

ABSTRACT
High-risk human papillomaviruses (HR-HPV) cause anogenital cancers, including cervical cancer, and head and neck cancers. Human papillomavirus 16 (HPV16) is the most prevalent HR-HPV. HPV oncogenesis is driven by two viral oncoproteins, E6 and E7, which are expressed through alternative splicing of a polycistronic RNA to yield four major splice isoforms (E6 full length, E6*I, E6*II, E6*X). The production of multiple mRNA isoforms from a single gene is controlled by serine/arginine-rich splicing factors (SRSFs), and HPV16 infection induces overexpression of a subset of these, SRSFs 1, 2, and 3. In this study, we examined whether these proteins could control HPV16 oncoprotein expression. Small interfering RNA (siRNA) depletion experiments revealed that SRSF1 did not affect oncoprotein RNA levels. While SRSF3 knockdown caused some reduction in E6E7 expression, depletion of SRSF2 resulted in a significant loss of E6E7 RNAs, resulting in reduced levels of the E6-regulated p53 proteins and E7 oncoprotein itself. SRSF2 contributed to the tumor phenotype of HPV16-positive cervical cancer cells, as its depletion resulted in decreased cell proliferation, reduced colony formation, and increased apoptosis. SRSF2 did not affect transcription from the P97 promoter that controls viral oncoprotein expression. Rather, RNA decay experiments showed that SRSF2 is required to maintain stability of E6E7 mRNAs. These data show that SRSF2 is a key regulator of HPV16 oncoprotein expression and cervical tumor maintenance.
IMPORTANCE Expression of the HPV16 oncoproteins E7 and E6 drives HPV-associated tumor formation. Although increased transcription may yield increased levels of E6E7 mRNAs, it is known that the RNAs can have increased stability upon integration into the host genome. SR splicing factors (SRSFs) control splicing but can also control other events in the RNA life cycle, including RNA stability. Previously, we demonstrated increased levels of SRSFs 1, 2, and 3 during cervical tumor progression. Now we show that SRSF2 is required for expression of E6E7 mRNAs in cervical tumor but not nontumor cells and may act by inhibiting their decay. SRSF2 depletion in W12 tumor cells resulted in increased apoptosis, decreased proliferation, and decreased colony formation, suggesting that SRSF2 has oncogenic functions in cervical tumor progression. SRSF function can be targeted by known drugs that inhibit SRSF phosphorylation, suggesting a possible new avenue in abrogating HPV oncoprotein activity.
INTRODUCTION
Human papillomaviruses (HPV) infect mucosal and cutaneous epithelia. At least 13 so-called “high-risk” HPV (HR-HPV) infect the anogenital epithelium and can cause persistent lesions that may progress to cancer (1). For example, around 500,000 women worldwide experience anogenital HPV infection, and nearly 300,000 die per annum from cervical cancer. Increasingly, HPV infection is also being linked to oropharyngeal cancer, whereby incidence of this disease is increasing rapidly (2). HPV16 is the most prevalent HR-HPV. HPV-associated tumorigenesis is driven by increased expression of the HPV E6 and E7 oncoproteins (3). E6 promotes ubiquitin-mediated degradation of p53 to inhibit apoptosis, modulates transcription of cell cycle-related genes, induces telomerase activity, controls cell shape and polarity, and activates cap-dependent translation (4). E7 binds and degrades Rb to promote S phase entry and cell division, controls transcription of cell cycle-related genes, and acts as a mitotic mutator (4).
HPV E6 and E7 oncoproteins are expressed from a polycistronic transcript that for HPV16 can potentially produce four different alternatively spliced mRNAs (E6 full length [E6fl], E6*I, E6*II, and E6*X [also called E6*III]) (5, 6). The putative E6* proteins all share the first 44 amino acids of full-length E6 with C-terminal truncations or frame shifts into the E7 open reading frame (5). E6*I is the most abundant isoform in cervical cell lines (7–10) and patient samples (11, 12) and has been suggested to encode E7 (6). Although detectable in tumor samples (12), the biological function of E6*II and E6*X has not been investigated.
Serine/arginine-rich (SR) proteins (SR splicing factors [SRSFs]) can regulate most of the processes in the life cycle of an mRNA, including transcription, RNA processing, RNA export, RNA stability, and translation (13). SR proteins are key players in the regulation of constitutive and alternative splicing. Constitutive splicing is the process whereby introns are removed from pre-mRNAs and exons are spliced together to form a protein-coding mRNA. Alternative splicing is a mechanism used by mammalian and viral genomes to maximize coding potential (14). A single gene is transcribed to give a single primary transcript, but from this precursor RNA different mature mRNA isoforms can be generated by differentiation inclusion or exclusion of exons and introns. Each isoform can encode a different protein. There are nine classical SR proteins, named SRSF1 to SRSF9. Apart from RNA processing-related functions, SR proteins have also been shown to be involved in chromatin remodelling, transcriptional regulation, genome stability maintenance, nucleolar stress, cell cycle progression, apoptosis control, and protein sumoylation (15–20). Unsurprisingly, due to their diverse functions, many SR proteins are overexpressed in a range of tumors (21–25). Importantly, SRSF1 (ASF/SF2), SRSF3 (SRp20), and SRSF9 (SRp30c) have been shown to possess oncogenic properties (22–31). Increased SRSF levels can result in the production of alternatively spliced RNA isoforms encoding key antiapoptotic, cell proliferation, and epithelial-mesenchymal transition (EMT)-inducing proteins (18).
HPV16 oncoprotein expression is controlled at several SRSF-regulated posttranscriptional levels, including constitutive and alternative RNA splicing, RNA stability, and translation (6, 32, 33). Increased expression levels of SRSFs 1, 2, and 3 in cervical tumor cells and samples from patients with HPV-positive cervical lesions (24) prompted an investigation of a possible oncogenic role of SR proteins in cervical cancer. Here, we examine the effect of SRSF depletion on E6E7 mRNA production in cervical tumor cells. Data revealed that SRSF1 and SRSF3 depletion had little effect on E6E7 expression. However, SRSF2 (SC35) depletion caused a marked reduction in levels of E6E7 mRNAs, leading to reduced levels of E7 protein and induction of p53, indicating a reduction in E6 protein levels. SRSF2 depletion resulted in reduced cell proliferation, decreased anchorage-independent growth, and induction of apoptosis in the tumor cells. SRSF2 did not control transcription of E6E7 RNAs but rather controlled their stability. These data suggest that SRSF2 is a key cellular factor controlling HPV16 oncoprotein expression by protecting the RNAs encoding them from decay. Since SR protein activity is a proven relevant antiviral drug target (34), these findings provide insight into new therapeutic avenues against HPV-associated oncogenesis.
MATERIALS AND METHODS
Cell culture and reagents.
W12E and W12G cells are clones 20863 and 20861, respectively, as described previously (35). W12t and W12ti cells were cloned in our laboratory from W12G cells and were originally named W12GPX and W12GPXY (36). W12ti, 293T, CaSki, and SiHa cells were passaged in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen), 10% fetal calf serum (FCS) (Invitrogen), and penicillin (50 U/ml)-streptomycin (50 μg/ml) (Invitrogen). W12E, W12G, and W12t cells (36) were cultured in the same medium but with the addition of 0.1 M cholera toxin and 0.4 μg/ml hydrocortisone. W12E and W12G cells were grown (2 × 105 cells per 10-cm plate) on mitomycin C-treated J2 3T3 mouse fibroblasts (35). Differentiation was achieved as previously described (10, 35). 3T3 cells were grown in DMEM and 10% donor calf serum with antibiotics as described above. All cells were cultured at 37°C in a 5% CO2 humidified incubator.
Transfection.
