on C6/36 cells), the 34,188,426 reads corresponding to the input reference sequence were aligned to cow (bovine serum medium supplement) and mosquito (C6/36 cells) as a filter to remove any non-dengue virus reads. The reads were filtered out using Bowtie2 (version 2.0.2) (37). The remaining reads were analyzed using a Trinity RNA-Seq de novo assembler (r2012-10-05) with a standard parameter set (38). Among the assembled contigs was one full-length (10,712-bp) DENV2 sequence, which was used as a reference in subsequent analyses (GenBank accession number KM587709).
Data from pooled and individual samples of passaged mouse serum were processed analogously, using more sensitive Bowtie2 parameters. SNPs were called using FreeBayes software, which uses a Bayesian model to derive the probability of an SNP at each position, or PSNP, given the read sequence and quality information (39).
The fraction of nonsynonymous mutations over all of the nucleotides in a protein versus the same quantity for synonymous mutations was calculated. At each position in the nucleotide alignments to the reference sequence, the fractions of wild-type sequence, synonymous SNPs, and nonsynonymous SNPs were calculated. The sum of these fractions over a given subprotein was used to calculate the overall ratio of nonsynonymous to synonymous SNPs.
Plaque assay for measuring infectious DENV titers in mice.
The infectious titer of DENV2 strain S221 in animal tissues was determined by plaque assay as previously described (40). Briefly, 2 × 105 BHK-21 cells were seeded in 24-well plates in 1× minimal essential medium (MEM; Gibco) supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and HEPES. Confluent monolayers were infected with 100 μl of diluted infected serum. After a 1-h incubation, BHK-21 cells were covered with 1 ml of 1% carboxymethyl cellulose in 1× MEM (2% FBS, penicillin, streptomycin, and HEPES). Four days later, cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet diluted in 20% ethanol. Plaques were scored visually and expressed as number of PFU per ml.
RESULTS
DENV does not acquire mutations that increase fitness during replication in vivo in the presence of UV-4B.
To determine if UV-4B places selective pressure on DENV replication in vivo, we sequentially passaged DENV2 strain S221 in STAT1−/−/2−/− mice treated orally (intragastrically, i.g.) with vehicle only or with 100 mg/kg UV-4B. The STAT1−/−/2−/− mice are the most susceptible mouse model to DENV infection so far described (36) and thus represent a highly stringent in vivo challenge system. During each passage, treatment with UV-4B or vehicle only was initiated at 1 h before infection and continued three times per day (TID) until harvest. In the first passage, three mice treated with vehicle only and three mice treated with UV-4B were each infected with 1 × 1010 GE of S221. Blood was harvested by cardiac puncture at 72 h postinfection (hpi) as this time point is the peak of viremia in DENV-infected mice, occurring just prior to potentially losing mice to severe disease (22). Nonstructural proteins are present in the tissues of infected mice within 6 h of infection (41), suggesting that another replication cycle could begin as early as 6 h postinfection. Based on studies of a related flavivirus, West Nile virus, flaviviral virions are typically released from cells starting at 8 to 10 h after infection, and a full replication cycle of flaviviruses may be as much as 12 to 16 h, depending on the cell type (42). Therefore, at least 4.5 replication cycles may have occurred during one 72-h infection. Serum was separated from the harvested blood and pooled for each group of three mice. A portion of the pooled serum was separated for RNA isolation to determine the DENV titer and for next-generation sequencing (NGS). The remaining serum was used to infect another two groups of three mice with the highest viral titer possible while still infecting each mouse with equal titers (1 × 108 GE/mouse for passages 2 to 4, 100-fold less than passage 1) under the same conditions (TID treatment with vehicle or UV-4B). This process was continued for a total of four passages (Fig. 1A). As expected from the 100-fold decrease in virus input from passage 1 to passage 2, there was a decrease in virus output harvested at 72 hpi (Fig. 1B). As seen in previous studies (22), treatment with UV-4B reduced viremia to 10 to 30% of that of vehicle-treated mice (Table 1; Fig. 1B). To determine the infectivity of the output virus, we determined the ratio of GE to PFU of the UV-4B- and vehicle-passaged samples. Serum from the first passage was used to perform BHK cell-based plaque assays since the virus would never pass directly from one mammal to another under normal conditions. The ratio of GE to PFU was similar between the UV-4B-passaged (5.56 × 104) and vehicle-passaged (7.69 × 104) samples. These results suggest that the mutations that increase viral fitness in the presence of UV-4B have not been acquired after the first passage. After further passaging, which equates to at least 18 replication cycles, the fitness of drug-treated viruses had still not reached that of viruses passaged in the absence of the drug (Fig. 1B). Although there was some fluctuation, UV-4B was still controlling viral replication to nearly the same level as seen in passage 1 (passage 4, 18.8%; passage 1, 11.9%) (Table 1). The variability is likely due to mouse-to-mouse variation, as can be seen in the single-passage experiment (Fig. 2A; see also Table 5).
FIG 1.

DENV passaged multiple times in mice with and without UV-4B treatment. (A) Schematic of experimental design. (B) Output serum titers after each passage in STAT1−/−/2−/− mice treated with vehicle or UV-4B. Sera from the three mice in each group were pooled and titrated by qRT-PCR to determine the GE/ml. (C) Schematic of mutated positions in multiple in vivo-passaged viral populations.
TABLE 1.
