

ABSTRACT
During uncoating, the conical capsid of HIV disassembles by dissociation of the p24 capsid protein (CA). Uncoating is known to be required for HIV replication, but the mechanism is poorly defined. Here, we examined the timing and effect of two capsid binding drugs (PF74 and BI2) on infectivity and capsid integrity in HIV-1-infected cells. The virus remained susceptible to the action of PF74 and BI2 for hours after uncoating as defined in parallel drug addition and cyclosporine (CsA) washout assays to detect the kinetics of drug susceptibility and uncoating, respectively. Resistance mutations in CA decreased the potency of these compounds, demonstrating that CA is the target of drug action. However, neither drug altered capsid integrity in a fluorescence microscopy-based assay. These data suggest that PF74 and BI2 do not alter HIV-1 uncoating but rather affect a later step in viral replication. Because both drugs bind CA, we hypothesized that a residual amount of CA associates with the viral complex after the loss of the conical capsid to serve as a target for these drugs. Superresolution structured illumination microscopy (SIM) revealed that CA localized to viral complexes in the nuclei of infected cells. Using image quantification, we determined that viral complexes localized in the nucleus displayed a smaller amount of CA than complexes at the nuclear membrane, in the cytoplasm, or in controls. Collectively, these data suggest that a subset of CA remains associated with the viral complex after uncoating and that this residual CA is the target of PF74 and BI2.
IMPORTANCE The HIV-1 capsid is a target of interest for new antiviral therapies. This conical capsid is composed of monomers of the viral CA protein. During HIV-1 replication, the capsid must disassemble by a poorly defined process called uncoating. CA has also been implicated in later steps of replication, including nuclear import and integration. In this study, we used cell-based assays to examine the effect of two CA binding drugs (PF74 and BI2) on viral replication in infected cells. HIV-1 was susceptible to both drugs for hours after uncoating, suggesting that these drugs affect later steps of viral replication. High-resolution structured illumination microscopy (SIM) revealed that a subset of CA localized to viral complexes in the nuclei of cells. Collectively, these data suggest that a subset of CA remains associated with the viral complex after uncoating, which may facilitate later steps of viral replication and serve as a drug target.
INTRODUCTION
Monomers of the viral capsid protein (CA) are arranged in a hexameric lattice to form the conical capsid of HIV-1. This structure contains the viral RNAs and associated viral proteins and is released into the cytoplasm of the cell after viral fusion. For the viral genome to ultimately integrate, the conical capsid must disassemble by a process called uncoating. During this time, in the reverse transcription complex (RTC) the viral RNA genome is reverse transcribed into double-stranded DNA. Once reverse transcription is completed, the viral complex becomes the preintegration complex (PIC) that is trafficked into the nucleus, where the double-stranded DNA integrates in to the chromosomal DNA of the cell to form a provirus. Uncoating is required for HIV-1 replication, but the mechanism of uncoating is not well defined. Specifically, it is not known how long the process of uncoating takes or whether all CA dissociates from the viral complex containing the genome during uncoating. From biochemical, microscopy, and cell-based assays, two viral factors have been implicated in uncoating: the CA protein and the process of reverse transcription. Mutations in CA can alter capsid stability and uncoating kinetics (1–6). Inhibition of reverse transcription delays uncoating in infected cells, indicating that this process facilitates capsid disassembly (7, 8).