Cells were seeded at 2 × 105 per well in a six-well plate 24 h prior to transfection in antibiotic-free medium. Small interfering RNA (siRNA; 10 μM) and Lipofectamine RNA interference (RNAi) MAX (Invitrogen) or plasmid (100 ng) and Lipofectamine (Invitrogen) were diluted in Opti-MEM serum-free medium (Invitrogen). siRNAs against SRSF2 were an siGENOME Dharmacon SMARTpool (product no. M-019711-00-0005) consisting of four siRNAs or a single siRNA whose efficacy has been previously demonstrated (37). SMARTpool siRNAs were also used to deplete SRSF3 (product no. M-030081-00-0005). SRSF1 was depleted using Sigma product no. SASI_Hs02_00313260. Cells were transfected for 48 h according to the manufacturer's protocol. Transfection efficiencies calculated by cotransfection with siGLO (Dharmacon) or a green fluorescent protein (GFP) expression plasmid were between 70 and 80% for W12E/G, CaSki, and C33A cells, 80 to 90% for W12ti cells, and >90% for 293T cells.
RNA extraction.
All protocols followed the manufacturer's instructions unless otherwise stated. Cells were scraped into TRIzol reagent (Invitrogen), and total RNA was extracted. Polyadenylated RNA was isolated using an oligo(dT)-based mRNA extraction kit (Oligotex; Qiagen). DNA was removed from all RNAs using the Promega RQ1 DNase kit. DNase-treated RNA was reverse transcribed using the Superscript III kit (Invitrogen).
PCR.
cDNA was amplified using 200 nM primers, 200 μM deoxynucleoside triphosphates (dNTPs), 1.5 mM MgCl2, and 2 units of Taq polymerase (Invitrogen). Primers were chosen at the 5′ end of the E6 open reading frame and the 3′ end of the E7 open reading frame so as to amplify all E6E7 isoforms: E6 forward, 5′-GAGAACTGCAATGTTTCAGGACCC-3′ (HPV16 genome nucleotides [nt] 94 to 117); E7 reverse, 5′-GAACAGATGGGGCACACAATTCC-3′ (HPV16 genome nt 845 to 823). PCR products were separated on a 6% polyacrylamide gel and poststained with ethidium bromide.
Reverse transcriptase quantitative PCR (RT-qPCR).
Standard curves were generated as recommended (Applied Biosystems instruction manual). cDNA (100 ng) was amplified for each reaction and carried out in triplicate. Primer and probe sequences used were as follows: E6 forward primer, 5′-TCATGTTTCAGGACCCACAG-3′; E6 reverse primer, 5′-CTGTTGCTTGCAACAGAGCTGC-3′; E6 probe sequence, 5′-CCACAGTTATGCACAGAGCTGC-3′. GAPDH was used as the internal standard control: GAPDH forward, 5′-CGCTCTCTGCTCCTCCTGTT-3′; GAPDH reverse, 5′-CCATGGTGTCTGAGCGATGT-3′; GAPDH probe, 5′-CAAGCTTCCCGTTCTCAGCC-3′. Reaction mixes (25 μl) contained 1× master mix (Stratagene), 900 nM primers, 100 nM probe, and 300 nM reference dye (Stratagene). qPCRs were performed and analyzed on an Applied Biosystems 7500 Fast System.
Western blotting.
Cells were harvested into NP-40 lysis buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris-HCl [pH 8.0], with protease and phosphatase inhibitor cocktails [Roche Diagnostics]). Lysates were cleared by centrifugation at 10,000 × g for 10 min at 4°C. Protein concentration was determined by Bradford assay. SDS-PAGE and Western blotting were carried out exactly as described previously (38). Primary antibodies were as follows: anti-phospho-SRSF (ATTC hybridoma supernatant clone MAb104, used neat), GAPDH (1:1,000; AMS Biotechnology), p53 (1:500; BD Pharmingen clone DO-7), Rb (1:2,000; Cell Signaling Technology clone 4H1), SRSF1 (1:1,000; Zymed clone 96), SRSF2, SRSF3 (1:1,000; Pharmingen), (1:250; Zymed clone 7B4), and HPV16 E6 (1:200; Santa Cruz sc1583). Secondary antibodies (Sigma) were used at a dilution of 1:2,000.
Annexin V assay.
Cells were transfected 48 h before harvesting for staining. Control cells were treated with UV light at 500 J/m2 24 h prior to harvesting for staining. Cells were trypsinized, counted, and resuspended at 1 × 106 cells/ml in 500 μl annexin binding buffer (100 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2). Next, 100 μl of cells was mixed with 5 μl annexin V at 488 nm (Invitrogen) and 1 μg/ml propidium iodide and incubated at room temperature for 15 min in the dark. Finally, 400 μl annexin binding buffer was added before analysis on a BD Biosciences FACScalibur machine.
Colony formation assay.
Cells were transfected 24 h prior to being plated on soft agar. Cells were counted, and 2 × 104 cells were resuspended in 1× DMEM, 10% FCS, and 0.35% agarose, and then 2.5 ml was plated out onto 6-cm plates containing 0.5% agarose in 1× DMEM and 10% FCS. The plates were incubated at 37°C in 5% CO2 for 12 to 14 days. The cells were stained with 0.5 ml 0.005% crystal violet for 1 h before being dried and photographed.
Luciferase assay.
293T cells were transfected as described above with a control vector, pGL3-promoter (Promega), or with an HPV16 long control region (LCR) expression vector (39) and either siGLO or SRSF2 siRNA. Cells were harvested after 48 h, and luciferase reporter activity (Promega luciferase assay kit) was measured using a Thermolabs Luminoskan Ascent plate reader.
mRNA stability assay.
W12ti cells were transfected with control siRNA or with SRSF2 siRNA for 24 h, and then actinomycin D at 10 μg/ml (or control vehicle [H2O] alone) was added to inhibit de novo RNA synthesis. RNA was harvested at 0, 1, and 4 h post-drug addition. DNase 1-treated RNA was converted to cDNA and amplified by semiquantitative RT-PCR using the E6 primers described in the PCR section. PCR products were separated on a 6% acrylamide gel and poststained with ethidium bromide.
RESULTS
HPV16 oncoproteins are expressed from a set of alternatively spliced mRNA isoforms in nontumor and tumor cervical epithelial cells.
HPV16 expresses at least four HPV16 E6 mRNA isoforms predicted to be generated by alternative splicing (Fig. 1A) (6). Some of these isoforms have been detected previously in cell lines and patient tissues (7, 12, 40–44). However, it is unclear whether all isoforms can be expressed simultaneously and/or are differentially expressed in nontumor versus tumor cells. To investigate expression of the E6 RNA isoforms, we used the W12 cell line, an epithelial cell line derived from an HPV16-infected low-grade cervical lesion (45). Previous studies demonstrated that W12 cells express at least some E6 RNA isoforms (10, 46). A subclone, W12E (clone 20863), contains ∼100 episomal copies of the HPV16 genome and, if treated as a noncontinuous cell line (i.e., not passaged in culture), maintains a nontumor phenotype capable of differentiation in monolayer culture (10, 35). Another subclone, W12G (clone 20861), also displays a nontumor phenotype but with reduced differentiation capacity. This is despite the cells containing HPV16 genome copies integrated into the host genome, often a hallmark of cervical cancer cells (35). Previously, we derived two tumor lines sequentially from W12G cells (36). W12t cells (formerly called W12GPX) are cervical tumor cells, while W12ti cells (formerly called W12GPXY) have an invasive tumor phenotype in vitro and in vivo (36). W12ti cells express 5-fold more total E6 RNA than W12G cells (Fig. 1B).
FIG 1.