Challenge dose and DENV output titers from passages 1 to 4 in vehicle- and UV-4B- treated STAT1−/−/2−/− mice at 72 hpi
| Passage no. | Amt of DENV in vehicle treatment group |
Amt of DENV in UV-4B treatment group |
Output ratio of UV-4B/vehicle (%) | ||
|---|---|---|---|---|---|
| Input (GE/mouse) | Output (GE/ml)a | Input (GE/mouse) | Output (GE/ml)a | ||
| 1 | 1 × 1010 | 8.94 × 109 | 1 × 1010 | 1.06 × 109 | 11.90 |
| 2 | 1 × 108 | 4.96 × 109 | 1 × 108 | 4.86 × 108 | 9.80 |
| 3 | 1 × 108 | 3.81 × 109 | 1 × 108 | 1.15 × 109 | 30.20 |
| 4 | 1 × 108 | 3.75 × 109 | 1 × 108 | 7.07 × 108 | 18.80 |
Determined in serum.
FIG 2.

UV-4B controls viral replication and increases selective pressure. (A) Viral load of single-passage samples. (B) Number of SNPs per protein accumulated in all 10 vehicle- and UV-4B-treated mice. (C) Ratio of nonsynonymous mutation rate per protein to the synonymous mutation rate per protein.
TABLE 5.
Viral titers and number of SNPs versus number of RMD in the single-passage experiment
| Treatment and sample no. | Titer (log copies/ml) | No. of RMDa | No. of significant SNPs | No. of significant SNPs/no. of RMD |
|---|---|---|---|---|
| Vehicle | ||||
| 1 | 10.79 | 391,060 | 1 | 0.000003 |
| 2 | 7.82 | 1,449 | 24 | 0.016563 |
| 3 | 9.62 | 13,760 | 9 | 0.000654 |
| 4 | 10.95 | 679,136 | 1 | 0.000001 |
| 5 | 10.6 | 240,096 | 1 | 0.000004 |
| 6 | 10.73 | 996,676 | 1 | 0.000001 |
| 7 | 10.47 | 107,713 | 1 | 0.000009 |
| 8 | 10.99 | 560,968 | 1 | 0.000002 |
| 9 | 10.78 | 397,347 | 1 | 0.000003 |
| 10 | 10.51 | 197,853 | 2 | 0.000010 |
| UV-4B | ||||
| 1 | 9.64 | 7,819 | 88 | 0.011255 |
| 2 | 9.61 | 6,626 | 88 | 0.013281 |
| 3 | 9.76 | 10,137 | 40 | 0.003946 |
| 4 | 10.02 | 41,596 | 7 | 0.000168 |
| 5 | 10.2 | 59,707 | 5 | 0.000084 |
| 6 | 10.03 | 34,831 | 11 | 0.000316 |
| 7 | 8.77 | 7,002 | 173 | 0.024707 |
| 8 | 9.29 | 8,909 | 62 | 0.006959 |
| 9 | 6.43 | 6,297 | 60 | 0.009528 |
| 10 | 9.33 | 10,361 | 86 | 0.008300 |
RMD, reads that map uniquely to DENV.
Fluctuation and accumulation of mutations in passaged virus in the presence of UV-4B or water vehicle treatment.
To identify mutations acquired during passaging, RNA sequencing was performed on the original input virus and on the pooled serum after each passage. Each passaged sample produced 4.5 to 10 million reads, with 90% mapping to the mouse genome and 0.17 to 1.2% mapping to DENV, resulting in 60- to 600-fold coverage of the dengue genome (Table 2).
TABLE 2.
Sequencing analysis and alignment of samples compared to the filter (mouse) and dengue virus genomes
| Treatment and passage no. | Total no. of reads | No. (%) of reads |
||
|---|---|---|---|---|
| Aligned to filter (mouse) | Aligned to DENV | Unaligned | ||
| Vehicle | ||||
| P1 | 10,069,281 | 8,880,427 (88.19) | 121,046 (1.20) | 1,067,808 (10.60) |
| P2 | 8,949,360 | 7,997,713 (89.37) | 15,194 (0.17) | 936,453 (10.46) |
| P3 | 7,436,803 | 6,571,910 (88.37) | 52,914 (0.71) | 811,979 (10.92) |
| P4 | 6,231,269 | 5,545,270 (88.99) | 12,046 (0.19) | 673,953 (10.82) |
| UV-4B | ||||
| P1 | 5,057,665 | 4,501,023 (88.99) | 12,958 (0.26) | 543,684 (10.75) |
| P2 | 4,451,789 | 3,970,184 (89.18) | 48,851 (1.10) | 432,754 (9.72) |
| P3 | 9,268,561 | 8,260,346 (89.12) | 21,653 (0.23) | 986,562 (10.64) |
| P4 | 6,263,807 | 5,646,343 (90.14) | 75,016 (1.20) | 542,448 (8.66) |
SNPs in the passaged samples were identified using FreeBayes to compare to the input virus. We found 33 significant SNPs (PSNP < 0.05) in either the UV-4B- or vehicle-treated samples in at least one time point. Eight SNPs were located in the untranslated region, and 12 SNPs in coding regions did not result in an amino acid change (synonymous SNPs). We focused our attention on the remaining 13 SNPs located in coding regions that lead to an amino acid change (nonsynonymous SNPs) (Table 3 and Fig. 1C). These 13 SNPs are shown in Table 3 with the ratio of the percentage of the viral population with the SNPs in UV-4B-treated mice to that of vehicle-treated mice. Some of the nonsynonymous SNPs appeared in early passages but dropped below the level of significance (positions 9692, 8807, 9144, and 832) or were undetectable (positions 4389, 4050, 7136, and 1283) by the fourth passage. These SNPs are possibly due to genetic drift and are less likely to have an impact on DENV fitness in the presence of drug. The remaining four nonsynonymous SNPs—at positions 8813, 842, 9134, and 6803—were prevalent in the UV-4B-treated samples throughout the four passages. However, 12 of the 13 nonsynonymous mutations were found in both the UV-4B- and vehicle-treated groups, indicating that the mutations are not due to specific positive selection. Instead, these mutations may be the result of mutagenesis or changes in the viral population structure due to the drug. Mutations that are nonspecific to the drug mechanism of action would not create escape mutants. Presumably, these nonspecific mutations improve viral fitness even in the absence of UV-4B.
