

ABSTRACT
Japanese encephalitis virus (JEV), which causes viral encephalitis in humans, is a serious risk to global public health. The JEV envelope protein mediates the viral entry pathway, including receptor-binding and low-pH-triggered membrane fusion. Utilizing mutagenesis of a JEV infectious cDNA clone, mutations were introduced into the potential receptor-binding motif or into residues critical for membrane fusion in the envelope protein to systematically investigate the JEV entry mechanism. We conducted experiments evaluating infectious particle, recombinant viral particle, and virus-like particle production and found that most mutations impaired virus production. Subcellular fractionation confirmed that five mutations—in I0, ij, BC, and FG and the R9A substitution—impaired virus assembly, and the assembled virus particles of another five mutations—in kl and the E373A, F407A, L221S, and W217A substitutions—were not released into the secretory pathway. Next, we examined the entry activity of six mutations yielding infectious virus. The results showed N154 and the DE loop are not the only or major receptor-binding motifs for JEV entry into BHK-21 cells; four residues, H144, H319, T410, and Q258, participating in the domain I (DI)-DIII interaction or zippering reaction are important to maintain the efficiency of viral membrane fusion. By continuous passaging of mutants, adaptive mutations from negatively charged amino acids to positively charged or neutral amino acids, such as E138K and D389G, were selected and could restore the viral entry activity.
IMPORTANCE Recently, there has been much interest in the entry mechanism of flaviviruses into host cells, including the viral entry pathway and membrane fusion mechanism. Our study provides strong evidence for the critical role of several residues in the envelope protein in the assembly, release, and entry of JEV, which also contributes to our understanding of the flaviviral entry mechanism. Furthermore, we demonstrate that the H144A, H319A, T410A, and Q258A mutants exhibit attenuated fusion competence, which may be used to develop novel vaccine candidates for flaviviruses.
INTRODUCTION
Japanese encephalitis virus (JEV) is a mosquito-borne zoonotic flavivirus that causes viral encephalitis in most of Asia, Papua New Guinea, and the Torres Strait of northern Australia (1, 2). The recent emergence of JEV in the Torres Strait islands and its spread onto the Cape York Peninsula pose a serious risk to public health in Australia and have elicited growing concern that this virus can spread throughout the world (3). JEV is one of the most important members of the JEV serological complex, which includes West Nile virus (WNV), St. Louis encephalitis virus (SLEV), and Murray Valley encephalitis virus (MVEV), causing approximately 67,900 cases of encephalitis annually in countries of Japanese encephalitis (JE) endemicity and having high morbidity and mortality rates (4, 5). The case fatality rate for JE is 20% to 30%, and 30% to 50% of survivors have severe neurological sequelae even years later (5, 6).
Similar to other members belonging to the family Flaviviridae, including dengue virus (DENV), yellow fever virus (YFV), and tick-borne encephalitis virus (TBEV), JEV is an enveloped, plus-sense, single-stranded RNA virus approximately 50 nm in diameter, containing an approximately 11-kb genome. The single open reading frame (ORF) in the genome encodes a single large polyprotein, which is cleaved by viral and host proteases into three structural proteins, capsid (C), precursor to membrane (prM), and envelope (E), and seven nonstructural (NS) proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 (7).
The entry of JEV into host cells involves two steps: receptor binding and low pH-triggered fusion of the viral and cellular membrane; both of these steps are mediated by the E protein (8–15). High-resolution structures of the E protein ectodomain of JEV reveal that it shares not only a high percentage of conserved amino acids but also a similar conformation with other flaviviruses (Fig. 1A and E) (16). The E protein is an ∼53-kDa elongated protein composed of predominantly β-strands and assembles as an antiparallel dimer in the crystal lattice; 90 E dimers are arranged on mature virions. The E protein has three distinct domains (Fig. 1A). The central domain I (DI), composed of a nine-strand mixed β-barrel and modified by a single asparagine-linked (N-linked) carbohydrate (N154), connects the extended domain II (DII) and the globular domain III (DIII) via short flexible loops. DII is composed of two extended loops protruding from DI and contains a highly conserved fusion loop at its tip. DIII is an immunoglobulin-like structure that is thought to interact with cellular receptors in the genus Flavivirus (17), and two loops of DIII, the DE and FG loops, exposed on the viral surface have been thought to be the receptor-binding motif. Loop 3 peptides (the DE loop) can prevent JEV infection by interfering with virus attachment to BHK-21 cells (18), and a peptide containing the E FG loop of DENV2 inhibited the binding of DIII to C6/36 cells (19). Furthermore, three mutants in the E FG loop (D390G, D390A, and D390H) of MVEV have different entry kinetics from those of the parent virus and increased dependence on glycosaminoglycans (GAGs) for attachment to different mammalian cells (20). Our recent study demonstrated that a peptide (P3) binding to the N terminus of E DIII near the BC and DE loops inhibits viral infection by blocking JEV attachment to host cells (21).
FIG 1.

Several amino acids in the E protein potentially involved in viral entry (Table 1). (A) Top view of the dimeric prefusion E protein ectodomain conformation of strain AT31, a homology model of the crystal structure of Protein Data Bank identification number (PDB ID) 3P54. In the ribbon diagram of the bottom E protein monomer, DI, DII, DIII, and the fusion peptide loop are shown in red, yellow, blue, and orange, respectively, and stick representations of four amino acids, R9, H144, H319, and E373, participating in the DI-DIII interaction are shown in magenta. In the ribbon diagram of the top E monomer, shown in gray, several amino acids in the receptor-binding motif or critical for membrane fusion are indicated by colored spheres. Residue 154 lacks a carbohydrate modification because the E ectodomain was purified from bacterial inclusion bodies in a previous study (16). (B) Side view of trimeric postfusion E protein ectodomain conformation, a homology model of the PDB ID 4FG0 docked H1 helix by Zdock server. The H1 helix is shown in blue. Only one E monomer is colored, and the others are shown in gray. Stick representations of five amino acids, W217, L221, Q258, F407, and T410, participating in the three-helix interaction of aA, aB, and H1 are shown in magenta. (C and D) Enlargement of the DI-DIII interaction and zippering reaction, respectively. (E) Structural comparison of the prefusion E conformation of JEV (PDB ID 3P54) and TBEV (PDB ID 1SVB). The three domains and fusion peptide of the E monomer of JEV are colored as described in panel A, and the E monomer of TBEV is shown in gray. The comparisons of a selected region or amino acids for mutagenesis are shown close to the E monomer. Structural comparisons of the prefusion E protein of JEV with other flaviviruses, WNV (PDB ID 2I69) and DENV (PDB ID 1OKE), are not shown.
Previous studies have shown that glycosaminoglycans (GAGs) can facilitate JEV attachment to mammalian cells, which has been clearly demonstrated in other flaviviruses (22, 23). A GAG-binding region (JEV E residues 279 to 297, I0 β-strand) (Fig. 1A) rich in positively charged residues and conserved among DENV serotypes and JEV serological complex has been identified with high affinity in a heparin-Sepharose column (22). This region has also been presumed to be involved in GAG binding in a crystal structure report of the DENV E protein (24). In addition, other cellular proteins, such as dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) (25), heat shock protein 70 (26), and vimentin (27), have been shown to participate in JEV infection. However, how the E protein participates in the interaction with these receptors is still poorly studied, and a more specific protein receptor is thought to be required.
After JEV is internalized into host cells through receptor-mediated endocytosis, the low pH within endosomes induces conformational changes in the E protein to mediate the fusion of the viral and endosomal membranes. A low-pH-dependent fusion model for flaviviral E protein has been well established according to the pre- and postfusion E protein ectodomain crystal structures (17, 24, 28–30). In the prefusion conformation, the E protein forms a horizontal, antiparallel homodimer with the fusion peptide at the tip of DII buried at the dimer interface. A hydrophobic pocket at the interface of DI and DII can be opened by a shift of the kl loop, allowing DII to hinge away from its dimer partner and expose the fusion peptide to interact with the target membrane (Fig. 1A) (29). Mutations around this pocket alter the pH threshold for fusion in flaviviruses (31–35). In the postfusion conformation, the E protein forms a vertical, parallel trimer with the fusion peptide exposed at its end via individual domain rearrangement: DIIIs run along the sides of the trimer toward the tip; the stem (H1 and H2) is embedded in a groove formed by neighboring DIIs and reaches the membrane (Fig. 1B) (28, 30). However, more experimental testing by structure-based mutagenesis may be necessary to analyze the critical amino acids in these two conformations.
In this study, we utilize the mutagenesis of an infectious cDNA clone to introduce mutations into the receptor-binding motif or in amino acids critical for membrane fusion. First, we find that most mutations impaired viral formation: five mutations—in I0, ij, BC, and FG and the R9A substitution—impair virus assembly, and five other mutations—in kl and the E373A, F407A, L221S, and W217A substitutions—impair virus release. Then, virus binding and entry assays show that N154 and the DE loop are not the only or major receptor-binding motifs involved in JEV entry into BHK-21 cells. Furthermore, we reveal that four mutations, H144A, H319A, T410A, and Q258A, result in significantly decreased entry activity, which may be due to impaired specific intramolecular interactions during the transition of the E protein from dimer to trimer. Finally, continuous passaging of the mutants shows that two mutations, E138K and D389G, repeatedly occur in several mutants and could restore the viral entry activity.
MATERIALS AND METHODS
Cells.
Baby hamster kidney cells (BHK-21) and human neuroblastoma cells (SH-SY5Y) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) in 5% CO2 at 37°C. Aedes albopictus C6/36 cells were propagated in RPMI 1640 medium supplemented with 10% FBS at 28°C in a closed culture system.
Infectious clones, replicons, and CprME constructs.
An infectious cDNA clone of JEV (pMWJEAT; strain AT31), kindly provided by T. Wakita, Tokyo Metropolitan Institute for Neuroscience, was used to engineer E protein mutations. A subclone, pGEM-E, containing the ApaI-BspEI fragment of pMWJEAT (nucleotides [nt] 114 to 3450 of the viral genome), was used to engineer mutations using a Fast Mutagenesis System (Transgen), as shown in Table 1, and the mutated DNA fragment was cloned back into pMWJEAT with ApaI and BspEI. The primers used for mutagenesis are listed in Table S1 in the supplemental material.