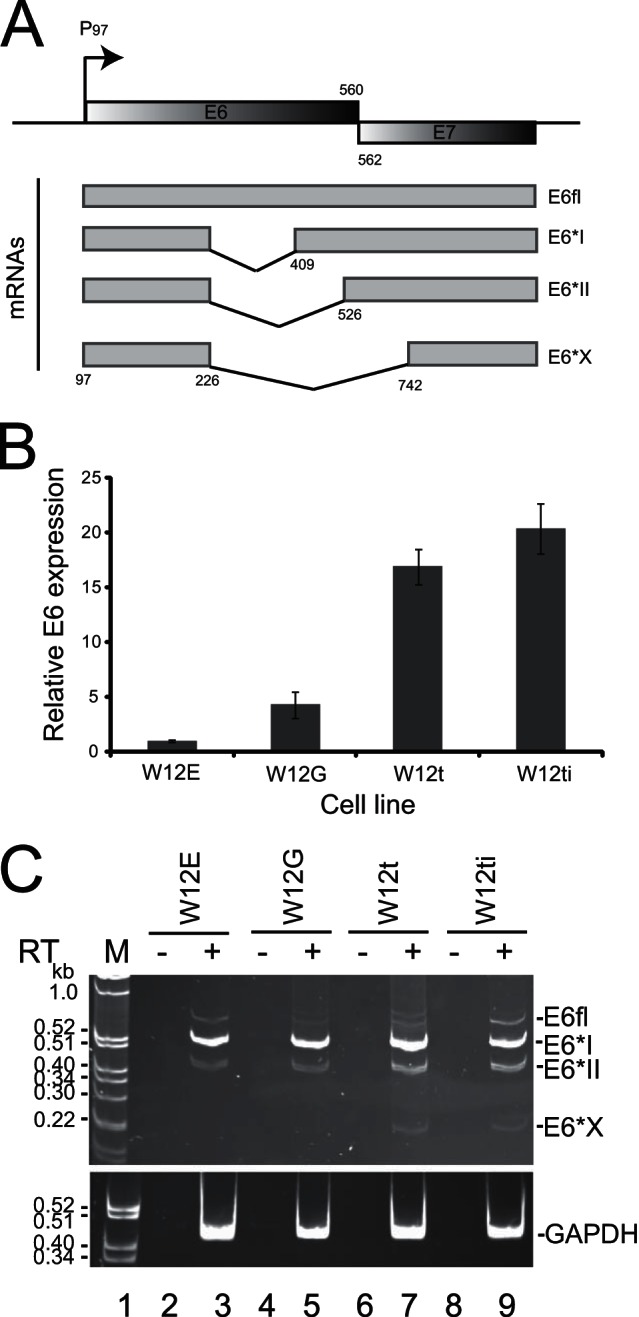
E6 splice isoform expression in epithelial cell transformation. (A) Diagram of the E6E7 region of the HPV16 genome. Shaded gray boxes, open reading frames; arrow, P97, HPV16 early promoter. The numbers (nucleotides) refer to the end of the E6 (560) and the beginning of the E7 (562) open reading frames. The four E6E7 mRNAs are shown below the genomic map. Numbers mark the nucleotide positions of one splice donor (226) and three possible splice acceptor sites (409, 526, 742). Gray boxes, exons; black lines, introns. (B) RT-qPCR quantification of the expression levels of total E6E7 RNA relative to levels of GAPDH in the four W12 cervical epithelial cell lines (W12E, W12G, W12t, W12ti). The graph shows the means and standard errors of the means from three separate experiments. (C) Ethidium bromide-stained polyacrylamide gel electrophoresis of RT-PCR products amplified from polyadenylated RNA isolated from the above-described W12 cell lines using primers that amplify all E6 isoforms. The various isoforms are indicated to the right of the gel (E6fl, E6*I, E6*II, E6*X). A second gel identical to that for fractionating GAPDH control PCR products from the same RNA/cDNA preparations was electrophoresed at the same time and is shown below. M, marker; RT, reverse transcriptase; −, RT-PCR in the absence of reverse transcriptase; +, RT-PCR in the presence of reverse transcriptase.
It proved very difficult to design appropriate cross-splice junction primers and probes with similar efficiencies by RT-qPCR for each of the E6 RNA isoforms. So a semiquantitative PCR strategy was designed to amplify simultaneously all E6-encoding mRNA isoforms. Two minor E6 isoforms, E6^E4 and E6^L1, were not included in the analysis, because neither is expressed in W12G, W12t, or W12ti cells that contain integrated HPV16 genomes. Lanes 3 and 5 in Fig. 1C show the result of amplification of E6E7 RNA isoforms from the nontumor W12E and W12G cell lines. Bands corresponding in size to E6fl, E6*I, and E6*II were detected. These three bands were also detected in the W12t and W12ti tumor cells. However, a band corresponding in size to E6*X was also detected (lanes 7 and 9). No products were amplified in the absence of reverse transcriptase. PCR products were sequenced to verify coding potential. The band above E6fl in lane 7 may represent the use of cryptic splice sites within the E6 gene region. These data suggest that all four E6 isoforms can be simultaneously expressed in HPV-positive cells.
E6 expression is controlled by SRSF2.
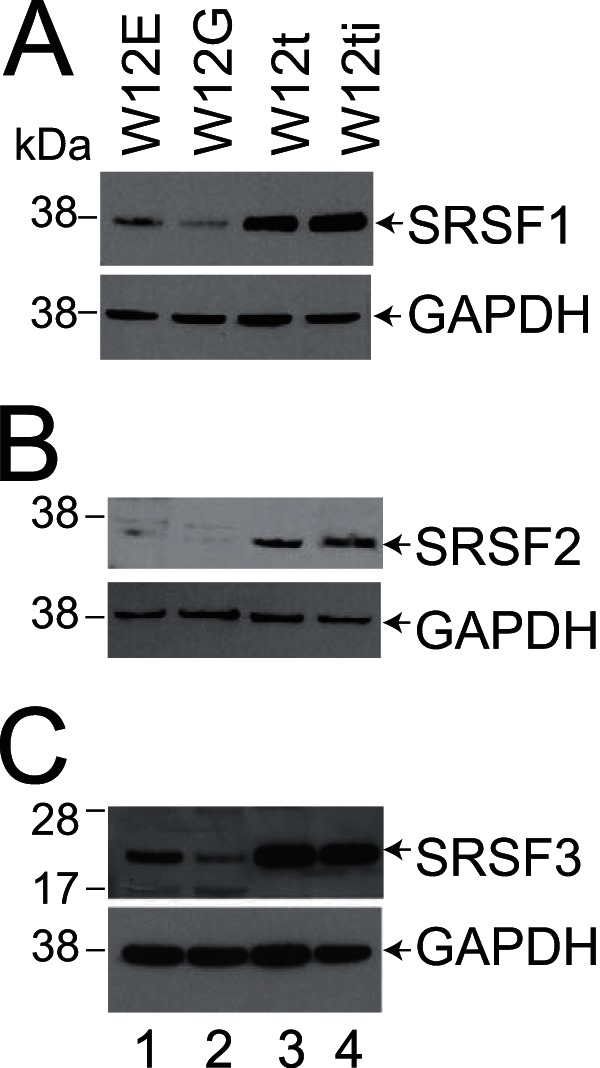
Previously, we demonstrated that the three smallest SR proteins, SRSF1, SRSF2, and SRSF3, are overexpressed during cervical tumor progression (24), suggesting that they may possess cervical tumor-promoting activities. Figure 2 shows that these three proteins are overexpressed in W12t and W12ti tumor cells (lanes 3 and 4), compared to a much lower expression level in the W12E and W12G nontumor cells (lanes 1 and 2). Increased SR protein levels could contribute to the increased levels of E6E7 RNAs in the tumor cells (Fig. 1B). To determine whether HPV16 oncoprotein expression was controlled by SRSF1, -2, or -3, expression of each SR protein was individually knocked down using commercially available siRNA pools in W12ti tumor cells. Levels of siRNA depletion of greater than 80% were achieved in every experiment (Fig. 3A). Knockdown of SRSF1 (Fig. 3B, lane 6) had little effect on E6E7 RNA isoform levels. SRSF3 depletion resulted in some reduction in the levels of all of the E6 RNA isoforms, as observed in another study (33) (Fig. 3B, lane 10). However, SRSF2 knockdown resulted in a significant reduction in the levels of all E6 RNA isoforms (Fig. 3B, lane 12). In case this was due to the siRNA pool used, the experiment was repeated using a single siRNA known to effectively deplete SRSF2 (37). Very similar results were obtained with a significant reduction in the levels of E6 RNAs (data not shown). The E6*X band is present but at low levels in control lanes 4 and 8. Its apparent absence in lanes 2, 6, and 10 could be due to the small amounts of E6E7 RNAs in these PCRs. RT-qPCR was used to quantify total E6 RNA levels in W12ti cells following SRSF depletion. Figure 3C shows that SRSF1 depletion caused around a 20% reduction in total E6 RNA levels. SRSF3 depletion reduced RNA levels by around 60%, while SRSF2 depletion reduced E6 RNA levels by over 90% compared to control transfected cells.
FIG 2.