Data from our laboratory suggest that uncoating occurs relatively early (less than 1 h) after viral fusion at some point when the reverse transcribing viral genome is trafficked toward the nucleus (7). For reference, completion of reverse transcription takes approximately 8 h (9). Our model is based on the characterization of viral complexes utilizing fluorescence microscopy and data from the cyclosporine (CsA) washout assay, in which the restriction factor TRIM-CypA is used to detect uncoating in HIV-infected cells (7, 9, 10). TRIM-CypA binds to multimerized CA in the conical capsid to inhibit HIV infectivity (11–13). In the CsA washout assay, OMK cells that endogenously express this factor are synchronously infected with a green fluorescent protein (GFP) reporter virus (HIV-GFP) in the presence of the drug CsA, which prevents TRIM-CypA binding (11). At various times postinfection, CsA is washed out, and any virus that is coated will be restricted for infection. Any virus that has uncoated to a sufficient extent to avoid TRIM restriction will be able to infect the cell. At 2 days postinfection, the percentage of infected cells is determined by flow cytometry, which is indicative of the percentage of uncoated viral complexes at each time point. Using this assay, we determined that uncoating occurs within an hour of viral fusion and is facilitated by reverse transcription (7). From this assay, we cannot determine if there is a rapid or progressive loss of CA during uncoating. The CsA washout assay is an indirect measure of uncoating, relying on the ability of the restriction factor TRIM-CypA to detect and restrict the infectivity of viral complexes that contain a sufficient amount of multimerized CA. However, the timing of the loss of sensitivity to TRIM-CypA restriction in the CsA washout assay correlates with the loss of CA in infected cells in a fluorescence microscopy-based uncoating assay and by Western blotting (7, 14, 15). A similar delay in uncoating due to inhibition of reverse transcription can also be detected by the CsA washout assay, a microscopy-based uncoating assay, and biochemically by Western blotting (7, 8). Therefore, data from multiple uncoating assays lend support to our model and indicate that the CsA washout assay is a good monitor for the process of uncoating in infected cells.
Recently CA, and by extension uncoating, has been implicated in other early steps of HIV-1 replication. Mutations in CA have been shown to alter nuclear import pathway usage and utilization of different nuclear import and pore proteins (16–20). CA mutations can also affect the ability of HIV-1 to infect nondividing cells (14, 21). Finally, mutations in CA have been shown to alter the proviral integration site (16, 17). According to our model of uncoating, all of these processes would occur after disassembly of the conical capsid. This raises the issue of how CA can influence aspects of viral replication such as integration site selection after the conical capsid is lost. One way to reconcile these data is a situation in which all molecules of CA do not dissociate from the RTC/PIC during uncoating. Therefore, we hypothesized that a subset of CA remains associated with the viral complex after disassembly of the conical capsid structure to facilitate downstream steps in viral replication. Here we have used cell-based assays, sensitivity to CA-targeted antiviral compounds, and high-resolution structured illumination microscopy (SIM) imaging to test this hypothesis.
MATERIALS AND METHODS
DNA plasmids.
To follow viral progress into the nucleus through the use of a GFP-tagged integrase, a Gag-integrase-GFP (GIG) expression vector was designed. The GIG vector was generated by BglI and XbaI restriction digestion of the Vpr-protease site-integrase-GFP expression vector (22), followed by ligation of the resulting integrase-GFP fragment into the Gag-mCherry expression vector in place of mCherry (23). The resulting Gag-integrase-GFP fusion protein contains a protease cleavage site between the Gag and integrase. During virion maturation, the viral protease will cleave the engineered site between Gag and integrase, leading to the generation of integrase-GFP.
Virus stocks.
Vesicular stomatitis virus (VSV) G-pseudotyped HIV GFP reporter virus was produced by polyethyleneimine (PEI) transfection of 293T cells with cytomegalovirus (CMV)-VSV G plasmid and wild-type or CA mutant proviral plasmid, as previously described (7). Viral stocks were tested for infectivity on HeLa P4R5 cells and assayed using by flow cytometry using an Accuri C6 flow cytometer (BD Biosciences). Virus containing the fluid-phase interdomain GFP (iGFP) marker was generated by PEI transfection of 293T cells with HIV Gag-iGFP ΔEnv, mCherry-Vpr, and CMV-VSV G plasmids. GIG virus for imaging experiments was produced by PEI transfection of 293T cells with 6 μg R9 Δenv proviral vector, 5 μg GIG expression vector, and 4 μg CMV-VSV G plasmid. The relative amounts of R9 Δenv proviral vector and GIG expression vector were adjusted in order to yield virus that was maximally infectious and with infectivity similar to that of viral preparations without GIG addition.
Cell lines and pharmaceuticals.