TABLE 3.
SNP location and frequency in multiple passages in UV-4- and vehicle-treated micea
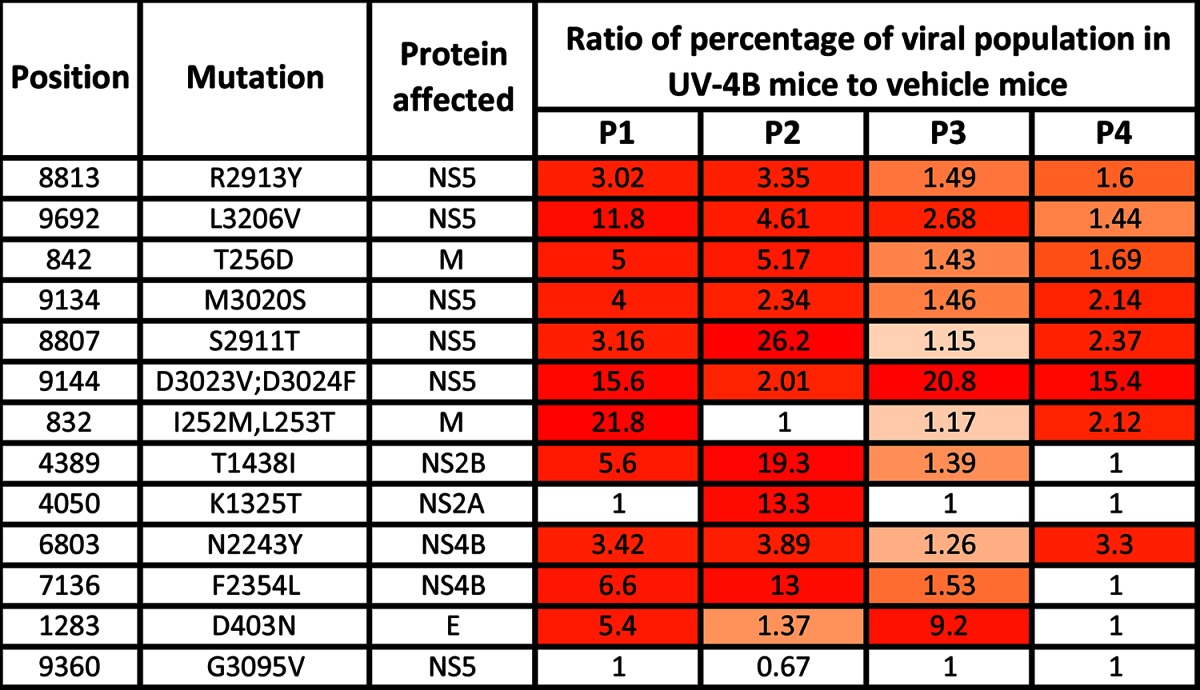
Positions are given in nucleotides. The mutation at position 4050 is the only mutation in the multiple-passage experiment that was found in only the drug-treated group. Frequency is expressed as the ratio of the percentage of the viral population with the mutation in UV-4B-treated mice to that in vehicle-treated mice. The color gradation from orange to red reflects increases in the ratio.
Although informative, the previous experiment has two notable limitations. First, DENV is normally passaged alternately between invertebrate (mosquito) and vertebrate (human) hosts; thus, a single passage in vertebrates (mice) is more physiologically relevant than studying multiple passages from vertebrate to vertebrate. Second, pooling samples for sequencing can dilute individual mouse-specific responses. To address these limitations, we next performed a single passage and sequenced viral RNA from individual mice.
Mutations are stochastic and highly variable between mice.
Two groups of 10 mice each were infected with 1 × 1010 GE of S221 and treated with either vehicle or UV-4B. After 72 h, we collected serum and performed qRT-PCR to determine the viral load. The average viral load in the UV-4B-treated mice was almost 10-fold less than that in the vehicle-treated animals (P = 0.0404, unpaired t test) (Fig. 2A), which is in agreement with the previous multiple-passage experiment.
As before, viral RNA in the serum samples was sequenced, and FreeBayes was used to identify SNPs in the passaged samples. Synonymous and nonsynonymous mutations were identified in each DENV protein (see Table S1 in the supplemental material). As UV-4B affects protein glycosylation, we focused on the 19 nonsynonymous mutations in glycosylated DENV proteins: membrane (M), envelope (E), and NS1. Only one mutation at position 842 was found in both experiments. However, because the mutation was found throughout passaging with the control in the multiple-passage experiment, it is presumed not to be specific to the drug mechanism. None of the other SNPs found in the multiple-passage experiment were observed in the single-passage experiment, including SNPs that were present in 30 to 50% of the population after a single passage. It is possible that these SNPs are present but not at high enough levels above the sequencing error rate to reach significance in the single-passage experiment. This result indicates that the viral response to antiviral pressure can be highly stochastic.