TABLE 1.
Mutations introduced into the JEV E protein
| Mutanta | Position(s) and regionb | Substitution(s)c | Potential function |
|---|---|---|---|
| I0 mutant | 284, 286, 288, 290, 293, 297 in DI | H → A, K → A, R → A, K → A, K → A, K → A | GAG binding motif |
| N154A | 154 in DI | N → A | DC-SIGN binding motif |
| BC mutant | 329–333 in DIII | SGSDG → AAAAA | Receptor binding motif |
| DE mutant | 363–367 in DIII | TSSAN → AAAAA | |
| FG mutant | 386–390 in DIII | GRGDK → AAAAA | |
| kl mutant | 270–272, 278, 280 in DII | IVV → SSS, V → S, L → S | Switch of hydrophobic pocket |
| ij mutant | 246, 249 in DII | H → A, K → A | Interaction with negatively charged amino acids in prM |
| R9A | 9 in DI | R → A | DI-DIII interaction |
| E373A | 373 in DIII | E → A | |
| H144A | 144 in DI | H → A | |
| H319A | 319 in DIII | H → A | |
| F407A | 407 in the stem | F → A | Zippering of the stem along DII |
| T410A | 410 in the stem | T → A | |
| L221S | 221 in DII | L → S | |
| W217A | 217 in DII | W → A | |
| Q258A | 258 in DII | Q → A |
Multiple amino acid mutations are named according to their located β-strand or loop structure nomenclature used for the class II proteins.
Amino acid position.
Substitutions are given in respective order to positions.
For recombinant viral particle (RVP) packaging, a complete CprME-coding fragment with a stop codon at the 3′ end was amplified from pMWJEAT and cloned into pCAGGS; the resulting plasmid was used to engineer E protein mutations using a Fast Mutagenesis System. All constructs were verified by DNA sequencing. A JEV luciferase-reporting replicon, SA14/U14163-Replicon, was constructed previously (36).
In vitro transcription, RNA transfection, and immunofluorescence assay (IFA).
The infectious clone plasmids of JEV were linearized with KpnI, and replicon cDNA plasmids were linearized with XhoI. The purified linearized cDNAs were then subjected to in vitro transcription using a T7 mMessage mMachine kit (Ambion). In vitro-transcribed genomic RNA was electroporated into BHK-21 cells in a 0.4-cm electrophoresis cuvette at 850 V and 25 μF with three pulses at 3-s intervals or transfected into BHK-21 cells using DMRIE-C (1,2-dimyristyloxypropyl-3-dimethyl-hydroxy ethyl ammonium bromide and cholesterol) reagent (Invitrogen). After the transfection of genome-length RNA, culture supernatants containing the viruses were collected every 24 h until day 5 and stored at −80°C in aliquots.
For IFA, the RNA-transfected cells were fixed in cold 5% acetic acid in methanol for 10 min at room temperature at 48 h posttransfection. The fixed cells were then incubated with mouse anti-E monoclonal antibody (MAB8744; Chemicon) recognizing the E protein conformation and rabbit prM antiserum (full-length prM proteins were expressed in Escherichia coli BL21 cells using pET30a expression vectors, and purified proteins were injected into rabbits to prepare polyclonal antibodies). The related animal experimentation was approved by the Institutional Review Board of Wuhan Institute of Virology (permit number WIVH25201201). The cells were washed and incubated with goat anti-mouse IgG conjugated with Texas-Red and goat anti-rabbit IgG conjugated with fluorescein isothiocyanate (FITC). Following washing and nuclear staining with 4′,6′-diamidino-2-phenylindole (DAPI), the cells were analyzed under a fluorescence microscope.
Virus production from infectious clones.
The culture supernatants from transfected cells were quantified by plaque assays at each time point posttransfection. Briefly, BHK-21 cells in 24-well plates were infected with a dilution series of viruses. The cells were kept in DMEM containing 2% FBS and 1% methylcellulose at 37°C. After 3 days of incubation, the cells were fixed with 3.7% formaldehyde and stained with 1% crystal violet.
The number of genome-containing particles (GCPs) was determined by quantitative PCR (qPCR). After the cell debris was removed by low-speed centrifugation, the viral RNA was extracted from the culture supernatants at each time point posttransfection using TRIzol reagent (Invitrogen). RNA samples were amplified with RNA-Direct Real-Time PCR Master Mix (Toyobo) according to the manufacturer's instructions. Following amplification, DNA melting curve analysis was performed to confirm the specificity of the PCR products. The number of genomic RNA copies was determined with a standard curve of the infectious clone plasmids. Primers for the qPCR targeting the NS5 gene were 5′-AGCTTCTAGATGGTGAACACCGCA-3′ and 5′-TCACGTCCATCACGGTCTTTCCTT-3′ (product length, 117 bp).
Production of RVPs.
To produce RVPs, BHK-21 cells were electroporated with in vitro-transcribed replicon RNA as described above. After incubation at 37°C for 24 h, the electroporated cells were transfected with wild-type (WT) or mutant pCAGGS-CprME using Lipofectamine 2000 reagent (Life Technologies). Culture supernatants collected at 48 h posttransfection were centrifuged to remove cellular debris, and 150 μl of supernatants containing RVPs was prepared for qPCR to determine the GCP numbers of the RVPs. Five-hundred microliters of the supernatants was mixed with equal volumes of 16% polyethylene glycol 8000 (PEG 8000)–1 M NaCl and precipitated at 4°C overnight. The RVPs were pelleted by centrifugation at 12,000 × g for 15 min at 4°C and resuspended in 50 μl of phosphate-buffered saline (PBS). Packaging cells were processed according to the instructions in the Renilla luciferase assay system at 24 and 48 h posttransfection and were also lysed using cell lysis buffer for Western blotting (Beyotime) at 48 h posttransfection. For Western blot analysis, RVP pellets and cell lysates were added to SDS-PAGE buffer, subjected to 12% SDS-PAGE, and transferred to polyvinylidene difluoride (PVDF) membranes. After a blocking step, the membranes were incubated with the appropriate dilutions of the primary antibodies overnight at 4°C: mouse or rabbit E DIII antiserum (purified E DIII proteins were injected into mouse or rabbit to prepare polyclonal antibodies), rabbit prM antiserum, rabbit NS5 antiserum (provided by C. J. Chen, Taichung Veterans General Hospital), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mouse monoclonal antibody (CW0100; Cwbiotech). The membrane was then washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. After the final wash, the signals were detected using enhanced chemiluminescence reagents (Promega). For the RVP infection assay, BHK-21 and SH-SY5Y cells in a 48-well plate were infected with equal volumes of culture supernatants containing WT or mutant RVPs. At 24 h postinfection, the infected cells were collected and processed according to the instructions in the Renilla luciferase assay system.
Subcellular fractionation.
Subcellular fractionation was performed as described previously (37, 38). BHK-21 cells electroporated with WT or mutant transcribed genomic RNA were trypsinized and washed in PBS at 48 h posttransfection. The cell pellets were resuspended in 1 ml of hypo-osmotic buffer (10 mM HEPES-NaOH, pH 7.8). The cells were allowed to swell on ice for 10 min and were then pelleted by centrifugation at 800 × g at 4°C for 2 min. The medium was returned to iso-osmoticity by the removal of 650 μl of the supernatant and the addition of 350 μl of hyperosmotic buffer (0.6 M sucrose, 10 mM HEPES-NaOH, pH 7.8). The cells were then disrupted by passage 30 times through a 25-gauge needle, and nuclei were removed by centrifugation for 30 min at 13,000 × g at 4°C. The supernatant was mixed with 700 μl of OptiPrep (60% iodixanol; D1556; Sigma) to generate a solution containing 30% iodixanol. Solutions containing 20% and 10% iodixanol were generated by mixing with the hypo-osmotic buffer. A total of 1,400 μl of these three solutions was layered in centrifuge tubes, which were centrifuged at 350,000 × g in an SW55 Ti rotor for 3 h at 4°C. Ten fractions of 420 μl were collected from the top and analyzed by Western blotting, qPCR, and plaque assay. Antibodies against calnexin (ab22595; Abcam), an endoplasmic reticulum (ER)-resident protein, were used as a marker for Western blotting.
VLPs, coimmunoprecipitation and endo-H digestion.
The plasmid pCAGC105E, containing the sequence of JEV prM/E (strain AT31), was kindly provided by Y. Matsuura, Osaka University, and was used for the production of virus-like particles (VLPs). The plasmid was mutagenized to engineer E protein mutations using a Fast Mutagenesis System, and all constructs were verified by DNA sequencing. To produce the VLPs, BHK-21 cells plated in a 24-well plate were transfected with pCAGC105E using Lipofectamine 2000, and the cells were incubated in the presence or absence of 20 mM NH4Cl at 37°C. Western blot analysis was performed at 48 h posttransfection as described above for the RVPs. For coimmunoprecipitation, the transfected BHK-21 cells were lysed using cell lysis buffer for Western blotting and immunoprecipitation (Beyotime) in the presence of 1 mM phenylmethylsulfonyl fluoride (PMSF) at 48 h posttransfection. Two-hundred microliters of cell lysates was incubated with 2 μl of mouse E DIII antiserum overnight at 4°C under constant rocking. After the addition of 20 μl of protein A+G agarose (P2012; Beyotime), the cell lysate-antiserum mixture was incubated for 3 h at 4°C under constant rocking. The beads were pelleted by centrifugation for 1 min at 1,000 × g and washed five times with PBS. The beads were resuspended in SDS-PAGE buffer and analyzed by Western blotting, and rabbit E DIII antiserum was used to avoid the interference of the antibody heavy chain. For endo-β-N-acetylglucosaminidase H (endo-H) (P0702S; New England BioLabs) digestion, E protein in the cell lysates was digested with endo-H according to the manufacturer's instructions. Undigested and digested samples were subjected to 10% SDS-PAGE and then analyzed by Western blotting with mouse E DIII antiserum as described above.