Levels of SRSF proteins 1, 2, and 3 are higher in the W12 tumor lines (W12t and W12ti) than in the nontumor lines (W12E and W12G). (A) Western blot showing SRSF1 levels in the four W12 lines; (B) Western blot showing SRSF2 levels in the four W12 lines; (C) Western blot showing SRSF3 levels in the four W12 lines. GAPDH was used as a loading control. Lane 1, W12E (nontumor cell line) protein extracts; lane 2, W12G (nontumor cell line) protein extracts; lane 3, W12t (tumor cell line) protein extracts; lane 4, W12ti (tumor cell line) protein extracts.
FIG 3.

The effect of SRSF depletion on E6E7 RNA expression in cervical tumor and nontumor cells. (A) SRSF protein levels before and after depletion by specific siRNA pools in the W12ti cells; (B) ethidium bromide-stained polyacrylamide gel electrophoresis of RT-PCR products amplified from polyadenylated RNA isolated from W12ti cells using primers that amplify all E6 isoforms. The effect of depletion of SRSF1 (lanes 5 and 6), SRSF3 (lanes 9 and 10), and SRSF2 (lanes 11 and 12) for 48 h on E6 isoform expression in W12ti cervical tumor cells is shown. The E6 RNA isoforms are indicated to the right of the gels. A second gel identical to that for fractionating GAPDH control PCR products from the same RNA/cDNA preparations was electrophoresed at the same time and is shown below each gel. RT, reverse transcriptase; −, RT-PCR in the absence of reverse transcriptase; +, RT-PCR in the presence of reverse transcriptase; no siRNA, cells transfected without siRNA; cntrl, cells transfected with a control siRNA, siGLO; SRSF, cells treated with SRSF siRNA for 48 h. The experiments were repeated at least three times, and similar data were obtained in each experiment. (C) qPCR quantification of total E6 RNA levels following SRSF depletion. The graph shows the means and standard deviations from the means from three separate experiments.
In case the effect of the knockdown of SRSF2 on E6 RNA levels was an artifact of the cell line used, experiments were repeated in SiHa (data not shown) and CaSki cells (Fig. 4B). SRSF2 depletion also reduced total E6 RNA levels in these cell lines. RT-qPCR analysis of total E6 levels in CaSki cells revealed around a 60% reduction following SRSF2 depletion (Fig. 4C). This demonstrates that SRSF2 control of E6 isoform expression is not restricted to the W12ti cell line. SRSF2 depletion in nontumor W12G cells (the parental line for W12ti), where it is not overexpressed (Fig. 2A and 3E), had no detectable effect on E6 RNA isoform expression (Fig. 4D), but RT-qPCR indicated a small increase rather than a decrease in total E6 RNA levels. These data suggest that overexpression of SRSF2 may be required to maintain the high levels of viral oncoprotein RNA expression seen in the cervical tumor cells.
FIG 4.

The effect of SRSF depletion on E6E7 RNA expression in CaSki cervical tumor and W12G nontumor cells. (A) SRSF protein levels before and after depletion by specific siRNA pools. The antibody used (MAb104) also detects other SR proteins, and an adjacent band corresponding to SFSR5 is shown as an internal control for protein levels and to show specificity of SRSF2 depletion. As expected, the starting levels of SRSF2 in W12G cells are much lower than in CaSki cells or W12ti cells than in the internal SRSF5 control band. (B and D) Ethidium bromide-stained polyacrylamide gel electrophoresis of RT-PCR products amplified from polyadenylated RNA isolated from cell lines using primers that amplify all E6 isoforms. The E6 RNA isoforms are indicated to the right of the gels. A second gel identical to that for fractionating GAPDH control PCR products from the same RNA/cDNA preparations was electrophoresed at the same time and is shown below each gel. RT, reverse transcriptase; −, RT-PCR in the absence of reverse transcriptase; +, RT-PCR in the presence of reverse transcriptase; no siRNA, cells transfected without siRNA; cntrl, cells transfected with a control siRNA, siGLO; SRSF, cells treated with SRSF siRNA for 48 h. The experiments were repeated at least three times, and similar data were obtained in each experiment. (B) The effect of SRSF2 depletion of E6 isoform expression in CaSki cervical tumor cells. (C) qPCR quantification of total E6 RNA levels in CaSki cells before and after SRSF2 depletion. The graph shows the means and standard deviations from the means from three separate experiments. (D) The effect of SRSF2 depletion on E6 isoform expression in W12G nontumor cervical cells. (E) qPCR quantification of total E6 levels in W12G cells before and after SRSF2 depletion. The graph shows the means and standard deviations from the means from three separate experiments.
SRSF2 knockdown results in increased p53 and induction of apoptosis.
E6 degrades the tumor suppressor p53 (47). Because it is difficult to detect the E6 oncoprotein using currently available antibodies, we assessed changes in p53 protein levels upon SRSF2 depletion as a readout for E6 protein levels. Levels of p53 increased significantly (P = 0.021) in W12ti cells upon SRSF2 depletion, suggesting that SRSF2 is required for E6 oncoprotein expression (Fig. 5A). E6 mRNA isoform E6*I encodes the E7 oncoprotein (6). Reflecting the loss of the RNA isoform E6*I (Fig. 3B), E7 protein levels also decreased in cells treated with siRNA against SRSF2 (Fig. 5B). However, no corresponding increase in pRb was detected (Fig. 5B).
FIG 5.

W12ti tumor cells exhibit decreased viral oncoprotein expression upon SRSF2 knockdown. (A) Western blot analysis of p53 protein levels with or without SRSF2 knockdown in W12ti cells. GAPDH is used as a loading control. SFSR2 depletion is shown with SRSF5 as the internal loading control. no siRNA, cells transfected without siRNA; cntrl, cells transfected with a control siRNA; SRSF2, cells transfected with SRSF2 siRNA for 48 h. Below the blots is a quantification of levels of p53 in untreated cells (no siRNA) or treated with the indicated siRNA. The data show the means and standard deviations from the means from three separate experiments. The asterisk indicates a significant change in protein expression (P ≤ 0.05) using a Student t test. (B) Western blot analysis of E7 and pRb protein levels with or without SRSF2 knockdown in W12ti cells. GAPDH is shown as the loading control. The same protein extracts as in panel A were used in this experiment, so the same level of SRSF2 depletion was achieved. no siRNA, cells transfected without siRNA; cntrl, cells transfected with a control siRNA; SRSF2, cells transfected with SRSF2 siRNA for 48 h. Below the blots is a quantification of levels of E7 in cells not treated (no siRNA) or treated with the indicated siRNAs. The data show the means and standard deviations from the means from three separate experiments. The asterisk indicates a significant change in protein expression (P ≤ 0.05) using a Student t test.
The loss of E6 expression in the tumor cells should stabilize p53, leading to apoptosis (4). Annexin V assays were carried out to determine if SRSF2 depletion caused cells to enter apoptosis (Fig. 6A). Cells were treated with control siRNA or siRNAs against SRSF2 (Fig. 6C) or siRNA against E6 itself and then stained with fluorescent annexin V and propidium iodide, followed by flow cytometry analysis. Proliferating and senescing cells do not take up either stain and are represented in the lower left-hand quadrant of the two-dimensional plots in Fig. 6A. Early apoptotic cells are present in the lower right-hand quadrant, because they display annexin staining but are impermeable to propidium iodide. Cells in the upper right-hand quadrant are stained with both annexin V and propidium iodide and are late apoptotic or dead cells. Material in the upper left-hand quadrant is likely dead cells/cell debris. As a positive control for apoptosis, cells were treated with 500 J/m2 UV radiation type B (UVB) 24 h prior to harvesting. After this treatment, the majority of cells were found in the early (48.8%) or late (43.4%) apoptotic quadrants (Fig. 6A, UVB). Treating W12ti cells with a nontargeting siRNA (siCntrl) resulted in some apoptosis, likely due to the toxic effects of the siRNA transfection reagent (Fig. 6A, siCntrl). Depletion of E6 (and therefore derepression of p53) with a specific E6 siRNA caused the majority of cells to enter apoptosis (31.5% in early apoptosis and 58.6% in late apoptosis), as expected (Fig. 6A, siE6). Following siRNA depletion of SRSF2, the majority of cells also entered the apoptotic quadrants (58.1% in early apoptosis, 34.3% in late apoptosis) (Fig. 6A, siSRSF2). Table 1 summarizes the data from three separate experiments. Figure 6B shows the means and standard deviations from the means of the percentages of live cells and apoptotic cells from three separate experiments. It is clear that depleting the tumor cells of SRSF2 mimicked the effect of direct E6 depletion by driving cells into apoptosis.