293T cells were obtained from the American Type Culture Collection. HeLa P4R5 cells were obtained from the AIDS Research and Reference Reagent Program. OMK cells and CHO-K1 pgsA745 cells were a gift from P. Bieniasz. All cell lines were maintained at 37°C and 5% CO2 and cultured in Dulbecco's modified Eagle medium (Cellgro), 10% fetal bovine serum (Gibco), and 1% penicillin-streptomycin-glutamine (Gibco). CHO cell medium also contained 1% minimal essential medium-nonessential amino acid solution (MEM NEAA) (Gibco). Cyclosporine (Calbiochem) was prepared in ethanol and used at a final concentration of 2.5 μM. PF74 was a gift from Chris Aiken, prepared in dimethyl sulfoxide (DMSO), and used at a final concentration of 10 μM. BI2 was a gift from Chris Aiken, prepared in DMSO, and used at a final concentration of 20 μM. Nevirapine was prepared in DMSO and used at a final concentration of 5 μM. Bafilomycin A was prepared in DMSO and used at a final concentration of 20 nM.
Capsid integrity assay.
The capsid integrity assay was conducted as previously described (24). HeLa cells stably expressing hemagglutinin (HA)-tagged recombinant human TRIM5α (rhTRIM5α) were seeded onto fibronectin-treated glass coverslips and exposed to labeled virus in the presence of DEAE-dextran, MG132, and either 10 μM PF74, 20 μM BI2, or DMSO for 6 h. Cells were then fixed with a 3.7% formaldehyde-PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] solution and stained using a rabbit anti-HA antibody (Sigma-Aldrich) and an AMCA-tagged anti-rabbit secondary antibody (Jackson ImmunoResearch). Cells were imaged on a DeltaVision wide-field microscope. Images were analyzed using an Interactive Data Language (IDL) program that first identified mCherry-labeled viral complexes which localized to TRIM bodies and then scored the presence of GFP. Twenty images for each condition were analyzed in 3 independent experiments. The data presented in Fig. 2 are the averages from three independent experiments, with error bars denoting standard error of the mean.