Virus binding and entry assay.
BHK-21 cells were plated in a 24-well plate and incubated with mutant viruses in triplicate at 10,000 GCPs per cell for 1 h at 4°C. For the virus binding assay, unbound viruses were removed by three rinses with cold PBS, and total RNA was extracted from cells using TRIzol reagent. qPCR was performed to quantify cell-associated viral RNA, and GAPDH was used as an internal control. The primers for GAPDH were as follows: 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-TGTAGACCATGTAGTTGAGGTCA-3′. For the virus entry assay, unbound viruses were removed by three rinses with cold PBS, and cells were kept in DMEM supplemented with 2% FBS in the presence or absence of 5 nM bafilomycin A1 or 50 μM chloroquine for 1 h at 37°C. After 1 h of incubation, the cells were trypsinized for 5 min at 37°C to remove the bound virus and then replated into a 24-well plate. qPCR was performed to quantify the cell-associated viral RNA at 16 h postinfection.
C6/36 cell-cell fusion-from-within assay.
The C6/36 cell-cell fusion assay of mutants was performed as previously described (35, 39). Briefly, C6/36 cells plated in a 24-well plate were infected with WT and mutant viruses at a multiplicity of infection (MOI) of 1 and then maintained in pH 7.7 culture medium at 28°C for 4 days. Fusion was triggered at room temperature by the application of serum-free medium buffered with morpholineethanesulfonic acid (MES) and acetic acid to a final pH 5.5 for 2 h. The cells were fixed in 4% paraformaldehyde for 10 min at room temperature, and the nuclei were stained with DAPI for 10 min. Numbers of the total nuclei and the nuclei of the syncytia in a microscopic field were counted (at least five microscopic fields per well) for calculation of the percent fusion (percent fusion = the number of nuclei of the syncytia/the total number of nuclei).
Continuous passaging of mutants and adaptive mutations.
The supernatants collected on day 4 posttransfection (passage 0 [P0]) of each mutant were continuously passaged in BHK-21 cells (3 to 4 days per passage) for five rounds to determine the stability of the engineered mutations or to recover the adaptive viruses. BHK-21 cells were infected with each mutant at an MOI of 1 in each round of passage. Viral RNA extracted from all of the recovered viruses was subjected to reverse transcription-PCR (RT-PCR) using Moloney murine leukemia virus (M-MLV) reverse transcriptase (28025; Invitrogen) and sequencing of the complete CprME gene to examine the adaptive mutations.
RESULTS
Structure-based prediction of several amino acids in the E protein potentially involved in viral entry.
We selected residues that were located on the different receptor-binding motifs for mutagenesis: (i) positively charged amino acids in the GAG-binding region in the I0 β-strand (H284/K286/R288/K290/K293/K297), (ii) a glycosylated residue (N154) that might interact with DC-SIGN, and (iii) residues located on the three loops of E DIII (BC, 329S/330G/331S/332D/333G; DE, 363T/364S/365S/366A/367N; FG, 386G/387R/388G/389D/390K) that were considered the receptor-binding motifs. Structure comparison (Fig. 1E) and sequence alignment (Fig. 2A) of the E proteins of various flaviviruses revealed that all of these residues were exposed on the surface of E proteins, but only the GAG-binding motif was conserved in other flaviviruses. Residues that were located on the three loops of E DIII were not conserved, which might be the reason why the E proteins of flaviviruses interacted with different receptors and usually resulted in different viral host range and tissue tropisms. Also we selected three types of residues that were critical for membrane fusion for mutagenesis: (i) hydrophobic residues located on the kl loop (270I/271V/272V/278V/280L) that might act as a switch in the dissociation of the E dimer, (ii) conserved residues participating in the DI-DIII interaction in the prefusion conformation (R9, H144, H319, and E373) (Fig. 1C), and (iii) conserved residues potentially participating in the zippering reaction of the stem along DII in the postfusion conformation (L221, W217, Q258, F407, and T410) (Fig. 1D). All of these residues were mutated as shown in Table 1. Multiple amino acid mutations are named according to the nomenclature used for the class II proteins, and the nomenclature for the secondary structure of the E protein was available from sequence alignments (Fig. 2A).
FIG 2.

The selected region or amino acids for mutagenesis in the JEV E protein. (A) Alignment of E protein sequences of Japanese encephalitis virus (JE), West Nile virus (WN), dengue virus 2 (DEN2), and tick-borne encephalitis virus (TBE). Conserved (.), more conserved (:), and most conserved (*) amino acids are shown above the alignment. The three domains and fusion peptide are indicated below the sequences and shown in red, yellow, blue, and orange, respectively; the DI/DIII and DIII/stem linkers are shown in magenta, and the stem and transmembrane region (H1, H2, TM1, and TM2) are shown in gray. The β-strands and a-helices are indicated with arrows and helices below the sequences, respectively, and labeled according to the nomenclature used for class II proteins. Mutations introduced into the E protein are boxed in black. (B) Schematic representation of a subclone construct used to introduce mutations into the E protein. The sequence of a putative E. coli promoter is shown at left (40).
Introducing mutations into the putative receptor-binding motif or in amino acids critical for membrane fusion impairs virus production.
To analyze the role of each amino acid in the viral life cycle, the selected residues were mutagenized in an infectious full-length JEV cDNA clone (pMWJEAT). We initially intended to construct a subclone, pGEM-E, containing the E sequence with SalI and BspEI restriction sites and then transfer the E mutations into pMWJEAT after site-directed mutagenesis. Unfortunately, we were not able to obtain the subclone due to the known toxicity of flavivirus cDNA to E. coli. To avoid a putative E. coli promoter, we substituted the ApaI restriction site for SalI and obtained a subclone, pGEM-CprME, successfully (Fig. 2B) (40).
After the transfection of BHK-21 cells with equal amounts of in vitro-transcribed RNA, the numbers of IFA-positive cells were compared between WT and the mutants at 48 h posttransfection. Three of the mutant viruses (H319A, T410A, and Q258A) had WT-like levels of virus spread (100% IFA-positive cells), whereas viral spread was obviously reduced for the N154A, DE, and H144A mutants and not observed for the rest of the mutants (I0, ij, kl, BC, FG, R9A, E373A, F407A, L221S, and W217A mutants) (Fig. 3A). To determine whether the different levels of viral spread were a result of the reduced production of infectious virus particles, the culture supernatants from transfected cells were quantified by plaque assay at each time point posttransfection. Consistent with the results of the IFA, the DE, N154A, H144A, H319A, T410A, and Q258A mutants yielded infectious viruses although the viral titers were lower than the WT titer at each time point, whereas the I0, ij, kl, BC, FG, R9A, E373A, F407A, L221S and W217A mutants did not yield any detectable viruses (Fig. 3B). Sequencing of the complete CprME gene of the WT and DE, N154A, H144A, H319A, T410A, and Q258A mutant viruses (collected on day 5 posttransfection), confirmed that only the engineered mutations were retained. Because all of the mutations target amino acids potentially involved in entry, they might yield viral particles deficient in either receptor binding or membrane fusion; thus, we detected the number of GCPs in the culture supernatants by qPCR. As shown in Fig. 3C, the GCP numbers of the DE, N154A, H144A, H319A, T410A, and Q258A mutants exhibited slightly slower growth kinetics than the that of the WT, but the mutations unable to yield infectious virus maintained a low GCP number in the supernatants at each time point posttransfection, indicating that these mutations did not yield entry-deficient virus particles. Next, the specific infectivities of WT and DE, N154A, H144A, H319A, T410A, and Q258A mutants were determined on the basis of the number of infectious viral particles per GCP. We observed that the range of the specific infectivities were as follows: WT, 20 to 1,000 (GCPs/PFU); DE mutant, 200 to 2,400; N154A, 130 to 1,900; H144A, 3,000 to 18,000; H319A, 750 to 3,600; T410A, 400 to 1,700; and Q258A, 700 to 4,900 (Fig. 3D). It was obvious that the viral particles of H144A, H319A, and Q258A were less infectious. Overall, the results indicated that most of the mutations impaired virus production except in the DE, N154A, H144A, H319A, T410A, and Q258A mutants.
FIG 3.

Infectious virus production in BHK-21 cells. In vitro-transcribed genomic RNA of the WT and each mutant was transfected into BHK-21 cells. (A) The RNA-transfected cells were analyzed by IFA at 48 h posttransfection, and mouse anti-E monoclonal antibody and rabbit prM antiserum were used as primary antibodies. The viral titer and GCP number of the supernatants collected at each time point posttransfection were quantified by plaque assay (B) and qPCR (C), respectively. (D) The specific infectivities of the WT and the mutants that produced infectious particles were calculated by the ratio of GCPs to infectious particles (GCPs/PFU). Results from one representative experiment out of three independent experiments are shown.
Because the lack of infectious particles in the supernatants might reflect a defect in viral assembly or the release of viral particles, we utilized a packaging system for RVPs in which a JEV luciferase-reporting replicon, SA14/U14163-Replicon, was supplemented with WT or mutant JEV structural proteins (CprME). BHK-21 cells were electroporated with in vitro-transcribed SA14/U14163-Replicon RNAs and then transfected with pCAGGS-CprME at 24 h postelectroporation. RVPs with a natural virus-like envelope and containing a replicon RNA were harvested at 48 h posttransfection. A Renilla luciferase reporter encoded by the replicon RNA permits the easy quantification of JEV expression in the transfected cells as well as the productive entry of the RVPs into new cells (38). Similar levels of luciferase activity were detected in the WT and mutation-expressing packaging cells at 24 h posttransfection, whereas higher levels of luciferase activity were detected in the WT and the DE, N154A, H144A, H319A, T410A, and Q258A mutation-expressing cells at 48 h posttransfection because the packaging cells were infected with the secreted RVPs, suggesting that none of the mutations affected the replication efficiency of the replicon RNA (Fig. 4A). Western blot analysis showed that most mutations were associated with a significant decrease of RVP release into the culture supernatants, consistent with the decrease in viral particles released in the electroporation experiments (Fig. 3 and 4B). Additionally, a qPCR assay was carried out to quantify the GCP number of the RVPs in the culture supernatants. As shown in Fig. 4C, the numbers of GCPs in most mutants were drastically reduced. To further test the effect of these mutations on the infectivity of the RVPs, we infected BHK-21 and SH-SY5Y cells with RVPs and measured the Renilla luciferase activity at 24 h postinfection. The lost luciferase signal of the I0, ij, kl, BC, FG, R9A, E373A, F407A, L221S, and W217A mutants could be explained by a significantly reduced yield of RVPs (data not shown). The infectivity of the WT and the DE, N154A, H144A, H319A, T410A, and Q258A mutants was determined by the Renilla luciferase activity per GCP. As shown in Fig. 4D, the H144A, H319A, T410A, and Q258A mutants, with similar number s of WT RVPs, had decreased entry activity, indicating the importance of these residues to virus entry into host cells. In contrast, the N154A and DE mutants maintained similar or even better entry activity than WT RVPs; thus, these residues may be not essential to viral entry (Fig. 4D). These data suggest that the mutations caused a major defect in the assembly or entry of viral particles but not in the replicon RNA replication.