In addition to the prM mutation at position 842, we identified 18 other nonsynonymous mutations in glycosylated proteins. These mutations were present at significant levels in at least 1 of 10 UV-4B-treated mice and did not appear in any control mice. Table 4 lists these mutations ranked by the average percentage at which they appeared in mice with the mutation. These mutations are present only in the UV-4B-passaged samples, and therefore each could be important in the development of UV-4B resistance and should be monitored in circulating viral populations.
TABLE 4.
Nonsynonymous SNPs present only in glycosylated proteins in drug-treated samples in the single-passage experiment
| Position (nt)a | Mutation(s) | Protein affected | Frequency of the mutation(s) (%) in viral population (avg) | % mice with mutation detected (significant and nonsignificant) | % mice with significant levels of mutation |
|---|---|---|---|---|---|
| 1810 | Y578Y, S579S | E | 15.7 | 90 | 50 |
| 2297 | I741V | E | 15.6 | 70 | 30 |
| 3102 | E1009G | NS1 | 14.9 | 100 | 50 |
| 1399 | I441I, K442K, I443V | E | 14.9 | 60 | 40 |
| 3213 | K1046R | NS1 | 14.4 | 20 | 10 |
| 2851 | N925N, S926S, L927L | NS1 | 14.2 | 70 | 10 |
| 2893 | T939T, N940N | NS1 | 13.0 | 60 | 10 |
| 2650 | V858V, K859K, L860 M | NS1 | 12.7 | 80 | 20 |
| 2082 | N669S | E | 12.1 | 50 | 20 |
| 1435 | E453E, L454L | E | 11.7 | 70 | 20 |
| 842 | T256D | prM | 11.7 | 50 | 10 |
| 870 | A265V | prM | 11.3 | 90 | 20 |
| 2905 | L943L, K944K | NS1 | 11.2 | 50 | 10 |
| 3080 | P1002S | NS1 | 10.8 | 80 | 20 |
| 2913 | R946K | NS1 | 10.1 | 50 | 10 |
| 2230 | V718V, F719F | E | 10.1 | 30 | 10 |
| 1358 | H428N | E | 10.0 | 100 | 20 |
| 857 | H261Y | prM | 9.8 | 50 | 10 |
| 1984 | I636I, V637V | E | 9.0 | 40 | 10 |
nt, nucleotides.
We next analyzed the number of mutations that accumulated in the single-passage experiment. Based on a PSNP value of <0.05 and a coverage threshold of 10, the DENV population from vehicle-treated mice had an average of 4.2 significant SNPs (range of 1 to 24), whereas viruses from UV-4B-treated mice had an average of 62 (range of 5 to 173) (Table 5; see also Table S1 in the supplemental material), a statistically significant difference (P = 0.0027, Mann-Whitney test assuming a nonlinear relationship). In accordance with previous and expected results, the vast majority of reads mapped to the mouse genome. The number of reads that map uniquely to DENV (RMD) varied greatly across the entire experiment. Most samples from the vehicle-treated mice had 10 to 100 times more RMD than those from the UV-4B-treated mice. This is to be expected, considering the differences in viral titers (Fig. 2A and Table 5).
In vehicle-treated samples with high numbers of RMD, the parent population likely makes up a majority of the quasispecies population. Therefore, in many of the vehicle-treated samples, the very low mutant populations are diluted because they did not occur significantly above the sequencing error rate. Alternatively, in situations where the parent population is controlled by some factor, the number of RMD remains low. These situations include the UV-4B-treated samples and two control samples (Table 5, vehicle 2 and vehicle 3). In these samples, virus growth is dampened by the combined effects of the antiviral treatment and individual mouse-specific differences. In vehicle 2 and -3 samples, the log GE/ml and RMD values are similar to the values of many of the treatment samples (Table 5, UV-4B-treated samples 1 to 3 and 7 to 10). In all of these samples, the parent population stays low and controlled, and many mutations are detectable.
Plotting the ratios of SNPs to RMD in each mouse showed that there are far more SNPs per RMD in the UV-4B-treated mice than in the vehicle-treated group (Fig. 2B). This result suggests that UV-4B promotes more viral evolution than vehicle alone. However, many of these mutations are present in both vehicle- and drug-treated samples, and none of the 10 UV-4B-treated mice showed signs of a successful escape mutation as the viral RNA levels remained controlled.
Given the increased prevalence of SNPs in DENV isolated from UV-4B-treated mice, we next sought to determine if any individual protein showed an increased number of SNPs. To do this we calculated the ratio of nonsynonymous to synonymous mutations for each protein. Using this ratio, we can compare the extent of positive (purifying) selection in DENV isolated from vehicle- and UV-4B-treated mice. Table 6 ranks DENV protein sequences isolated from UV-4B-treated mice from the highest to lowest level of positive selection, based on the ratio of nonsynonymous to synonymous mutations. Two of the four glycosylated proteins (marked with an X), membrane (0.30) and NS1 (0.26), have the high values. The other two glycosylated proteins, premembrane and envelope, rank fifth and seventh, respectively, out of the 12 viral proteins. Under vehicle-only conditions, the average across all proteins is 0.0017; under UV-4B conditions, the average is 0.11, demonstrating that there is much more positive selection going on in most proteins under the UV-4B treatment condition (Table 6 and Fig. 2C). In particular, much of this selective pressure seems to be targeting the glycosylated proteins. While 18.8% of nonsynonymous mutations in nonglycosylated proteins are found in both vehicle- and drug-treated samples, only 5% of nonsynonymous mutations in glycosylated proteins are found in both. This indicates that there is more nonspecific selective pressure inflicted by UV-4B in nonglycosylated proteins. However, that a portion of the mutations are present in both groups suggests that there is positive selection that is not specific to the mechanism of action of UV-4B. Alternatively, the drug acts as a mutagen or otherwise interferes with viral population structure that leads to an increased accumulation of mutations. However, the observation that the ratio of nonsynonymous to synonymous mutations observed was consistently higher in the drug-treated samples argues for selection of these mutations. A possible explanation is that these mutations are favorable for an aspect of viral replication in vivo and that this same aspect becomes even more crucial in the presence of the drug. This interpretation is consistent with our observation that an even greater difference between the UV-4B and vehicle treatments is found in mutations in untranslated regions (UTRs; 13-fold) than in coding regions (6-fold) (Table 7). However, even under this heightened positive pressure, a mutant that is as fit as the virus in samples treated with vehicle only has not been identified (Table 1 and Fig. 1B).