FIG 2.
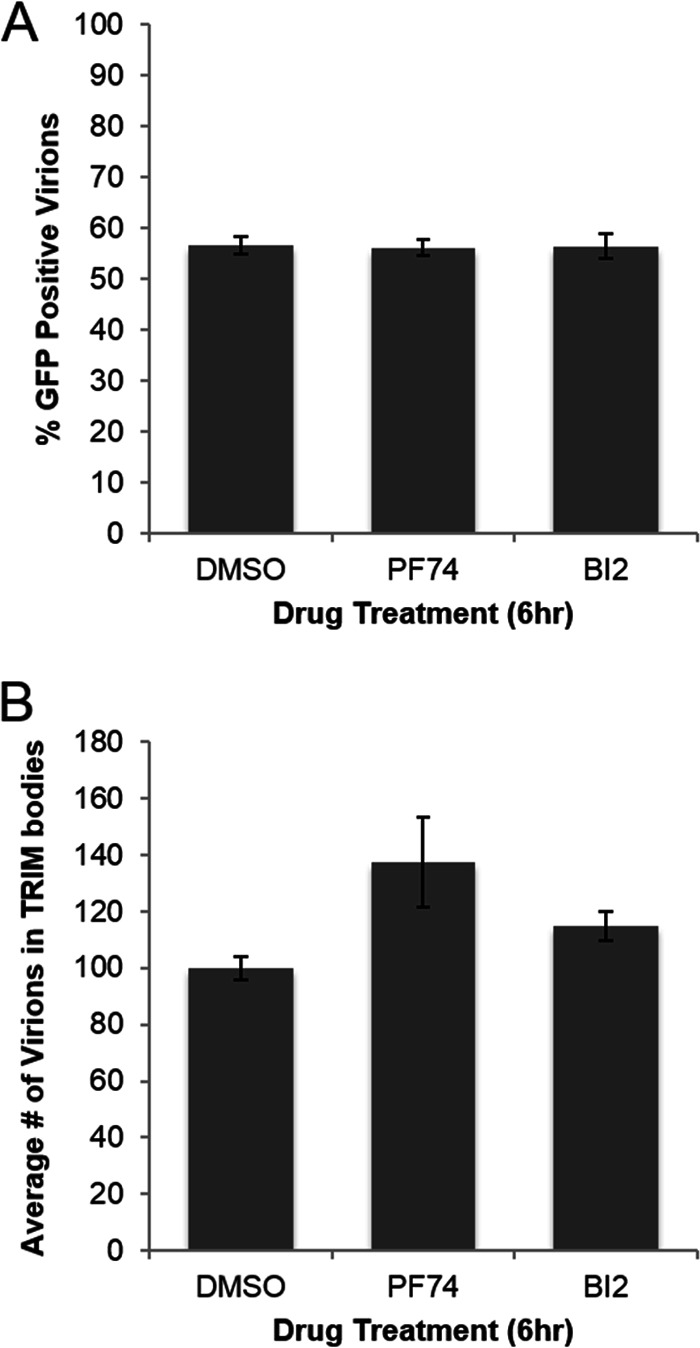
Effect of PF74 and BI2 on HIV capsid integrity. VSV G-pseudotyped virus labeled with mCherry-VPR and a fluid-phase GFP marker was incubated with cells expressing HA-tagged TRIM5α in the presence of PF74, BI2, or the carrier control DMSO. mCherry-Vpr labeled virus localizing to TRIM bodies was counted and scored for GFP signal in 20 images from each condition. The percentage of viral complexes retaining GFP (A) and total number of viral complexes localizing to TRIM bodies (B) averaged from three independent experiments are presented, with error bars denoting standard error of the mean.
CsA washout assay.
The CsA washout assay was conducted as previously described (7, 10). Briefly, OMK cells were plated in 96-well dishes, with each experimental or control reaction performed on triplicate wells. Cells were spinoculated with VSV G-pseudotyped HIV-GFP in the presence of CsA and 10 μg/ml Polybrene for 1.5 h at 16°C. The inoculation medium was then exchanged for warm medium, and CsA was washed out of the time zero reaction by medium exchange. Washout continued at various times postinfection. The negative control was ethanol washout. At 2 days postinfection, cells were harvested with 100 μl trypsin and fixed by addition of 100 μl fixative (1× phosphate-buffered saline [PBS]–10% formaldehyde; 4:1). The percentage of GFP-positive cells was determined by flow cytometry using the Accuri C6 flow cytometer (BD Biosciences) and averaged for each triplicate reaction, and the standard error was calculated. The data presented in Fig. 1A and 4A are from a representative experiment of three experiments, with error bars denoting standard error of the mean. The repeat experiments were done within the same week to avoid variability in cell sensitivity to infection. The half-life of uncoating was determined by a best-fit line and averaged from three independent experiments.
FIG 1.