FIG 4.

RVP production in BHK-21 cells. In vitro-transcribed replicon RNA was electroporated into BHK-21 cells, and the WT and each mutant pCAGGS-CprME construct were transfected at 24 h postelectroporation. (A) The luciferase activities of the packaging cells were determined at 24 and 48 h posttransfection. (B) The packaging cell lysates and the RVP pellets precipitated from cell culture supernatants were analyzed by Western blotting at 48 h posttransfection, and mouse E DIII antiserum, rabbit prM antiserum, rabbit NS5 antiserum, and anti-GAPDH mouse monoclonal antibody were used as primary antibodies. (C) The GCP numbers of RVPs contained in the supernatants collected at 48 h posttransfection were quantified by qPCR. (D) BHK-21 and SH-SY5Y cells were infected with equal volumes of supernatants, and the luciferase activities of the infected cells were determined at 24 h postinfection. The specific infectivities of the WT and the mutants were calculated by the Renilla luciferase activity per GCP. Asterisks denote a statistically significant reduction in specific infectivities in BHK-21 or SH-SY5Y cells compared to WT values (*, P < 0.05; **, P < 0.01). Results from one representative experiment out of at least two independent experiments are shown, and the means and error bars were from duplicate experiments. RLU, relative light units; NC, negative control; M, membrane protein.
Mutations impair virus production by preventing virus assembly and release.
To further confirm that the failure of infectious viruses and RVP formation in most mutations was due to a major defect in virus assembly or release, subcellular fractionation of genomic RNA-electroporated BHK-21 cell homogenates was performed utilizing gradient centrifugation. Ten fractions from the top to the bottom were collected. First, we examined the number of intracellular infectious particles and the viral genome number contained in each fraction. The infectious particles yielded by some mutations (the DE mutant, N154A, H144A, H319A, T410A, and Q258A) mainly accumulated in fractions 4 to 7. In addition, there were two peaks in the number of viral genomes in fractions 4 and 7 in both the mutations capable of yielding infectious particles and those incapable of creating infectious virus (Fig. 5). Then, we analyzed the localization of the E and prM proteins in these 10 fractions by Western blotting using calnexin, an ER-resident protein, as a marker. For the DE, N154A, H144A, H319A, T410A, and Q258A mutants, the majority of the E and prM proteins accumulated in fractions 4 to 7, whereas for mutations failing to yield infectious particles, the majority of the E and prM proteins accumulated in fractions 7 to 10 (Fig. 5). In fact, previous studies have demonstrated that fraction 7, with a peak in the viral genome number, represented the viral assembly sites, and both the C and E proteins could be detected in this fraction (37, 38). As shown in Fig. 5, the lack of a peak in the viral genome number in fraction 7 in the five I0, ij, BC, FG, and R9A mutants indicates the absence of virus particle assembly, and a peak in fraction 4 colocalizing with the calnexin protein might represent an RNA replication site derived from the ER membrane, termed a vesicle packet (VP) (41, 42). For the four kl, E373A, L221S, and W217A mutants, with two peaks in the viral genome number but structural protein localization different from that of the WT, we presumed that the assembled viral particles are not able to be released from the assembly sites to the lumen of the ER (Fig. 5). For a special mutation, F407A, which exhibits WT-like structural protein localization, we presumed that the assembled viral particles were released from the assembly sites into the lumen of the ER, but the failure to yield infectious particles might have occurred because the particles were not able to be released into the secretory pathway (Fig. 5).
FIG 5.

Subcellular fractionation of in vitro-transcribed genomic RNA electroporated BHK-21 cells. Ten fractions were collected from the top, and the virus titer of each fraction was quantified by plaque assay (black columns). The viral genome number of JEV contained in each fraction was quantified by qPCR (black line); each fraction was also evaluated by Western blotting for the presence of E and prM proteins using calnexin (CNX) as a marker. Data are shown for only the WT and seven representative mutations.
VLPs have been shown to be a model system with which to identify viral assembly and release defects and can be produced efficiently by the coexpression of the prM and E proteins (43). To analyze the roles of mutations in the assembly and release of VLPs, site-directed mutagenesis was carried out as shown in Table 1 in a JEV prM/E expression construct, pCAGC105E. The amounts of prM proteins of each mutation in prM/E-transfected cell lysates were similar to the amount in the WT, whereas the amounts of E proteins of some mutations, such as in the DE, FG, R9A, and E373A mutants, in the cell lysates were obviously reduced (Fig. 6A). Because the prM and E proteins were derived from a single open reading frame-encoded polyprotein, we did not think that substitutions greatly affected the expression of prM/E proteins but, rather, altered the stability of some mutant E proteins. The amounts of prM/E proteins in pellets compared to those of the WT were consistent with the defect in RVP production experiments, and most mutations impaired VLP release into the culture supernatants except in the DE, N154A, H144A, H319A T410A, and Q258A mutants although the DE and N154A mutants exhibited a significant decrease in VLP release (Fig. 6B, top). The pr peptide derived from furin cleavage of the prM protein in the secretory pathway plays a role in silencing flavivirus fusion activity during virus secretion by binding the virus at low pH and inhibits virus-membrane interaction. Thus, failure of the mutants to bind to pr would result in their undergoing membrane fusion in the acidic environment of the trans-Golgi compartment (44, 45). To determine whether mutant VLP release was inhibited by the lack of pr protection in the secretory pathway, we added 20 mM NH4Cl to the transfected cells to neutralize the acidic pH in the trans-Golgi compartment and prevent the processing of prM to pr. As shown in Fig. 6B (bottom), the cells in the presence of NH4Cl produced immature VLPs without furin cleavage of the prM protein in the secretory pathway, but the amounts of prM/E proteins in the pellets were comparable to those in the absence of NH4Cl. These data suggest the importance of these residues in maintaining the assembly function of VLPs before the VLPs traffic through the secretory pathway.
FIG 6.

VLP production in BHK-21 cells. BHK-21 cells were transfected with the WT and each mutant pCAGC105E. (A) The cell lysates were analyzed by Western blotting at 48 h posttransfection using mouse E DIII antiserum, rabbit prM antiserum, and anti-GAPDH mouse monoclonal antibody. (B) The VLP pellets precipitated from cell culture supernatants in the presence or absence of 20 mM NH4Cl were analyzed by Western blotting at 48 h posttransfection. Results from one representative experiment out of three independent experiments are shown.
The explanation for the reduced assembly of VLPs might be that the mutations impair the interaction between prM and E as a proper prM/E heterodimeric interaction is important in flavivirus assembly (46). To investigate whether these mutations affect the prM/E heterodimeric interaction, immunoprecipitated proteins utilizing mouse E DIII antiserum from prM/E-transfected cell lysates were analyzed by Western blotting. The ratio of prM protein relative to E protein for each mutation was similar to that observed for the WT transfection, suggesting that the reduced VLP assembly was not due to the impairment of prM/E heterodimerization (Fig. 7A).
FIG 7.

The prM/E heterodimeric interaction and endo-H digestion. (A) The transfected BHK-21 cell lysates as described in the VLP experiments were immunoprecipitated (IP) with mouse E DIII antiserum at 48 h posttransfection and then analyzed by Western blotting for the presence of the E and prM proteins. Normal mouse IgG (Nor-IgG) was used for immunoprecipitation as a negative control; rabbit E DIII antiserum was used for Western blotting to avoid the interference of antibody heavy chain. (B) The cell lysates at 48 h posttransfection were digested with endo-H or left undigested and analyzed by Western blotting using mouse E DIII antiserum. The long exposures for the I0, ij, BC, and DE mutants are shown below. Results of representative experiment out of at least two independent experiments are shown.
To further confirm whether the mutant E proteins were able to traffic through the secretory pathway, the E proteins in prM/E-transfected cell lysates were treated with endo-H, an enzyme that can cleave immature high-mannose glycans from proteins. Sensitivity to endo-H indicated that the proteins were mainly present in a compartment prior to the trans-Golgi apparatus (38, 47). As shown in Fig. 7B, there were two bands of the E proteins with different molecular weights in the WT and H144A, H319A, T410A, and Q258A mutations. The top band of the E protein (endo-H resistant) indicated that the E proteins were mostly distributed in the trans-Golgi apparatus. We observed that the WT and T410A and Q258A mutant E proteins were mostly distributed in the trans-Golgi apparatus; the H144A and H319A mutant E proteins were distributed in roughly equivalent proportions between the Golgi apparatus and a compartment prior to the trans-Golgi apparatus. The rest of the mutant E proteins were mainly present in a compartment prior to the trans-Golgi apparatus (the N154A substitution resulted in no glycosylation). A smaller amount of E proteins in the Golgi compartment could therefore explain the reduced accumulation or release of immature mutant viruses in the cells.
Significantly reduced entry activity of several mutants.
We were able to characterize the effect of six representative mutants—the DE mutant, N154A, H144A, H319A, T410A, and Q258A—on viral entry using high-titer virus preparations despite the failure of the other mutations to yield infectious virus. The N154A or DE mutant might target DC-SIGN- or other receptor-binding motifs, respectively; H144A and H319A targeted the DI-DIII interaction in the prefusion conformation of the E monomer, and T410A and Q258A might target the zippering reaction of the stem along DII in the postfusion conformation (Fig. 1).