TABLE 6.
Calculation of the ratio of nonsynonymous to synonymous mutations in DENV proteins under vehicle and UV-4B treatment conditions
| Protein | Glycosylated | Nonsynonymous/synonymous mutations by treatmenta |
|
|---|---|---|---|
| Vehicle | UV-4B | ||
| M | X | 0.00 | 0.30 |
| NS1 | X | 0.00 | 0.26 |
| NS5 | 0.01 | 0.21 | |
| NS4A | 0.00 | 0.20 | |
| prM | X | 0.00 | 0.14 |
| NS2A | 0.00 | 0.11 | |
| E | X | 0.01 | 0.09 |
| NS3 | 0.00 | 0.02 | |
| NS4B | 0.00 | 0.01 | |
| C | 0.00 | 0.00 | |
| NS2B | 0.00 | 0.00 | |
| 2K peptide | 0.00 | 0.00 | |
| Avg | 0.00 | 0.11 | |
The ratio of nonsynonymous to synonymous mutations is derived by dividing the fraction of nonsynonymous mutations over all of the nucleotides in that protein by the same quantity for synonymous mutations.
TABLE 7.
Coding and noncoding mutations in treatment and control groups
| Protein mutation type | Length (nt)a | Avg no. of mutations (×103) |
Avg no. of mutations in treatment group/avg no. of mutations in control group | ||
|---|---|---|---|---|---|
| Overall | Control group | Treatment group | |||
| Coding | 10,184 | 0.88 | 0.25 | 1.51 | 6.1 |
| Noncoding | 538 | 0.23 | 0.03 | 0.43 | 13.1 |
nt, nucleotides.
DISCUSSION
In this study, we developed an experimental approach to monitor the evolution of DENV in response to the host-targeting antiviral UV-4B. While the presence of some mutations in both treated and untreated (vehicle-only) populations suggest that these mutations can be attributed to mouse passaging, our results demonstrate that there is a group of mutations present in glycosylated proteins only under UV-4B treatment conditions which are likely attributed to the specific mechanism of the drug. Even in the presence of mutations that we attribute to the activity of UV-4B, we find no evidence of a true escape mutant after four serial passages consisting of at least 18 rounds of replication in mice. This finding supports the theory that there is a high genetic barrier for the evolution of escape mutants in the presence of a host-based antiviral.
In general, we find more positive selection in DENV when infected mice are treated with UV-4B than when they are treated with vehicle only. The fact that a subset of these mutations are present in both drug- and vehicle-treated mice suggests that some of the positive selective pressure can be attributed to a nonspecific cause unrelated to the mechanism of action of the drug. It is possible that the drug itself is acting as a mutagen or otherwise interfering with viral population structure, leading to an increased accumulation of mutations. However, given that the ratio of nonsynonymous to synonymous mutations observed was consistently higher in the drug-treated samples argues that these mutations are selected for. A possible explanation is that these mutations are favorable for an aspect of replication of the virus in vivo and that this same aspect becomes even more crucial in the presence of the drug.
A different subset of mutations are present in only the drug-treated samples, and many of these mutations are present on glycosylated viral proteins, which is unsurprising given that the drug decreases the folding efficiency of glycosylated proteins (21). While 18.8% of nonsynonymous mutations in nonglycosylated proteins are found in both vehicle- and drug-treated samples, only 5% of nonsynonymous mutations in glycosylated proteins are found in both. This indicates that there is more nonspecific selective pressure inflicted by UV-4B in nonglycosylated proteins. We highlight several mutations in glycosylated proteins that may contribute to drug resistance in Table 4. In addition, multiple mutations in the NS5 protein suggest that viral replication may be broadly impacted by UV-4B. Regardless, there is no evidence of a true escape mutant even after four serial passages consisting of at least 18 rounds of replication in mice. This finding supports the theory that there is a high genetic barrier for the evolution of escape mutants in the presence of a host-based antiviral.
Virus evolution under pressure from antivirals has been studied mostly in the context of direct antiviral agents. As such, the antiviral activity can often be overcome by a single mutation. For example, individual SNPs are responsible for resistance of HCV to daclatasvir and of influenza A virus to oseltamivir (14, 15). In contrast to the published reports on directly acting antivirals, it is possible that the virus in this study did not undergo enough replication cycles to allow a resistant mutant to develop.