FIG 6.

W12ti tumor cells undergo apoptosis upon SRSF2 knockdown. (A) FACS plots of annexin V-propidium iodide staining of W12ti cells transfected with the indicated siRNAs above the plots or irradiated with UVB as a positive control for cell death. The percentages of cells occupying each quadrant in one experiment are shown. The lower right-hand quadrant contains early apoptotic cells, while the upper right-hand quadrant contains late apoptotic/dead cells. W12ti cells were treated with control siRNA (siCntrl) or siRNA pool against SRSF2 (siSRSF2) or siRNA against HPV16 E6 (siE6) or treated with UVB irradiation (UVB) for 24 h as a positive indicator of apoptosis. Very similar results were obtained in three separate experiments (see Table 1). (B) Graph of the percentage of W12ti cells from three independent experiments occupying the live and early apoptotic quadrants of the annexin V and propidium iodide staining plots shown in panel A. (C) Western blot showing the levels of SRSF2 in mock-transfected cells (no siRNA) or cells transfected with control siRNA (cntrl) or transfected with siRNA against SRSF2 (SRSF2). SRSF5 is shown as an internal loading control.
TABLE 1.
Flow cytometry analysis of apoptosis of W12ti cells upon depletion of SRSF2
| Treatment | % of cells positive for annexin V-propidium iodide ± SDa |
||
|---|---|---|---|
| Live compartment | Early apoptosis | Late apoptosis | |
| Control siRNA | 31.2 ± 5.8 | 20.2 ± 5.1 | 20.4 ± 2.1 |
| SRSF2 siRNA | 8.0 ± 2.7 | 55.1 ± 5.8 | 36.4 ± 2.1 |
| E6 siRNA | 7.0 ± 2.3 | 28.1 ± 3.5 | 49.7 ± 8.9 |
| UVB | 6.1 ± 4.4 | 45.0 ± 4.3 | 44.3 ± 5.7 |
Percentage of cells positive for annexin V-propidium iodide upon treatment with control siRNA, siRNA against SRSF2 or E6, or UVB (500 J/m2 24 h prior to harvesting). The numbers are the means and standard deviations from the mean from three separate experiments.
SRSF2 depletion causes a reduction in tumor cell proliferation and anchorage-independent growth.
If SRSF2 is required to maintain high expression levels of the HPV16 oncoprotein and its depletion causes apoptosis, SRSF2 depletion could be expected to cause a reduction in cell proliferation. Cell proliferation was thus measured by counting live cells (trypan blue exclusion) over a 72-h time course in W12G (HPV-positive nontumor), W12ti (HPV-positive tumor), and C33a (HPV-negative tumor) cells after transfection with siRNA against SRSF2. Following SRSF2 depletion, the proliferation rates of the W12ti and C33a tumor cells decreased by over 50% (Fig. 7A, Table 2), while there was no significant effect on W12G cell growth (Fig. 7B, Table 2). However, flow cytometry analysis indicated that SRFS2 knockdown did not significantly inhibit cell cycle progression. Only a small increase in G1 phase cells, with a corresponding decrease in G2 phase cells, was noted (Table 3).
FIG 7.
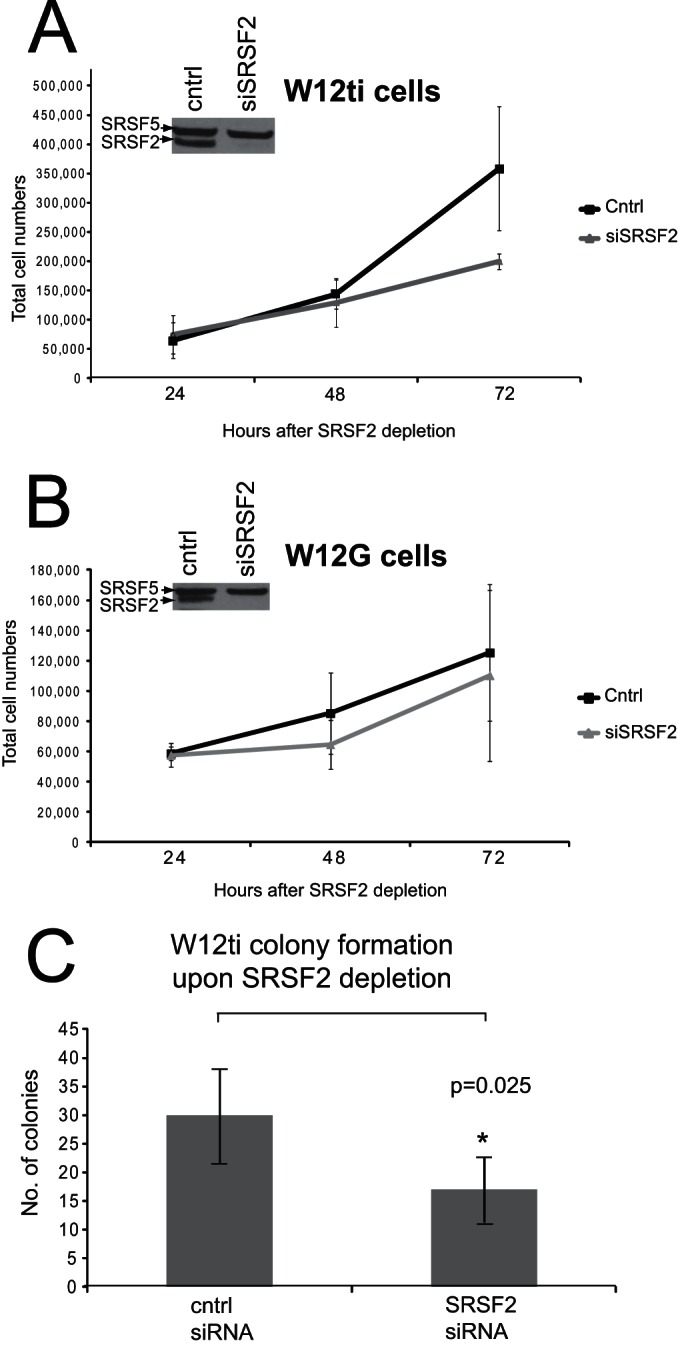
The effect of SRSF2 depletion on W12 cell proliferation and colony formation. (A) Proliferation rates over 72 h of W12ti tumor cells treated with control siRNA (cntrl) or treated with an siRNA pool against SRSF2 (siSRSF2). (B) W12G nontumor cells treated with control siRNA (cntrl) or treated with an siRNA pool against SRSF2 (siSRSF2). The inset Western blots in panels A and B show levels of SRSF2 depletion and SRSF5 levels as an internal loading control. (C) Colony formation assay of W12ti cells with (SRSF2 siRNA) or without (cntrl siRNA) SRSF2 knockdown. The graph shows means and standard deviations from the means of numbers of colonies from three independent experiments. Asterisk indicates a significant change in colony formation (P ≤ 0.05) using a Student t test.
TABLE 2.
Actual numbers of W12G, W12ti, and C33a cells at 24 and 72 h posttransfection with control siRNA or with siRNA against SRSF2
| Hours posttransfection | No. of cells ± SDa |
|||||
|---|---|---|---|---|---|---|
| W12G |
W12ti |
C33a |
||||
| Control | siSRSF2 | Control | siSRSF2 | Control | siSRSF2 | |
| 24 | 5.87 × 104 ± 0.29 × 104 | 5.77 × 104 ± 0.62 × 104 | 6.47 × 104 ± 0.37 × 104 | 7.5 × 104 ± 0.2 × 104 | 1.76 × 105 ± 0.09 × 105 | 1.73 × 105 ± 0.1 × 105 |
| 72 | 1.25 × 105 ± 0.37 × 105 | 1.1 × 105 ± 0.46 × 105 | 3.58 × 105 ± 0.86 × 105 | 1.3 × 105 ± 0.09 × 105 | 2.81 × 106 ± 0.2 × 106 | 9.57 × 105 ± 0.14 × 105 |
W12G, HPV-positive nontumor cells; W12ti, HPV-positive tumor cells; C33a, HPV-negative tumor cells; control, control transfection with siGLO; siSRSF2, transfection with siRNA against SRSF2.