To examine the effects of these mutations on virus binding and entry activity, we exploited relative qPCRs to measure the viral genome RNA associated with the cells. First, BHK-21 cells were incubated with equal amounts of the WT or mutants (10,000 GCPs per cell) normalized by qPCR as described above, and we measured the bound viral genome RNA following 1 h of incubation at 4°C. According to the results, these mutations in E proteins altered the viral binding competence to the host cells. Even though we added the same number of GCPs, we detected different amounts of viral genome RNA binding to the cells. As shown in Fig. 8A (bottom), the DE, H319A, and T410A mutants had binding activity similar to that of the WT, N154A and H144A exhibited approximately half of the binding activity of the WT, and Q258A had higher binding activity. Then, we initialized viral internalization into BHK-21 cells for 1 h at 37°C and removed the remaining virus from the cell surface by trypsinizing the cells. To detect productive viral entry, we measured viral genome RNA synthesis in these cells by qPCR at 16 h postinfection since both the positive and negative strands of viral genome RNA remained at the same levels within 10 h postinfection (data not shown). Consistent with the RVP entry assay, viral RNA synthesis in the H144A, H319A, T410A, and Q258A mutants was significantly reduced compared to that of the WT, and the N154A and DE mutants maintained equal or even greater entry activity (Fig. 8A, top). The ratio of synthetic viral RNA to bound viral RNA was defined as viral entry activity, and the values for H144A, H319A, T410A, and Q258A were 6.8%, 14.1%, 59.8%, and 9.9%, respectively, whereas those for the WT and the DE and N154A mutants were 100%, 203.3%, and 92.5%, respectively (Fig. 8B). For the DE and N154A mutants, which maintained entry activity equal to or even greater than that of the WT, we presumed that these residues are not essential to virus binding and entry into BHK-21 cells. For the H144A, H319A, T410A, and Q258A mutants, which may disturb the DI-DIII interaction and zippering reaction during the transition of the E protein from dimer to trimer, the reduced entry activity was most likely due to a defect in viral membrane fusion.
FIG 8.
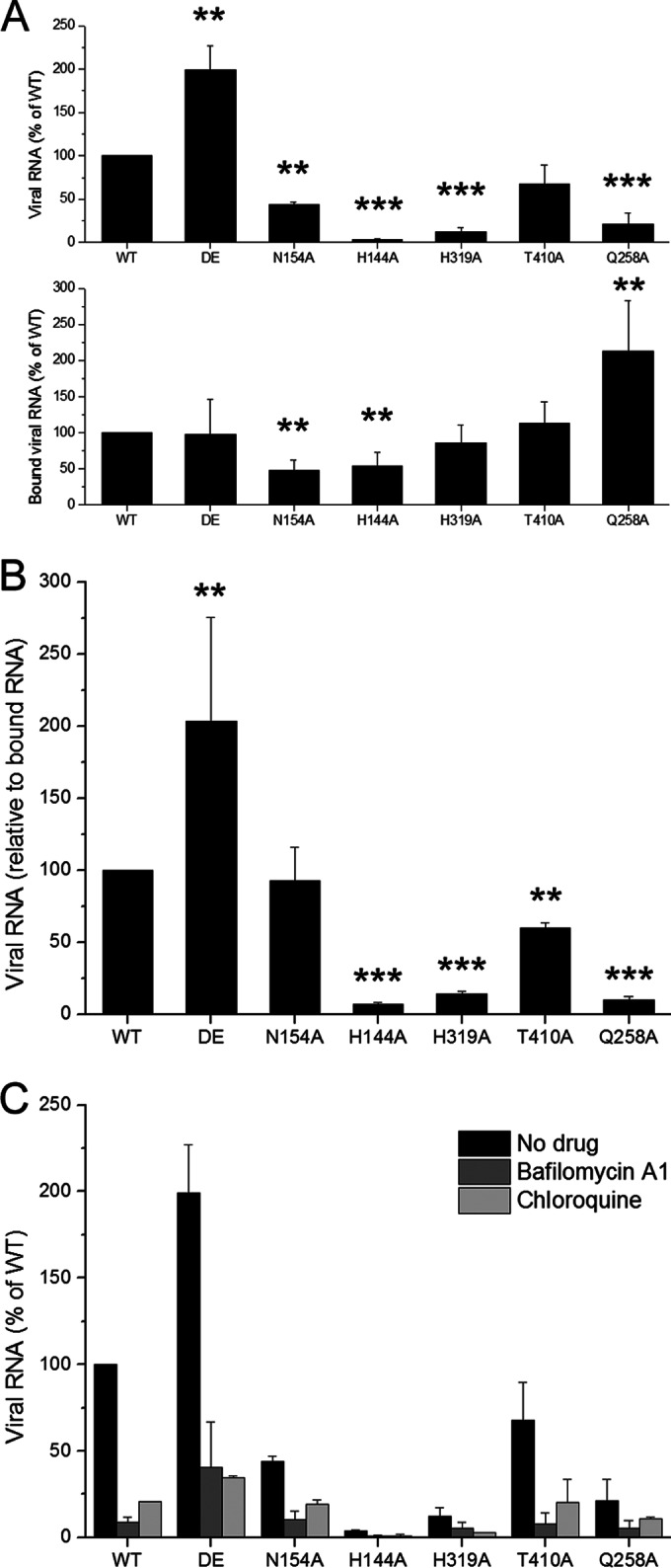
Binding and entry activity of several mutant viruses. BHK-21 cells were incubated with WT or mutant viruses (DE mutant, N154A, H144A, H319A, T410A, and Q258A) at 10,000 GCPs per cell for 1 h at 4°C. (A) The bound viral genome RNA of each mutant relative to that of WT was analyzed by qPCR using GAPDH as an internal control (bottom). After the unbound virus was removed, the cells were kept at 37°C for 1 h and then trypsinized and replated into a 24-well plate. To detect productive virus entry, the synthetic viral genome RNA of each mutation relative to that of WT was determined by qPCR at 16 h postinfection (top). (B) The entry activity of each mutation was measured by the ratio of synthetic viral RNA to bound viral RNA. Asterisks denote a statistically significant reduction in entry activity compared to that of the WT (**, P < 0.01; ***, P < 0.001). (C) qPCR was performed as described for panel A (top) in the presence of bafilomycin A1 or chloroquine. Means and standard errors are from three independent experiments.
A previous study indicated that histidine residues play a critical role as sensors to trigger conformational changes in fusion proteins that drive pH-dependent membrane fusion (48). We proposed that replacing histidine with alanine would change virus entry in a pH-independent manner, and we tested whether the entry of H144A, H319A, T410A, and Q258A mutants could be inhibited by the neutralization of endosomal compartments using bafilomycin A1 or chloroquine. We observed that infection with either histidine mutant was significantly inhibited by bafilomycin A1 or chloroquine (Fig. 8C). These results are consistent with a previous study indicating that the 50% inhibitory concentrations (IC50s) of NH4Cl are similar for the WT WNV and histidine mutants; the authors of that study suggested that no single histidine residue is required for the E protein-mediated entry of WNV and that other histidine or nonhistidine residues may coordinate to cause conformational changes in the fusion proteins (49). In our opinion, we cannot exclude the possibility that bafilomycin A1 might inhibit infection by blocking the traffic of the endocytosis pathway, but not pH neutralization, because a previous study has shown that ATPase inhibitors not only raise the endosomal pH but also block endosomal maturation (50).
To further examine the membrane fusion competence of the H144A, H319A, T410A, and Q258A mutants, a C6/36 cell-cell fusion-from-within assay was performed. The cells were inoculated with the WT and the mutants at an MOI of 1. The pH of the medium was kept at 7.7 at all times to prevent fusion. The E protein is not expressed on the cell surface, so the cell-cell fusion was actually induced by the virus particles secreted into the cell culture supernatant. Based upon the growth curves of infectious viruses, all the viruses could reach the highest titer at 96 h postinfection (Fig. 9A). So fusion of the infected cells was performed at 96 h postinfection. Although the titer of the H144A mutant was lower than the titers of other viruses, the GCP numbers of the H144A mutant were similar to those of other viruses (Fig. 9B). So we could rule out that reduced syncytium formation was due to a decreased amount of virus particles present in the cell culture supernatants. Finally, the results showed that these mutants formed fewer syncytia than the WT, indicating reduced fusion competence (Fig. 9C and D).
FIG 9.

C6/36 cell-cell fusion from within. C6/36 cells were infected with the WT and mutant viruses at an MOI of 1. The cell culture supernatants were harvested every 24 h, and then the viral titer and GCP number were calculated by plaque assays (A) and qPCR (B), respectively. (C) At 4 days postinfection, fusion of the infected cells was triggered by serum-free medium at pH 5.5 for 2 h, and then the nuclei were stained with DAPI for 10 min. The percent fusion was determined by the numbers of the nuclei of the syncytia relative to the total number of nuclei. Asterisks denote a statistically significant reduction in fusion activity compared to that of the WT (**, P < 0.01; ***, P < 0.001). (D) Presentation of the syncytia formed by WT and other mutants. Syncytia were usually formed by dozens of cells, and a representative syncytium is indicated by a white arrow.
Adaptive mutations in BHK-21 cells.