The most thorough studies of a host-based antiviral have focused on the mutagenic effects of ribavirin. A study of the evolution of HCV in patients treated with ribavirin or interferon or both used consensus sequencing (43), which does not identify mutants that exist in <20% of the population. Another study used deep sequencing to identify strains of HCV that evolved to be as fit in the presence of ribavirin as wild-type virus in the absence. The relevance of this and other in vitro studies to infections in vivo is not always clear (44).
In individuals infected with HCV, virions with each single mutation and every possible double mutation combination are made daily and are likely to exist in the population at all times (45, 46). These calculations are based on the error rate of the RNA polymerase, the genome size, and the number of virions produced daily. The estimated error rate for the DENV RNA polymerase is similar to that of HCV (10−5 to 10−6 per copied nucleotide). While the genome size of DENV is similar to that of HCV at ∼11 kb, we do not know the number of virions that are produced each day in human patients or in our mouse model. Therefore, we cannot extrapolate the same calculations to DENV. Work on HCV and other RNA viruses has demonstrated that the mutant spectrum of RNA viruses can be extensive. Yet after multiple passages, each with multiple viral replication cycles, a DENV with superior fitness in the presence of UV-4B did not evolve. This finding suggests that high levels of resistance to UV-4B could require numerous mutations in multiple genes.
In the single-passage experiment, which may be more physiologically relevant than the multiple-passage experiment, more SNPs occurred under pressure from UV-4B, indicating that antiviral therapy increases viral genetic diversity. High genetic diversity can alter the population biology of RNA viruses and influence their fitness in dynamic environments and when they face population bottlenecks. The effect that this altered genetic diversity has on the fitness of the passaged virus is an important question that can be addressed in future studies using reverse genetics.
It is possible that by focusing on nonsynonymous SNPs, we are ignoring changes related to splicing or mRNA transport. However, it is clear that the sum total of all the changes, synonymous and nonsynonymous, has not resulted in a population that is as fit in the presence of UV-4B as the wild type in its absence.
Four successive passages through a vertebrate host is not physiologically relevant as natural DENV transmission alternates between human and mosquito. However, we sought to apply consistent selective pressure to force mutations potentially resulting in resistance to UV-4B. By doing so, we were able to identify specific mutations that may already be contributing to the level of replication we see after one passage, or at least 18 rounds of replication. The ratio of nonsynonymous to synonymous mutations suggests that higher levels of positive selection are occurring in specific proteins, particularly glycosylated proteins, especially considering that these mutations are present only in the UV-4B-treated samples. As UV-4B begins clinical trials, clinicians can take into consideration the mutations and proteins we have identified as contributing to viral fitness in the presence of UV-4B. The mutations can be sought in circulating DENV strains, and differences in viral replication in UV-4B-treated patients could be attributed to the presence of mutations in glycosylated proteins NS1 and membrane. It is possible that a combination of many mutations is required to reach a virus that is as fit in the presence of UV-4B as in its absence. However, the genetic barrier to achieve this is likely high because in this study, after 18 in vivo rounds of replication, we could not demonstrate generation of such escape as a consequence of UV-4B treatment in vivo.
As UV-4B is now entering clinical trials, the amino acid changes found in the single-passage experiment provide a framework for predicting potential mutations in patients. We found that treating mice with UV-4B applies both nonselective and selective pressure, resulting in enhanced genetic diversity. More specific positive selection was observed, as expected, in glycosylated proteins and, unexpectedly, in NS5, which suggests that UV-4B perhaps has a broad impact on viral replication. Despite continuous forced drug pressure over 18 replication cycles resulting in increased accumulation of mutations, a virus with increased fitness was not observed. We predict that it is unlikely that UV-4B-resistant viruses will emerge in humans.
Supplementary Material
ACKNOWLEDGMENT
This research was funded in part by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract number HHS272201100030C.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00028-15.
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gibbons RV, Vaughn DW. 2002. Dengue: an escalating problem. BMJ 324:1563–1566. doi: 10.1136/bmj.324.7353.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabchareon A, Wallace D, Sirivichayakul C, Limkittikul K, Chanthavanich P, Suvannadabba S, Jiwariyavej V, Dulyachai W, Pengsaa K, Wartel TA, Moureau A, Saville M, Bouckenooghe A, Viviani S, Tornieporth NG, Lang J. 2012. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. Lancet 380:1559–1567. doi: 10.1016/S0140-6736(12)61428-7. [DOI] [PubMed] [Google Scholar]