TABLE 3.
Flow cytometry
| Stage in cell cycle | % of W12ti cells ± SDa |
||
|---|---|---|---|
| Mock | Control siRNA | siSRSF2 | |
| Avg G1 | 65.5 ± 3.2 | 66.1 ± 2.2 | 71.9 ± 6.0 |
| Avg S | 10.4 ± 0.8 | 12.9 ± 2.9 | 13.8 ± 5.3 |
| Avg G2 | 19.9 ± 3.1 | 15.4 ± 1.8 | 12.8 ± 3.2 |
The mean percentage (and standard deviation from the mean) of W12ti cells in each stage of the cell cycle in three independent experiments. Mock, cells with no siRNA; control siRNA, cells transfected with control siGLO RNA; siSRSF2, cells transfected with an siRNA pool against SRSF2.
Anchorage-independent growth is characteristic of tumor cells and can be measured by observing colony formation of the tumor cells in soft agar. To determine whether SRSF2 overexpression is required to maintain anchorage-independent growth, W12ti tumor cells were treated with control siRNA or siRNA against SRSF2 and then grown in soft agar, and the number and sizes of the colonies were determined. SRSF2 depletion resulted in a significant reduction (P = 0.025) in the ability of the W12ti tumor cells to form colonies (Fig. 7C). These data implicate SRSF2 in the maintenance of the W12ti tumor phenotype.
SRSF2 knockdown does not affect transcription of the E6E7 gene region but is required to stabilize E6E7 oncoprotein RNAs.
SRSF2 can regulate transcription elongation directly (48), but it can also regulate expression of key transcription factors such as SP1, which transactivates the HPV16 P97 promoter (33). Therefore, we tested the hypothesis that the depletion of E6E7 RNAs upon SRSF2 knockdown in W12ti cells was a result of SRSF2-mediated downregulation of E6E7 transcription from the viral P97 promoter. 293T cells were transfected with either control siRNA or siRNA against SRSF2 together with a luciferase reporter construct under the control of the HPV16 long control region (LCR) (genome nt 7101 [−803] to +137; this region contains the P97 promoter and a transcription initiation site) (39). The HPV16 LCR was less transcriptionally active than the control SV40 promoter in the pGL3 promoter plasmid. However, there was no detectable effect of SRSF2 depletion on luciferase production from the test HPV16 P97 promoter, indicating neither transcription initiation nor elongation was attenuated (Fig. 8A).
FIG 8.

SRSF2 does not regulate HPV16 P97 promoter activity but may stabilize E6E7 RNAs. (A) Graph of luciferase activity in 293T cells transiently transfected (48 h) with either a luciferase reporter plasmid containing the control SV40 promoter (pGL3-promoter; Promega) or the HPV16 long control region encompassing the P97 promoter (HPV16 LCR) (39). Cells transfected with the HPV16 LCR construct were also transfected with either control siRNA (cntrl siRNA) or siRNA against SRSF2 (siSRSF2). The means and standard deviations from the means from three separate experiments are shown. RT-PCR analysis of E6E7 RNAs in actinomycin D-treated W12ti cells 24 h following transfection with siRNA against SRSF2 (B) or control siRNA (cntrl siRNA) (C). The number of hours of actinomycin D treatment is shown above the gels. GAPDH RNA levels are stable over this time period and are shown as a loading control.
An alternative explanation for the reduction in E6 mRNA production in the absence of SRSF2 could be that SRSF2 is required to protect E6 RNA against decay. HPV16 oncogene expression in cervical tumor cells is enhanced by increased stability of the E6E7 mRNAs (49). Moreover, SRSF2 can stabilize the mRNA encoding the microtubule protein Tau by binding to an SRSF2 binding site in exon 10 (50). In order to test whether SRSF2 regulates E6E7 RNA stability, the protein was siRNA depleted in W12ti cells, followed by treatment 24 h posttransfection with 10 μg/ml actinomycin D, which blocks de novo RNA synthesis, allowing a time course analysis of the decay of steady-state levels of RNA already present in the cell. E6 RNA isoform levels were detected by semiquantitative PCR (in order to detect all isoforms simultaneously, which is not possible with qPCR) over a 4-h time course of drug treatment in W12ti cells depleted of SRSF2. Although starting levels of E6 mRNA isoforms were very low when levels of SRSF2 were reduced, as expected (35 PCR cycles were performed for Fig. 8B, compared to 30 cycles for Fig. 8C), the data in Fig. 8B reveal that in the absence of SRSF2, E6E7 transcript stability is significantly reduced between 1 and 4 h after the addition of actinomycin D compared to that of E6 RNAs in cells with undepleted levels of SRSF2 (Fig. 8C). All E6 RNA isoforms were equally depleted over the 4-h period (Fig. 8B). E6*X was not detected at this level of amplification in the absence of SRSF2. GAPDH levels were not significantly reduced upon actinomycin D treatment (Fig. 8C) or upon depletion of SRSF2 (Fig. 8B). Taken together, these observations suggest that in cervical tumor cells, SRSF2 enhances E6E7 RNA stability.
DISCUSSION
SR proteins control viral and cellular splicing as well as other RNA processing events, such as nuclear export, RNA stability, and translation (13). Moreover, SRSF1, -3, and -9 are overexpressed in a number of cancers and possess oncogenic properties (22, 26, 31). Previously, we reported increased levels of the three smallest SR proteins, SRSFs 1 to 3, in our W12 model of cervical tumor progression and in patient samples (24). Another study has revealed that SRSF3 can control E6E7 expression in cervical cancer cells and in undifferentiated keratinocytes (33) and has oncogenic properties in HPV18-positive HeLa cells (22). Here, we investigated if SRSFs 1, 2, or 3 regulated the expression of viral oncoprotein RNA isoforms in HPV16-positive W12 cervical cancer cells. Because these proteins can control constitutive and alternative splicing, a change in the splice isoform production was expected. However, the depletion of none of these factors altered the relative levels of oncoprotein RNA isoforms. SRSF1 knockdown had only a small effect on E6E7 RNA levels. SRSF3 depletion caused a 60% reduction in E6E7 RNA levels, as observed previously (33). However, SRSF2 depletion resulted in very significantly reduced levels of all E6 mRNA isoforms, suggesting that although SRSF3 can control viral oncoprotein expression, possibly by stimulating production of transcription factors that can transactivate the HPV16 P97 promoter (33), SRSF2 makes a greater contribution and is absolutely required for viral oncoprotein expression.
E6E7 RNA levels were also reduced in CaSki cells when SRSF2 was depleted. Although the reduction was less than that in W12ti cells, the large number of HPV16 integrant genomes in CaSki cells (600 copies) may result in high E6E7 RNA expression levels that are not able to be as fully repressed as in W12ti cells. Furthermore, SRSF2 depletion in W12G cells, where there is much reduced SRSF2 expression compared to that in W12ti cells (24) (Fig. 2), did not result in any reduction in E6 RNA levels. Indeed, a small increase (0.5-fold) was observed, perhaps due to SRSF1 regulation of the transcription factors that control the P97 viral promoter or to mRNA decay proteins. These data suggest that SRSF2 control of E6/E7 RNAs is tumor cell specific and may be due to the high levels of the splicing factor that are present in cervical cancer cells.