To investigate the function and stability of the engineered mutations in the virus life cycle, we selected adaptive viruses from DE, N154A, H144A, H319A, F407A, T410A, and Q258A mutants using continuous passage on BHK-21 cells. After five passages, each mutant virus reached a peak viral titer similar to that of the WT, but the plaque morphology of several cell-passaged mutants, H319A, F407A, T410A, and Q258A, exhibited a substantial reduction in size compared to that of their corresponding parental strains (the small plaque morphology of the H319A adaptive mutant was not very clear [data not shown]). Sequence analysis was performed on BHK-21 cell-passaged adaptive mutations to reveal changes in the viral structural proteins, C, prM, and E. Notably, each mutation was conserved, but we observed other mutations in the E protein gene, whereas no mutations were found in the C and M protein genes (Fig. 10A). Most of the adaptive mutations were from negatively charged amino acids to positively charged or neutral amino acids: E306A and N393H in DE, A295V in H144A, E138K and D389G in H319A, A72V and E138K in F407A, and D389G in both T410A and Q258A. We also observed that two mutations, E138K and D389G, repeatedly occurred in several mutants with a small-plaque phenotype, including H319A, F407A, T410A, and Q258A (Fig. 10A). Next, the BHK-21 cells were incubated with equal numbers of the WT or adaptive mutants, and the binding and entry activity of each adaptive mutant was determined by qPCR. The binding activity of the adaptive mutants was no more than three times higher than that of parental strains, but the entry activity increased significantly compared to the activities of the parental strains (Fig. 10B). The ratios of synthetic viral RNA/bound viral RNA in the DE, H144A, H319A, F407A, T410A, and Q258A mutants (P5) were 109.1%, 151.0%, 62.2%, 121.3%, 141.4%, and 101.4%, respectively (Fig. 10C). These data showed that adaptive mutants that formed small-sized plaques increased the efficiency of virus entry into BHK-21 cells. In addition, H319A adaptive mutants that did not form clear small plaques had lower ratios of synthetic viral RNA/bound viral RNA than the WT.
FIG 10.

Continuous passage of mutations for five rounds in BHK-21 cells. (A) Summary of sequencing results for mutant viruses (P0 and P5). (B and C) Binding and entry activities of the revertants were determined as described in the legend of Fig. 8A and B. Asterisks denote a statistically significant reduction in entry activity compared to that of the WT (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Means and standard errors are from three independent experiments. (D) BHK-21 cells were transfected with mutant pCAGC105E, F407A, F407A D189G, F407A A72V, and T410A. The cell lysates and VLP pellets were analyzed by Western blotting for the E and prM proteins at 48 h posttransfection. Both the D189G and the A72V mutations were selected by continuous passage of the F407A mutant in duplicate experiments. Results from one representative experiment out of three independent experiments are shown.
Finally, we focused on a mutation at position 72 from alanine to valine in the F407 mutant. The residue A72 is located at the tip of DII and on the surface of the E protein and is conserved in WNV and TBEV. Because the F407 mutation failed to yield any detectable virus but might form assembled viral particles, we presumed that A72V was a compensatory mutation to increase F407 mutant virus release into the culture supernatant. To confirm this hypothesis, we introduced this mutation into the pCAGC105E (F407A) mutant. The intracellular amount of E protein was unaffected by the A72V mutation, but this mutation compensated for the F407A mutation by restoring VLP production to an extent comparable to that of the T410 mutation, which resulted in WT-like VLP production competence (Fig. 10D). Future experiments to introduce this compensatory mutation into the F407A mutant may further understanding of the mechanism of its release defect and yield infectious virus for entry activity study.
DISCUSSION
Mutational analysis of the E protein of flavivirus has been used to investigate the role of E protein in virus assembly, release, or entry (35, 38, 51–53). In this study, utilizing mutagenesis of an infectious cDNA clone of JEV, we introduced mutations into the receptor-binding motif or the amino acids critical for membrane fusion to systematically study and reveal the JEV entry mechanism. It is not surprising that these motifs are normally located in a region conserved among different types of JEVs and even among the family Flaviviridae, indicating their important role in the viral life cycle (Fig. 2A). Infectious particle and RVP production showed that most mutations impaired virus production. Subcellular fractionation and VLP production showed that five mutations, in I0, ij, BC, and FG and R9A, impaired viral assembly, and the assembled viral particles of five other mutations, in kl and E373A, F407A, L221S, and W217A, could not be released into the secretory pathway. A virus binding and entry assay showed that N154 and the DE loop are not the only or the major receptor-binding motifs in JEV entry into BHK-21 cells, but four amino acids, H144, H319, T410, and Q258, are important for maintaining the efficiency of viral membrane fusion (Table 2). Finally, continuous passaging of mutants showed that adaptive mutations from negatively charged amino acids to positively charged or neutral amino acids, such as E138K and D389G, can restore viral entry activity.
TABLE 2.
Summary of the effects of mutations on virus assembly, release, binding, and entrya
| Mutant | Assembly | Release | Binding | Entry |
|---|---|---|---|---|
| I0 mutant | × | × | ND | ND |
| ij mutant | × | × | ND | ND |
| BC mutant | × | × | ND | ND |
| FG mutant | × | × | ND | ND |
| R9A | × | × | ND | ND |
| kl mutant | √ | × | ND | ND |
| E373A | √ | × | ND | ND |
| F407Ab | √ | × | ND | ND |
| L221S | √ | × | ND | ND |
| W217A | √ | × | ND | ND |
| DE mutant | √ | ↓↓ | √ | ↑ |
| N154A | √ | ↓ | ↓ | √ |
| H144A | √ | √ | ↓ | ↓↓ |
| H319A | √ | √ | √ | ↓↓ |
| T410A | √ | √ | √ | ↓ |
| Q258A | √ | √ | ↑ | ↓↓ |
Symbols are as follows: ×, impairment; √, no detectable effects; ND, not done; ↓ and ↓↓, reduction of at least 2-fold or more than 10-fold, respectively; ↑, increase of at least 2-fold.
A mutation, A72V, can restore the release of F407A mutant virus into culture supernatants.
Potential receptor-mediated entry.
It has been well accepted that receptor molecule usage in flaviviruses is cell type dependent (54), and several proteins, including GAGs, DC-SIGN, heat shock protein 70, and vimentin, have been identified as the receptor candidates for JEV. Five potential receptor-binding motifs potentially involved in JEV entry into mammalian or mosquito cells, including I0 (GAGs binding motif), N154 (DC-SIGN binding motif), the BC loop, the DE loop, and the FG loop in DIII, have been studied. Due to their important roles in E protein folding or virus assembly, only two of them (N154A and the DE loop) yield infectious viruses. In fact, a previous study has shown that the WNV BC loop plays key roles in E protein DIII folding, virus infectivity, virulence, and antigenicity (55). Because the FG loop in DIII contains an arginine-glycine-aspartic acid (RGD) motif that plays an essential role in integrin-ligand interactions (56), its interaction with integrin αvβ3, a receptor for WNV (57, 58), should be evaluated. The N154A and DE mutants significantly reduced virus release into the culture supernatants, but we could still test their effect on virus entry using high-titer virus preparations harvested at 4 days posttransfection. We observed that these residues are not essential to virus binding and entry into BHK-21 cells.
DC-SIGN can facilitate JEV infection in a lymphoid cell line that was originally not susceptible to infection, which also has been well studied as a flavivirus attachment protein that interacts with mannose glycans on the E protein (59–61). As shown in Fig. 7B, the E protein of the N154A mutation had only one protein band with a small molecular weight in the presence or absence of endo-H, indicating that the N154A mutation abolished the carbohydrate modification in the E protein. So we thought that N154 is not essential to virus binding and entry into BHK-21 cells because N-linked carbohydrates specifically interact with DC-SIGN, which is not expressed on BHK-21 cells; further study of dendritic cell infection may be necessary. Our finding with respect to the DE mutation is inconsistent with a previous study indicating that loop 3 peptides (the DE loop) can prevent JEV infection by interfering with virus attachment to BHK-21 cells (18). A similar phenomenon was observed in DENV2 virus, where a peptide containing the FG loop inhibits E DIII protein binding to C6/36 cells (19), but the deletion of the DIII FG loop in DENV2 does not affect the infection of C6/36 cells (52). A possible explanation for this phenomenon is that these loops are not the only or the major receptor-binding motifs (51). We found that the DE mutant is capable of infecting C6/36 cells (data not shown), but it is unclear whether this mutant can grow in mosquitoes as C6/36 cells may be not a useful model for predicting virus replication in live adult mosquitoes (52, 53). Further study should focus on whether the DE loop contributes to host range or tissue tropism.
DI-DIII interaction in the E monomer in the prefusion conformation.
Prefusion E protein ectodomain crystal structures show that the region of the DI-DIII interaction in the E monomer is highly conserved across the contact interface, and Van der Waals contacts, salt bridges, and direct and water-mediated hydrogen bonds form an intricate intramolecular network. During the transition of the E protein from dimer to trimer, the DI-DIII interaction provided by conserved residues, such as Arg9 in DI, Glu373 in DIII, H144 in DI, and H319 in DIII in JEV, has to be broken (Fig. 1C and E). So it seems necessary to break the salt bridge formed by R9 and E373 to allow viral membrane fusion (28). However, we presume that the salt bridge maintaining the stabilization of the DI/DIII interface is also essential to viral assembly because introducing mutations to R9 and E373 indeed resulted in the impairment of virus production. The protonation of H144 and H319 is likely important in the destabilization of the DI/DIII interface under low-pH conditions for the initiation of viral membrane fusion (28, 62). In fact, H323 in DIII of TBEV, equivalent to H319 in DIII in JEV, has been identified as the critical pH sensor in initializing the fusion of VLPs with synthetic liposomes. Fritz et al. also showed that some other conserved histidines were completely dispensable for the early stage of membrane fusion (48). However, Nelson et al. indicate that no single histidine residue, but other histidine or nonhistidine residues, may coordinate to cause conformational changes in the E proteins in WNV (49). In this study, we show that the H144A and H319A mutations result in a significant reduction of virus entry activity, which is most likely due to a defect in membrane fusion. This result may contribute to an understanding of the different functions of the histidine in the genus Flavivirus and also to the development of novel vaccines or antiflaviviral strategies based on the DI-DIII interaction in the E prefusion conformation.
Speculative zippering reaction of the stem in postfusion conformation.