- 4.Capeding MR, Tran NH, Hadinegoro SR, Ismail HI, Chotpitayasunondh T, Chua MN, Luong CQ, Rusmil K, Wirawan DN, Nallusamy R, Pitisuttithum P, Thisyakorn U, Yoon IK, van der Vliet D, Langevin E, Laot T, Hutagalung Y, Frago C, Boaz M, Wartel TA, Tornieporth NG, Saville M, Bouckenooghe A, the CYD14 Study Group. 2014. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet 384:1358–1365. doi: 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- 5.Chin-inmanu K, Suttitheptumrong A, Sangsrakru D, Tangphatsornruang S, Tragoonrung S, Malasit P, Tungpradabkul S, Suriyaphol P. 2012. Feasibility of using 454 pyrosequencing for studying quasispecies of the whole dengue viral genome. BMC Genomics 13(Suppl 7):S7. doi: 10.1186/1471-2164-13-S7-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruiz-Jarabo CM, Arias A, Baranowski E, Escarmis C, Domingo E. 2000. Memory in viral quasispecies. J Virol 74:3543–3547. doi: 10.1128/JVI.74.8.3543-3547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biebricher CK, Eigen M. 2005. The error threshold. Virus research 107:117–127. doi: 10.1016/j.virusres.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Gnadig NF, Beaucourt S, Campagnola G, Borderia AV, Sanz-Ramos M, Gong P, Blanc H, Peersen OB, Vignuzzi M. 2012. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc Natl Acad Sci U S A 109:E2294–E2303. doi: 10.1073/pnas.1204022109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vignuzzi M, Andino R. 2012. Closing the gap: the challenges in converging theoretical, computational, experimental and real-life studies in virus evolution. Curr Opin Virol 2:515–518. doi: 10.1016/j.coviro.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adiwijaya BS, Herrmann E, Hare B, Kieffer T, Lin C, Kwong AD, Garg V, Randle JC, Sarrazin C, Zeuzem S, Caron PR. 2010. A multi-variant, viral dynamic model of genotype 1 HCV to assess the in vivo evolution of protease-inhibitor resistant variants. PLoS Comput Biol 6:e1000745. doi: 10.1371/journal.pcbi.1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verbinnen T, Van Marck H, Vandenbroucke I, Vijgen L, Claes M, Lin TI, Simmen K, Neyts J, Fanning G, Lenz O. 2010. Tracking the evolution of multiple in vitro hepatitis C virus replicon variants under protease inhibitor selection pressure by 454 deep sequencing. J Virol 84:11124–11133. doi: 10.1128/JVI.01217-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charpentier C, Dwyer DE, Mammano F, Lecossier D, Clavel F, Hance AJ. 2004. Role of minority populations of human immunodeficiency virus type 1 in the evolution of viral resistance to protease inhibitors. J Virol 78:4234–4247. doi: 10.1128/JVI.78.8.4234-4247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miura M, Maekawa S, Sato M, Komatsu N, Tatsumi A, Takano S, Amemiya F, Nakayama Y, Inoue T, Sakamoto M, Enomoto N. 2014. Deep sequencing analysis of variants resistant to the non-structural 5A inhibitor daclatasvir in patients with genotype 1b hepatitis C virus infection. Hepatol Res 44:E360–E367. doi: 10.1111/hepr.12316. [DOI] [PubMed] [Google Scholar]
- 15.Pollara CP, Piccinelli G, Rossi G, Cattaneo C, Perandin F, Corbellini S, Tomasi DD, Bonfanti C. 2013. Nosocomial outbreak of the pandemic Influenza A (H1N1) 2009 in critical hematologic patients during seasonal influenza 2010-2011: detection of oseltamivir resistant variant viruses. BMC Infect Dis 13:127. doi: 10.1186/1471-2334-13-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitby K, Pierson TC, Geiss B, Lane K, Engle M, Zhou Y, Doms RW, Diamond MS. 2005. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J Virol 79:8698–8706. doi: 10.1128/JVI.79.14.8698-8706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapel C, Garcia C, Bartosch B, Roingeard P, Zitzmann N, Cosset FL, Dubuisson J, Dwek RA, Trepo C, Zoulim F, Durantel D. 2007. Reduction of the infectivity of hepatitis C virus pseudoparticles by incorporation of misfolded glycoproteins induced by glucosidase inhibitors. J Gen Virol 88:1133–1143. doi: 10.1099/vir.0.82465-0. [DOI] [PubMed] [Google Scholar]
- 18.Block TM, Jordan R. 2001. Iminosugars as possible broad spectrum anti hepatitis virus agents: the glucovirs and alkovirs. Antivir Chem Chemother 12:317–325. doi: 10.1177/095632020101200601. [DOI] [PubMed] [Google Scholar]
- 19.Chang J, Schul W, Butters TD, Yip A, Liu B, Goh A, Lakshminarayana SB, Alonzi D, Reinkensmeier G, Pan X, Qu X, Weidner JM, Wang L, Yu W, Borune N, Kinch MA, Rayahin JE, Moriarty R, Xu X, Shi PY, Guo JT, Block TM. 2011. Combination of alpha-glucosidase inhibitor and ribavirin for the treatment of dengue virus infection in vitro and in vivo. Antiviral Res 89:26–34. doi: 10.1016/j.antiviral.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu X, Pan X, Weidner J, Yu W, Alonzi D, Xu X, Butters T, Block T, Guo JT, Chang J. 2011. Inhibitors of endoplasmic reticulum alpha-glucosidases potently suppress hepatitis C virus virion assembly and release. Antimicrob Agents Chemother 55:1036–1044. doi: 10.1128/AAC.01319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courageot MP, Frenkiel MP, Dos Santos CD, Deubel V, Despres P. 2000. Alpha-glucosidase inhibitors reduce dengue virus production by affecting the initial steps of virion morphogenesis in the endoplasmic reticulum. J Virol 74:564–572. doi: 10.1128/JVI.74.1.564-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry ST, Buck MD, Plummer EM, Penmasta RA, Batra H, Stavale EJ, Warfield KL, Dwek RA, Butters TD, Alonzi DS, Lada SM, King K, Klose B, Ramstedt U, Shresta S. 2013. An iminosugar with potent inhibition of dengue virus infection in vivo. Antiviral Res 98:35–43. doi: 10.1016/j.antiviral.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Margeridon-Thermet S, Shulman NS, Ahmed A, Shahriar R, Liu T, Wang C, Holmes SP, Babrzadeh F, Gharizadeh B, Hanczaruk B, Simen BB, Egholm M, Shafer RW. 2009. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J Infect Dis 199:1275–1285. doi: 10.1086/597808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitsuya Y, Varghese V, Wang C, Liu TF, Holmes SP, Jayakumar P, Gharizadeh B, Ronaghi M, Klein D, Fessel WJ, Shafer RW. 2008. Minority human immunodeficiency virus type 1 variants in antiretroviral-naive persons with reverse transcriptase codon 215 revertant mutations. J Virol 82:10747–10755. doi: 10.1128/JVI.01827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. 2007. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res 17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghedin E, Laplante J, DePasse J, Wentworth DE, Santos RP, Lepow ML, Porter J, Stellrecht K, Lin X, Operario D, Griesemer S, Fitch A, Halpin RA, Stockwell TB, Spiro DJ, Holmes EC, St George K. 2011. Deep sequencing reveals mixed infection with 2009 pandemic influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. J Infect Dis 203:168–174. doi: 10.1093/infdis/jiq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher R, van Zyl GU, Travers SA, Kosakovsky Pond SL, Engelbrech S, Murrell B, Scheffler K, Smith D. 2012. Deep sequencing reveals minor protease resistance mutations in patients failing a protease inhibitor regimen. J Virol 86:6231–6237. doi: 10.1128/JVI.06541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solmone M, Vincenti D, Prosperi MC, Bruselles A, Ippolito G, Capobianchi MR. 2009. Use of massively parallel ultradeep pyrosequencing to characterize the genetic diversity of hepatitis B virus in drug-resistant and drug-naive patients and to detect minor variants in reverse transcriptase and hepatitis B S antigen. J Virol 83:1718–1726. doi: 10.1128/JVI.02011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Svarovskaia ES, Martin R, McHutchison JG, Miller MD, Mo H. 2012. Abundant drug-resistant NS3 mutants detected by deep sequencing in hepatitis C virus-infected patients undergoing NS3 protease inhibitor monotherapy. J Clin Microbiol 50:3267–3274. doi: 10.1128/JCM.00838-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armenia D, Vandenbroucke I, Fabeni L, Van Marck H, Cento V, D'Arrigo R, Van Wesenbeeck L, Scopelliti F, Micheli V, Bruzzone B, Lo Caputo S, Aerssens J, Rizzardini G, Tozzi V, Narciso P, Antinori A, Stuyver L, Perno CF, Ceccherini-Silberstein F. 2012. Study of genotypic and phenotypic HIV-1 dynamics of integrase mutations during raltegravir treatment: a refined analysis by ultra-deep 454 pyrosequencing. J Infect Dis 205:557–567. doi: 10.1093/infdis/jir821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsibris AM, Korber B, Arnaout R, Russ C, Lo CC, Leitner T, Gaschen B, Theiler J, Paredes R, Su Z, Hughes MD, Gulick RM, Greaves W, Coakley E, Flexner C, Nusbaum C, Kuritzkes DR. 2009. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 4:e5683. doi: 10.1371/journal.pone.0005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Muh U, Welker M, Wincheringer D, Zhou Y, Chu HM, Lin C, Weegink C, Reesink H, Zeuzem S, Kwong AD. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 33.Perry ST, Prestwood TR, Lada SM, Benedict CA, Shresta S. 2009. Cardif-mediated signaling controls the initial innate response to dengue virus in vivo. J Virol 83:8276–8281. doi: 10.1128/JVI.00365-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zellweger RM, Prestwood TR, Shresta S. 2010. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe 7:128–139. doi: 10.1016/j.chom.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prestwood TR, Prigozhin DM, Sharar KL, Zellweger RM, Shresta S. 2008. A mouse-passaged dengue virus strain with reduced affinity for heparan sulfate causes severe disease in mice by establishing increased systemic viral loads. J Virol 82:8411–8421. doi: 10.1128/JVI.00611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perry ST, Buck MD, Lada SM, Schindler C, Shresta S. 2011. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog 7:e1001297. doi: 10.1371/journal.ppat.1001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marth GT, Korf I, Yandell MD, Yeh RT, Gu Z, Zakeri H, Stitziel NO, Hillier L, Kwok PY, Gish WR. 1999. A general approach to single-nucleotide polymorphism discovery. Nat Genet 23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- 40.Shresta S, Kyle JL, Snider HM, Basavapatna M, Beatty PR, Harris E. 2004. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T- and B-cell-dependent immunity are less critical. J Virol 78:2701–2710. doi: 10.1128/JVI.78.6.2701-2710.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prestwood TR, May MM, Plummer EM, Morar MM, Yauch LE, Shresta S. 2012. Trafficking and replication patterns reveal splenic macrophages as major targets of dengue virus in mice. J Virol 86:12138–12147. doi: 10.1128/JVI.00375-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brinton MA. 2014. Replication cycle and molecular biology of the West Nile virus. Viruses 6:13–53. doi: 10.3390/v6010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asahina Y, Izumi N, Enomoto N, Uchihara M, Kurosaki M, Onuki Y, Nishimura Y, Ueda K, Tsuchiya K, Nakanishi H, Kitamura T, Miyake S. 2005. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J Hepatol 43:623–629. doi: 10.1016/j.jhep.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 44.Feigelstock DA, Mihalik KB, Feinstone SM. 2011. Selection of hepatitis C virus resistant to ribavirin. Virol J 8:402. doi: 10.1186/1743-422X-8-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rong L, Dahari H, Ribeiro RM, Perelson AS. 2010. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med 2:30ra32. doi: 10.1126/scitranslmed.3000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domingo E, Sheldon J, Perales C. 2012. Viral quasispecies evolution. Microbiol Mol Biol Rev 76:159–216. doi: 10.1128/MMBR.05023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.