SRSF2 depletion resulted in decreased levels of E6E7 RNA but also gave a functionally significant E6 oncoprotein-associated effect: p53 levels increased after SRSF2 knockdown, indicating reduced levels of E6 protein. As expected in the presence of increased p53, cells treated with SRSF2 siRNA displayed significantly increased levels of apoptotic cells compared to mock-transfected cells. Knockdown of SRSF2 has been shown to arrest the cell cycle at the G2/M checkpoint in non-virally infected cells (15). Our data did not indicate significant cell cycle arrest even in the face of an SRSF2 depletion-mediated decreased in E7 levels (Table 3). A reduction in E7 expression would normally result in increased levels of the cell cycle checkpoint protein pRb, leading to G1 cell cycle arrest. Surprisingly, levels of pRb were not increased after SRSF2 depletion (Fig. 4B). This can be explained if SRSF2 controls pRb expression directly. SRSF2 siRNA-mediated reduction in pRb levels would stimulate cell cycle progression and could ameliorate the effects of increased p53 caused by decreased E6 levels. This may explain why SRSF2 depletion resulted in fewer late apoptotic/dead cells than siRNA depletion of E6 itself (Fig. 6A). Alternatively, if, like other SR proteins, SRSF2 can exert oncogenic effects by altering the levels of tumor-promoting alternatively spliced isoforms of cellular mRNAs, then SRSF2 depletion could potentially result in changes in other cellular proteins that could also antagonize the apoptotic effect of reduced levels of the viral E6 oncoprotein.
As well as its roles in splicing regulation, SRSF2 can control transcription elongation either directly by affecting recruitment of the RNA polymerase II C-terminal domain Ser-2 kinase complex, P-TEFb (48), or indirectly by regulating expression of transcription factors, for example, SP1. However, as seen here, SRSF2 depletion did not significantly alter transcription of a luciferase reporter gene under the control of the viral P97 promoter that regulates expression of E6 and E7. SR proteins can control other steps in the life cycle of an mRNA, and SRSF2 is known to regulate RNA stability. This can be accomplished directly by SRSF2 binding to an SRSF2 cognate binding site, for example, in exon 10 of the mRNA encoding the microtubule protein Tau (50), or indirectly through recruitment of RNA surveillance pathways, for example, to SRSF2-regulated alternatively spliced isoforms of its own RNAs, to degrade the transcripts (51). Our results suggest that SRSF2 is involved in processing of the viral oncoprotein RNAs at some stage in the RNA biogenesis pathway, because loss of the protein caused a non-transcriptionally regulated disappearance of the RNAs. The most likely mechanism is that SRSF2 is required for E6E7 RNA stability. The E6E7 bicistronic mRNA is unusual in that it does not possess a 5′ untranslated region and there are several splice acceptor sites close to stop codons, meaning that it should normally be targeted for nonsense-mediated decay (NMD) (52). SR proteins can regulate targeting of RNAs to the NMD pathway (53). Therefore, SRSF2 may recruit factors to E6E7 RNAs in the nucleus to protect from premature stop codon-mediated decay in the cytoplasm. Alternatively, during cervical tumor progression, when the HPV genome is inserted into the host genome, 3′ processing of the oncoprotein RNAs no longer occurs at the viral polyadenylation site but rather at some genomic polyadenylation site downstream from the point of genome integration. It is possible that SRSF2 could direct alternative splicing of the chimeric viral/genomic mRNAs by selecting a downstream exon or 3′ untranslated region (54) that would confer stability on these E6E7 mRNAs as previously noted (49). SRSF2 could also regulate E6E7 RNAs indirectly by controlling expression of an intermediary factor that controls E6E7 RNA stability. Indeed, the lower numbers of cells that entered late apoptosis upon treatment with SRSF2 siRNA than upon treatment with E6 siRNA might suggest a temporal difference in response of E6E7 expression to depletion of SRSF2 due to an intermediary regulatory event. However, the full mechanism of SRSF2-mediated stabilization of E6 RNA isoforms requires further study.
Our data demonstrate that SRSF2 has oncogenic properties in HPV16-positive cervical cancer cells. When the splicing factor was depleted in W12ti cells, there was around a 50% reduction in anchorage-independent growth and an initiation of apoptosis. While high levels of SRSF2 in the tumor cells are clearly required for viral oncoprotein expression, we cannot discount the possibility that the increased levels of endogenous SRSF2 seen in cervical tumor progression (24) could change the profile of cellular mRNA alternative splicing, leading to production of oncogenic or antiapoptotic protein isoforms, as has been demonstrated for SRSF1 (28). It is highly likely that SRSF2 plays a role in cervical tumor progression by acting directly on E6E7 mRNA expression, but our data, showing that SRSF2 depletion reduces the proliferation of HPV-negative C33a cells, underlining the fact that SRSF2 depletion also impacts cellular gene expression.
This is the first report, to our knowledge that SRSF2 is an essential cellular regulator of HPV oncogenic expression. This suggests it could be a valid target for therapeutic inhibition in cervical tumors. SR protein function is regulated by phosphorylation, and drugs that regulate SR kinases have been used successfully to inhibit the replication of HIV and hepatitis C virus (34). Development of small-molecule inhibitors against SR kinases could have the potential to abrogate HPV oncoprotein expression in the early stages of cervical cancer progression.
ACKNOWLEDGMENTS
We thank Margaret Stanley (University of Cambridge) for the original W12 cell line, Paul Lambert and Denis Lee (University of Wisconsin) for the W12E and G clones, and Trond Aasen, Mike Edward, and Malcolm Hodgins (University of Glasgow) for collaborations on developing and characterizing the W12t and W12ti cells lines. Iain Morgan (Virginia Commonwealth University) and Ed Dornan (University of Glasgow) kindly supplied the HPV16 LCR plasmid, and the E2 expression plasmid was obtained from Peter Howley (Harvard Medical School). We are grateful to Diane Vaughan for help with flow cytometry and fluorescence-activated cell sorting (FACS) analysis.
M.M. was funded by an MRC Doctoral Training Award. The study was also funded by grant number 08-0159 from the Association for International Cancer Research and Wellcome Trust grant number Wtd004098 to S.V.G.
REFERENCES
- 1.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Goon PKC, Stanley MA, Ebmeyer J, Steinsträsser L, Upile T, Jerjes W, Bernal-Sprekelsen M, Görner M, Sudhoff HH. 2009. HPV & head and neck cancer: a descriptive update. Head Neck Oncol 1:36. doi: 10.1186/1758-3284-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moody C, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 4.McLaughlin-Drubin ME, Munger K. 2009. Oncogenic activities of human papillomaviruses. Virus Res 143:195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shally M, Alloul N, Jackman A, Muller M, Gissmann L, Sherman L. 1996. The E6 variant proteins E6I-E6IV of human papillomavirus 16: expression in cell free systems and bacteria and study of their interaction with p53. Virus Res 42:81–96. doi: 10.1016/0168-1702(96)01301-9. [DOI] [PubMed] [Google Scholar]
- 6.Tang S, Tao M, McCoy JP, Zheng Z-M. 2006. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol 80:4249–4263. doi: 10.1128/JVI.80.9.4249-4263.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sherman L, Alloul N, Golan I, Dürst M, Baram A. 1992. Expression and splicing patterns of human papillomavirus type 16 mRNAs in precancerous lesions and carcinoma of the cervix, in human keratinocytes immortalized by HPV 16 and in cell lines established from cervical cancers. Int J Cancer 50:356–364. doi: 10.1002/ijc.2910500305. [DOI] [PubMed] [Google Scholar]
- 8.Hummel M, Hudson JB, Laimins LA. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol 66:6070–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozbun MA, Meyers C. 1997. Characterisation of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J Virol 71:5161–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milligan SG, Veerapraditsin T, Ahamat B, Mole S, Graham SV. 2007. Analysis of novel human papillomavirus type 16 late mRNAs in differentiated W12 cervical epithelial cells. Virology 360:172–181. doi: 10.1016/j.virol.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cricca M, Venturoli S, Leo E, Costa S, Musiani M, Zerbini M. 2009. Molecular analysis of HPV 16 E6I/E6II spliced mRNAs and correlation with the viral physical state and the grade of the cervical lesion. J Med Virol 81:1276–1282. doi: 10.1002/jmv.21496. [DOI] [PubMed] [Google Scholar]