In the postfusion conformation, final zippering of the stem along DII forces the juxtaposition of the fusion peptides and the transmembrane segments; thus, the H1 helix in the stem will contact both the DII aA and aB helices. Mutations introduced into the H1 and aA helices in TBEV indicate that the interaction of F403 (equivalent to F407 in the H1 helix in JEV) with a hydrophobic pocket formed by W219 and L223 (equivalent to W217 and L221, respectively, in the aA helix in JEV) is critical for initiating the zippering reaction though the F403I mutation retains a WT-like fusion activity (63). However, the crystal structure of the DENV E protein late-stage fusion intermediate shows that F402, which is equivalent to F403 in TBEV, indeed lies against a conserved hydrophobic surface formed by the side chains of L216, L218, and M260 instead of W212 and L216, which are equivalent to W219 and L223 in TBEV, respectively (64). Five mutations of conserved amino acids, F407 and T410 in the H1 helix, W217 and L221 in the aA helix, and Q258 in the aB helix in JEV, likely participating in the zippering reaction are analyzed in this study (Fig. 1D and E). Three of the mutations, F407A, L221S, and W217A, are lethal in BHK-21 cells, and no infectious viruses were obtained posttransfection due to their important roles in virus assembly and release as described above; the other two mutations, T410A and Q258A, result in a significant reduction in viral membrane fusion activity. High-resolution structures of the E protein ectodomain usually lack the stem region; thus, the intramolecular interaction between the stem region and the E protein ectodomain in the postfusion conformation is still disputed. T406 in TBEV, equivalent to T410 in JEV, is predicted to form hydrogen bonds with Q260 at the beginning of the aB helix (Q258 in JEV) (28), but our data show that the entry activity of T410A relative to that of the WT was reduced by only 40%, much less than that with Q258A. However, in a homology model of the trimeric postfusion E protein, T410, likely exposed on the trimer surface, is apart from Q258, and Q258 may interact with the other residues at the end of the H1 helix (Fig. 1D). However, these results have suggested that proper initiation of the zippering reaction of the stem might be critical for JEV infection in mammalian cells; in fact, peptides derived from the E protein stem can inhibit flavivirus entry (65, 66).
Positive-charge mutations recover the reduced entry activity.
We found that adaptive viruses from H144A, H319A, T410A, and Q258A mutants arising by serial passage in BHK-21 cells can recover the reduced entry competence to WT-like levels, and most adaptive mutations were from negatively charged amino acids to positively charged or neutral amino acids. In particular, the E138K and D389G mutations repeatedly occur in several mutants and have exhibited a small-plaque phenotype. Serial passage of the N154A mutant in BHK-21 cells did not result in any changes in the E protein. Because the N154A mutation did not have any effect on virus entry activity, we presume that the positive-charge mutations are selected in response to reduced entry activity. In fact, a previous study has shown that serial passage of the WT Nakayama strain of JEV in BHK cells also did not result in any changes in the E protein (67). We also found that all of the adaptive mutations were located on the surface of the E protein. For example, residue 389 lies at the DIII FG loop, which also provides further evidence for the FG loop acting as a receptor-binding motif. In this study, we observed that the entry activity of several positive-charge revertants has been recovered compared to activity levels of the parental strains. We calculated the entry activity relative to bound viral RNA (Fig. 10C), so the recovered entry activity was likely not due to the higher virus binding competence of the revertants than that of the WT. Previous studies showed that the positive-charge mutations can enhance the binding affinity to GAGs and increase the efficiency of virus internalization into host cells, and the revertants were most likely a result of altered receptor usage for virus binding-entry with the involvement of GAGs in the continuous passaging process (20, 67, 68). In our opinion, the positive-charge revertants could internalize in the host cells more efficiently utilizing the GAGs on the cell surface, which is likely the reason that the revertants can recover the reduced entry activity into BHK-21 cells.
Critical regions in the envelope protein participating in the flavivirus entry pathway, including the receptor binding domain, the stem region, and the hydrophobic pocket, have been shown to be suitable as drug targets (13). In fact, many useful DENV-targeting compounds blocking these regions of the viral entry pathway have been identified (69). Because JEV shares a high percentage of amino acids in the E protein with the other flaviviruses, more work on the identification and characterization of critical amino acids in the E protein may provide a novel theoretical basis for drug and vaccine design for flaviviruses. Our study has revealed the critical role of several amino acids in the envelope protein in the assembly, release, and entry of JEV. In addition, we show for the first time that the H144A, H319A, T410A, and Q258A mutants are fusion-attenuated viruses, which may be useful for developing a novel vaccine for flaviviruses.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Key Scientific Program (973)-Nanoscience and Nanotechnology (2011CB933600) and the National Natural Science Foundation of China (grant no. 31400157).
We thank Takaji Wakita for providing the infectious plasmid clone of JEV and the public technology service center, Wuhan Institute of Virology, for technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00293-15.
REFERENCES
- 1.Mackenzie JS, Gubler DJ, Petersen LR. 2004. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med 10:S98–109. doi: 10.1038/nm1144. [DOI] [PubMed] [Google Scholar]
- 2.Endy TP, Nisalak A. 2002. Japanese encephalitis virus: ecology and epidemiology. Curr Top Microbiol Immunol 267:11–48. doi: 10.1007/978-3-642-59403-8. [DOI] [PubMed] [Google Scholar]
- 3.Yun SI, Lee YM. 2014. Japanese encephalitis: the virus and vaccines. Hum Vaccin Immunother 10:263–279. doi: 10.4161/hv.26902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Hurk AF, Ritchie SA, Mackenzie JS. 2009. Ecology and geographical expansion of Japanese encephalitis virus. Annu Rev Entomol 54:17–35. doi: 10.1146/annurev.ento.54.110807.090510. [DOI] [PubMed] [Google Scholar]
- 5.Campbell GL, Hills SL, Fischer M, Jacobson JA, Hoke CH, Hombach JM, Marfin AA, Solomon T, Tsai TF, Tsu VD, Ginsburg AS. 2011. Estimated global incidence of Japanese encephalitis: a systematic review. Bull World Health Org 89:766–774, 774A–774E. doi: 10.2471/BLT.10.085233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misra UK, Kalita J. 2010. Overview: Japanese encephalitis. Prog Neurobiol 91:108–120. doi: 10.1016/j.pneurobio.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Unni SK, Ruzek D, Chhatbar C, Mishra R, Johri MK, Singh SK. 2011. Japanese encephalitis virus: from genome to infectome. Microbes Infect 13:312–321. doi: 10.1016/j.micinf.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Nawa M, Takasaki T, Yamada K, Kurane I, Akatsuka T. 2003. Interference in Japanese encephalitis virus infection of Vero cells by a cationic amphiphilic drug, chlorpromazine. J Gen Virol 84:1737–1741. doi: 10.1099/vir.0.18883-0. [DOI] [PubMed] [Google Scholar]
- 9.Das S, Chakraborty S, Basu A. 2010. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J Neurochem 115:537–549. doi: 10.1111/j.1471-4159.2010.06951.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhu YZ, Xu QQ, Wu DG, Ren H, Zhao P, Lao WG, Wang Y, Tao QY, Qian XJ, Wei YH, Cao MM, Qi ZT. 2012. Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J Virol 86:13407–13422. doi: 10.1128/JVI.00903-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalia M, Khasa R, Sharma M, Nain M, Vrati S. 2013. Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol 87:148–162. doi: 10.1128/JVI.01399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kielian M, Rey FA. 2006. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol 4:67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierson TC, Kielian M. 2013. Flaviviruses: braking the entering. Curr Opin Virol 3:3–12. doi: 10.1016/j.coviro.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kielian M. 2006. Class II virus membrane fusion proteins. Virology 344:38–47. doi: 10.1016/j.virol.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. 2004. Conformational changes of the flavivirus E glycoprotein. Structure 12:1607–1618. doi: 10.1016/j.str.2004.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luca VC, AbiMansour J, Nelson CA, Fremont DH. 2012. Crystal structure of the Japanese encephalitis virus envelope protein. J Virol 86:2337–2346. doi: 10.1128/JVI.06072-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Zhang LY, Sun MX, Li PP, Huang L, Wei JC, Yao YL, Isahg H, Chen PY, Mao X. 2012. Inhibition of Japanese encephalitis virus entry into the cells by the envelope glycoprotein domain III (EDIII) and the loop 3 peptide derived from EDIII. Antiviral Res 94:179–183. doi: 10.1016/j.antiviral.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Hung JJ, Hsieh MT, Young MJ, Kao CL, King CC, Chang W. 2004. An external loop region of domain III of dengue virus type 2 envelope protein is involved in serotype-specific binding to mosquito but not mammalian cells. J Virol 78:378–388. doi: 10.1128/JVI.78.1.378-388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee E, Lobigs M. 2000. Substitutions at the putative receptor-binding site of an encephalitic flavivirus alter virulence and host cell tropism and reveal a role for glycosaminoglycans in entry. J Virol 74:8867–8875. doi: 10.1128/JVI.74.19.8867-8875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zu X, Liu Y, Wang S, Jin R, Zhou Z, Liu H, Gong R, Xiao G, Wang W. 2014. Peptide inhibitor of Japanese encephalitis virus infection targeting envelope protein domain III. Antiviral Res 104:7–14. doi: 10.1016/j.antiviral.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Wu SC, Chiang JR, Lin CW. 2004. Novel cell adhesive glycosaminoglycan-binding proteins of Japanese encephalitis virus. Biomacromolecules 5:2160–2164. doi: 10.1021/bm0498068. [DOI] [PubMed] [Google Scholar]
- 23.Chiou SS, Liu H, Chuang CK, Lin CC, Chen WJ. 2005. Fitness of Japanese encephalitis virus to Neuro-2a cells is determined by interactions of the viral envelope protein with highly sulfated glycosaminoglycans on the cell surface. J Med Virol 76:583–592. doi: 10.1002/jmv.20406. [DOI] [PubMed] [Google Scholar]