- 12.Schmitt M, Dalstein V, Waterboer T, Clavel C, Gissmann L, Pawlita M. 2011. The HPV16 transcriptome in cervical lesions of different grades. Mol Cell Probes 25:260–265. doi: 10.1016/j.mcp.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Long JC, Caceres JF. 2009. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 14.Singh RK, Cooper TA. 2012. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18:472–482. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Wang J, Manley JL. 2005. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev 19:2705–2714. doi: 10.1101/gad.1359305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Manley JL. 2005. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122:365–378. doi: 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. 2007. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell 26:867–881. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 18.Das S, Krainer AR. 2014. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res 12:1195–1204. doi: 10.1158/1541-7786.MCR-14-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loomis RJ, Naoe Y, Parker JB, Savic V, Bozovsky MR, Macfarlan T, Manley JL, Chakravarti D. 2009. Chromatin binding of SRp20 and ASF/SF2 and dissociation from mitotic chromosomes is modulated by histone H3 serine 10 phosphorylation. Mol Cell 33:450–461. doi: 10.1016/j.molcel.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fregoso O, Das S, Akerman M, Krainer A. 2013. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol Cell 50:56–66. doi: 10.1016/j.molcel.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen-Eliav M, Golan-gerstl R, Siegfried Z, Andersen CL, Thorsen K, Ømtoft TF, Mu D, Karni R. 2012. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol 229:630–639. doi: 10.1002/path.4129. [DOI] [PubMed] [Google Scholar]
- 22.Jia R, Li C, McCoy JP, Deng CX, Zheng Z. 2010. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci 6:806–826. doi: 10.7150/ijbs.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stickeler E, Kittrell F, Medina D, Berget S. 1999. Stage-specific changes in SR splicing factors and alternative splicing in mammary tumorigenesis. Oncogene 18:3574–3582. doi: 10.1038/sj.onc.1202671. [DOI] [PubMed] [Google Scholar]
- 24.Mole S, McFarlane M, Chuen-Im T, Milligan SG, Millan D, Graham SV. 2009. RNA splicing factors regulated by HPV16 during cervical tumour progression. J Pathol 219:383–391. doi: 10.1002/path.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fay J, Kelehan P, Lambkin H, Schwartz S. 2009. Increased expression of cellular RNA-binding proteins in HPV-induced neoplasia and cervical cancer. J Med Virol 81:897–907. doi: 10.1002/jmv.21406. [DOI] [PubMed] [Google Scholar]
- 26.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. 2007. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14:185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karni R, Hippo Y, Lowe SW, Krainer AR. 2008. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A 105:15323–15327. doi: 10.1073/pnas.0801376105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R. 2012. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 19:220–229. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das S, Anczuków O, Akerman M, Krainer AR. 2012. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 1:110–117. doi: 10.1016/j.celrep.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He X, Ee PL, Coon JS, Beck WT. 2004. Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin Cancer Res 10:4652–4660. doi: 10.1158/1078-0432.CCR-03-0439. [DOI] [PubMed] [Google Scholar]
- 31.Fu Y, Huang B, Shi Z, Han J, Wang Y, Huangfu J, Wang W. 2013. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing β-catenin biosynthesis. EMBO Mol Med 5:737–750. doi: 10.1002/emmm.201202218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johansson C, Schwartz S. 2013. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat Rev Micro 11:239–251. doi: 10.1038/nrmicro2984. [DOI] [PubMed] [Google Scholar]
- 33.Jia R, Liu X, Tao M, Kruhlak M, Guo M, Meyers C, Baker CC, Zheng ZM. 2009. Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J Virol 83:167–180. doi: 10.1128/JVI.01719-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez-Lopez HR, Graham SV. 2012. Alternative splicing in tumour viruses: a therapeutic target? Biochem J 445:145–156. doi: 10.1042/BJ20120413. [DOI] [PubMed] [Google Scholar]
- 35.Jeon S, Allen-Hoffman BL, Lambert PF. 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol 69:2989–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aasen T, Hodgins MB, Edward M, Graham SV. 2003. The relationship between connexins, gap junctions, tissue architecture and tumour invasion, as studied in a novel in vitro model of HPV-16-associated cervical cancer progression. Oncogene 22:7969–7980. doi: 10.1038/sj.onc.1206709. [DOI] [PubMed] [Google Scholar]
- 37.Gabut M, Mine M, Marsac C, Brivet M, Tazi J, Soret J. 2005. The SR protein SC35 is responsible for aberrant splicing of the E1α pyruvate dehydrogenase mRNA in a case of mental retardation with lactic acidosis. Mol Cell Biol 25:3286–3294. doi: 10.1128/MCB.25.8.3286-3294.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacDonald AI, Sun P, Hernandez Lopez H, Aasen T, Hodgins MB, Edward M, Roberts S, Massimi P, Thomas M, Banks L, Graham SV. 2012. A functional interaction between the MAGUK protein hDlg and the gap junction protein connexin 43 in cervical tumour cells. Biochem J 446:9–21. doi: 10.1042/BJ20111144. [DOI] [PubMed] [Google Scholar]
- 39.Taylor ER, Dornan ES, Boner W, Connolly JA, McNair S, Kannouches P, Lehmann AR, Morgan IM. 2003. The fidelity of HPV16 E1/E2-mediated DNA replication. J Biol Chem 278:52223–52230. doi: 10.1074/jbc.M308779200. [DOI] [PubMed] [Google Scholar]
- 40.Schwarz E, Freese U, Gissman L, Mayer W, Roggenbuck B, Stremlau A, zur Hausen H. 1985. Structure and transcription of human papillomavirus sequences in cervical carinoma cells. Nature 314:111–114. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- 41.Shirasawa H, Tanzawa H, Matsunaga T, Simizu B. 1991. Quantitative detection of spliced E6-E7 transcripts of human papillomavirus type 16 in cervical premalignant lesions. Virology 184:795–798. doi: 10.1016/0042-6822(91)90455-K. [DOI] [PubMed] [Google Scholar]
- 42.McNicol P, Guijon F, Wayne S, Hidajat R, Paraskevas M. 1995. Expression of human papillomavirus type 16 E6-E7 open reading frame varies quantitatively in biopsy tissue from different grades of cervical intraepithelial neoplasia. J Clin Microbiol 33:1169–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitt M, Dalstein V, Waterboer T, Clavel Gissman CL, Pawlita M. 2010. Diagnosing cervical cancer and high grade precursors by HPV16 transcription patterns. Cancer Res 70:249–256. doi: 10.1158/0008-5472.CAN-09-2514. [DOI] [PubMed] [Google Scholar]
- 44.Schmitt M, Pawlita M. 2011. The HPV transcriptome in HPV16 positive cell lines. Mol Cell Probes 25:108–113. doi: 10.1016/j.mcp.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Stanley MA, Browne HM, Appelby M, Minson AC. 1989. Properties of a nontumourigenic human cervical keratinocyte cell line. Int J Cancer 43:672–676. doi: 10.1002/ijc.2910430422. [DOI] [PubMed] [Google Scholar]
- 46.Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley MA, Crawford L. 1990. Detection of novel splicing patterns in a HPV-16-containing keratinocyte cell line. Virology 178:254–262. doi: 10.1016/0042-6822(90)90401-C. [DOI] [PubMed] [Google Scholar]
- 47.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 48.Lin A, Coutinho-Mansfield G, Wang D, Pandit S, Fu X-D. 2008. The splicing factor SC35 has an active role in transcription elongation. Nat Struct Mol Biol 15:819–826. doi: 10.1038/nsmb.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeon S, Lambert PF. 1995. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A 92:1654–1658. doi: 10.1073/pnas.92.5.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qian W, Iqbal K, Grundke-Iqbal I, Gong C-X, Liu F. 2011. Splicing factor SC35 promotes Tau expression through stabilisation of its mRNA. FEBS Lett 585:875–880. doi: 10.1016/j.febslet.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J. 2001. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20:1785–1796. doi: 10.1093/emboj/20.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kervestin S, Jacobson A. 2012. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Z, Krainer AR. 2004. Involvement of SR proteins in mRNA surveillance. Mol Cell 16:597–607. doi: 10.1016/j.molcel.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 54.Smits H, Cornelissen M, Jebbink M, van den Tweel J, Struyk A, Briët M, ter Schegget J. 1991. Human papillomavirus type 16 transcripts expressed from viral-cellular junctions and full-length viral copies in CaSki cells and in a cervical carcinoma. Virology 182:870–873. doi: 10.1016/0042-6822(91)90632-L. [DOI] [PubMed] [Google Scholar]