- 24.Modis Y, Ogata S, Clements D, Harrison SC. 2005. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol 79:1223–1231. doi: 10.1128/JVI.79.2.1223-1231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimojima M, Takenouchi A, Shimoda H, Kimura N, Maeda K. 2014. Distinct usage of three C-type lectins by Japanese encephalitis virus: DC-SIGN, DC-SIGNR, and LSECtin. Arch Virol 159:2023–2031. doi: 10.1007/s00705-014-2042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S, Laxminarayana SV, Chandra N, Ravi V, Desai A. 2009. Heat shock protein 70 on Neuro2a cells is a putative receptor for Japanese encephalitis virus. Virology 385:47–57. doi: 10.1016/j.virol.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 27.Liang JJ, Yu CY, Liao CL, Lin YL. 2011. Vimentin binding is critical for infection by the virulent strain of Japanese encephalitis virus. Cell Microbiol 13:1358–1370. doi: 10.1111/j.1462-5822.2011.01624.x. [DOI] [PubMed] [Google Scholar]
- 28.Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J 23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Modis Y, Ogata S, Clements D, Harrison SC. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A 100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Modis Y, Ogata S, Clements D, Harrison SC. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 31.Lee E, Weir RC, Dalgarno L. 1997. Changes in the dengue virus major envelope protein on passaging and their localization on the three-dimensional structure of the protein. Virology 232:281–290. doi: 10.1006/viro.1997.8570. [DOI] [PubMed] [Google Scholar]
- 32.Beasley DW, Aaskov JG. 2001. Epitopes on the dengue 1 virus envelope protein recognized by neutralizing IgM monoclonal antibodies. Virology 279:447–458. doi: 10.1006/viro.2000.0721. [DOI] [PubMed] [Google Scholar]
- 33.Hurrelbrink RJ, McMinn PC. 2001. Attenuation of Murray Valley encephalitis virus by site-directed mutagenesis of the hinge and putative receptor-binding regions of the envelope protein. J Virol 75:7692–7702. doi: 10.1128/JVI.75.16.7692-7702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monath TP, Arroyo J, Levenbook I, Zhang ZX, Catalan J, Draper K, Guirakhoo F. 2002. Single mutation in the flavivirus envelope protein hinge region increases neurovirulence for mice and monkeys but decreases viscerotropism for monkeys: relevance to development and safety testing of live, attenuated vaccines. J Virol 76:1932–1943. doi: 10.1128/JVI.76.4.1932-1943.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butrapet S, Childers T, Moss KJ, Erb SM, Luy BE, Calvert AE, Blair CD, Roehrig JT, Huang CY. 2011. Amino acid changes within the E protein hinge region that affect dengue virus type 2 infectivity and fusion. Virology 413:118–127. doi: 10.1016/j.virol.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 36.Li XD, Li XF, Ye HQ, Deng CL, Ye Q, Shan C, Shang BD, Xu LL, Li SH, Cao SB, Yuan ZM, Shi PY, Qin CF, Zhang B. 2014. Recovery of a chemically synthesized Japanese encephalitis virus reveals two critical adaptive mutations in NS2B and NS4A. J Gen Virol 95:806–815. doi: 10.1099/vir.0.061838-0. [DOI] [PubMed] [Google Scholar]
- 37.Boson B, Granio O, Bartenschlager R, Cosset FL. 2011. A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog 7:e1002144. doi: 10.1371/journal.ppat.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Wispelaere M, Yang PL. 2012. Mutagenesis of the DI/DIII linker in dengue virus envelope protein impairs viral particle assembly. J Virol 86:7072–7083. doi: 10.1128/JVI.00224-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guirakhoo F, Hunt AR, Lewis JG, Roehrig JT. 1993. Selection and partial characterization of dengue 2 virus mutants that induce fusion at elevated pH. Virology 194:219–223. doi: 10.1006/viro.1993.1252. [DOI] [PubMed] [Google Scholar]
- 40.Pu SY, Wu RH, Yang CC, Jao TM, Tsai MH, Wang JC, Lin HM, Chao YS, Yueh A. 2011. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J Virol 85:2927–2941. doi: 10.1128/JVI.01986-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatel-Chaix L, Bartenschlager R. 2014. Dengue virus- and hepatitis C virus-induced replication and assembly compartments: the enemy inside—caught in the web. J Virol 88:5907–5911. doi: 10.1128/JVI.03404-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin SR, Zou G, Hsieh SC, Qing M, Tsai WY, Shi PY, Wang WK. 2011. The helical domains of the stem region of dengue virus envelope protein are involved in both virus assembly and entry. J Virol 85:5159–5171. doi: 10.1128/JVI.02099-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. 2008. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319:1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- 45.Yu IM, Holdaway HA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. 2009. Association of the pr peptides with dengue virus at acidic pH blocks membrane fusion. J Virol 83:12101–12107. doi: 10.1128/JVI.01637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allison SL, Stiasny K, Stadler K, Mandl CW, Heinz FX. 1999. Mapping of functional elements in the stem-anchor region of tick-borne encephalitis virus envelope protein E. J Virol 73:5605–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freeze HH, Kranz C. 2010. Endoglycosidase and glycoamidase release of N-linked glycans. Curr Protoc Protein Sci Chapter 12:Unit 12.4. doi: 10.1002/0471140864.ps1204s62. [DOI] [PubMed] [Google Scholar]
- 48.Fritz R, Stiasny K, Heinz FX. 2008. Identification of specific histidines as pH sensors in flavivirus membrane fusion. J Cell Biol 183:353–361. doi: 10.1083/jcb.200806081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson S, Poddar S, Lin TY, Pierson TC. 2009. Protonation of individual histidine residues is not required for the pH-dependent entry of West Nile virus: evaluation of the “histidine switch” hypothesis. J Virol 83:12631–12635. doi: 10.1128/JVI.01072-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J Virol 85:4198–4211. doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roehrig JT, Butrapet S, Liss NM, Bennett SL, Luy BE, Childers T, Boroughs KL, Stovall JL, Calvert AE, Blair CD, Huang CY. 2013. Mutation of the dengue virus type 2 envelope protein heparan sulfate binding sites or the domain III lateral ridge blocks replication in Vero cells prior to membrane fusion. Virology 441:114–125. doi: 10.1016/j.virol.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 52.Erb SM, Butrapet S, Moss KJ, Luy BE, Childers T, Calvert AE, Silengo SJ, Roehrig JT, Huang CY, Blair CD. 2010. Domain-III FG loop of the dengue virus type 2 envelope protein is important for infection of mammalian cells and Aedes aegypti mosquitoes. Virology 406:328–335. doi: 10.1016/j.virol.2010.07.024. [DOI] [PubMed] [Google Scholar]
- 53.Huang CY, Butrapet S, Moss KJ, Childers T, Erb SM, Calvert AE, Silengo SJ, Kinney RM, Blair CD, Roehrig JT. 2010. The dengue virus type 2 envelope protein fusion peptide is essential for membrane fusion. Virology 396:305–315. doi: 10.1016/j.virol.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 54.Perera-Lecoin M, Meertens L, Carnec X, Amara A. 2014. Flavivirus entry receptors: an update. Viruses 6:69–88. doi: 10.3390/v6010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang S, Bovshik EI, Maillard R, Gromowski GD, Volk DE, Schein CH, Huang CY, Gorenstein DG, Lee JC, Barrett AD, Beasley DW. 2010. Role of BC loop residues in structure, function and antigenicity of the West Nile virus envelope protein receptor-binding domain III. Virology 403:85–91. doi: 10.1016/j.virol.2010.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruoslahti E. 1997. Integrins as signaling molecules and targets for tumor therapy. Kidney Int 51:1413–1417. doi: 10.1038/ki.1997.193. [DOI] [PubMed] [Google Scholar]
- 57.Chu JJ, Ng ML. 2003. Characterization of a 105-kDa plasma membrane associated glycoprotein that is involved in West Nile virus binding and infection. Virology 312:458–469. doi: 10.1016/S0042-6822(03)00261-7. [DOI] [PubMed] [Google Scholar]
- 58.Chu JJ, Ng ML. 2004. Interaction of West Nile virus with αvβ3 integrin mediates virus entry into cells. J Biol Chem 279:54533–54541. doi: 10.1074/jbc.M410208200. [DOI] [PubMed] [Google Scholar]
- 59.Navarro-Sanchez E, Altmeyer R, Amara A, Schwartz O, Fieschi F, Virelizier JL, Arenzana-Seisdedos F, Despres P. 2003. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep 4:723–728. doi: 10.1038/sj.embor.embor866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boonnak K, Slike BM, Burgess TH, Mason RM, Wu SJ, Sun P, Porter K, Rudiman IF, Yuwono D, Puthavathana P, Marovich MA. 2008. Role of dendritic cells in antibody-dependent enhancement of dengue virus infection. J Virol 82:3939–3951. doi: 10.1128/JVI.02484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pokidysheva E, Zhang Y, Battisti AJ, Bator-Kelly CM, Chipman PR, Xiao C, Gregorio GG, Hendrickson WA, Kuhn RJ, Rossmann MG. 2006. Cryo-EM reconstruction of dengue virus in complex with the carbohydrate recognition domain of DC-SIGN. Cell 124:485–493. doi: 10.1016/j.cell.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 62.Kampmann T, Mueller DS, Mark AE, Young PR, Kobe B. 2006. The Role of histidine residues in low-pH-mediated viral membrane fusion. Structure 14:1481–1487. doi: 10.1016/j.str.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 63.Pangerl K, Heinz FX, Stiasny K. 2011. Mutational analysis of the zippering reaction during flavivirus membrane fusion. J Virol 85:8495–8501. doi: 10.1128/JVI.05129-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klein DE, Choi JL, Harrison SC. 2013. Structure of a dengue virus envelope protein late-stage fusion intermediate. J Virol 87:2287–2293. doi: 10.1128/JVI.02957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt AG, Yang PL, Harrison SC. 2010. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLoS Pathog 6:e1000851. doi: 10.1371/journal.ppat.1000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hrobowski YM, Garry RF, Michael SF. 2005. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol J 2:49. doi: 10.1186/1743-422X-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee E, Hall RA, Lobigs M. 2004. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J Virol 78:8271–8280. doi: 10.1128/JVI.78.15.8271-8280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee E, Lobigs M. 2002. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray Valley encephalitis virus. J Virol 76:4901–4911. doi: 10.1128/JVI.76.10.4901-4911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De La Guardia C, Lleonart R. 2014. Progress in the identification of dengue virus entry/fusion inhibitors. Biomed Res Int 2014:825039. doi: 10.1155/2014/825039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.