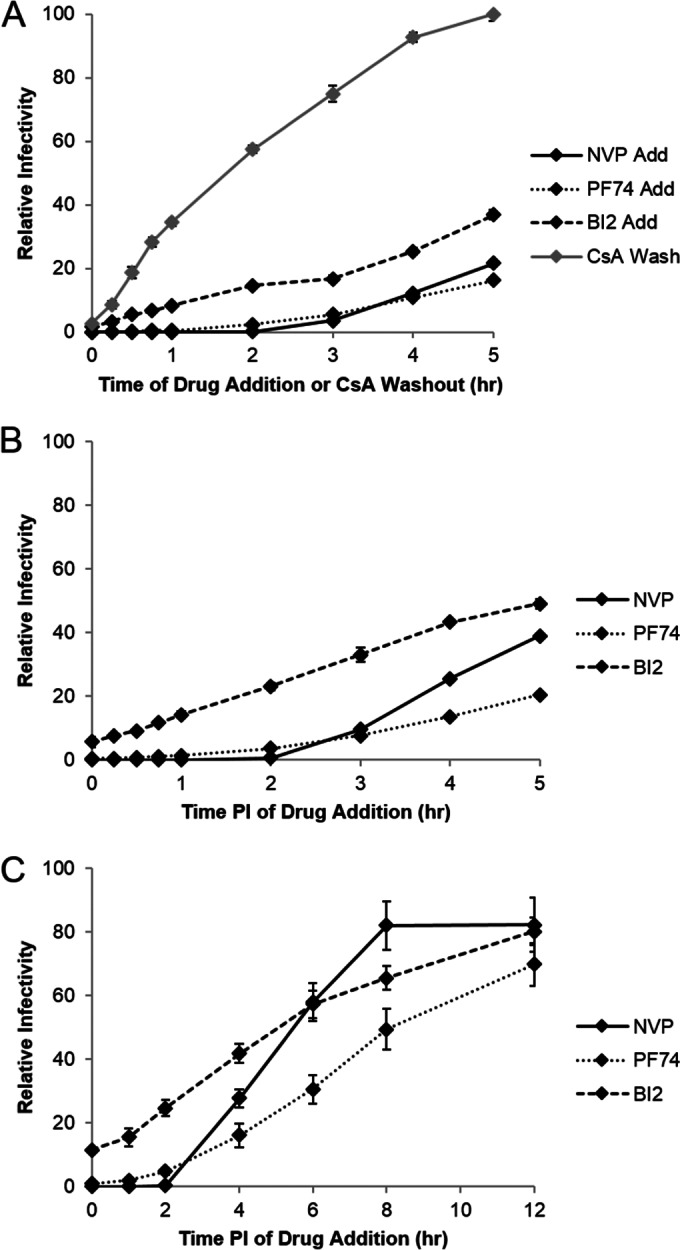
Comparison of HIV uncoating and drug susceptibility. (A) To examine the relationship between drug susceptibility and uncoating, the CsA washout assay was performed in parallel with PF74 and BI2 drug addition assays in OMK cells. Nevirapine (NVP) addition was used as a drug control. Data were normalized to maximal infectivity for the CsA washout assay and to the no-drug control for the drug addition assays. Shown are results from a representative experiment, with error bars representing standard error of the mean. (B) Susceptibility of HIV to PF74 and BI2 over time was tested in HeLa P4R5 cells, which lack TRIM-CypA. Data were normalized to the no-drug control. Shown are results from an average of four experiments, with error bars representing standard error of the mean. (C) HIV susceptibility to PF74 and BI2 was tested up to 12 h in HeLa P4R5 cells. Data were normalized to the no-drug control. Shown are results from an average of three experiments, with error bars representing standard error of the mean.
FIG 4.
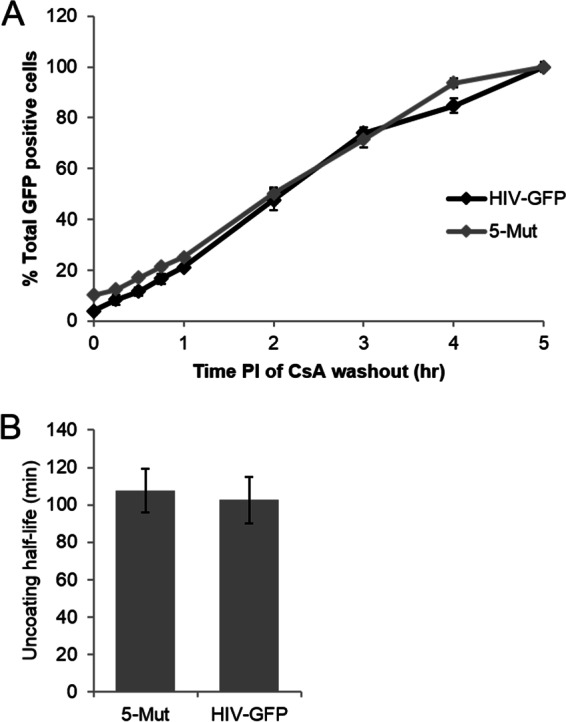
Uncoating kinetics of the CA mutant virus 5Mut. The uncoating of the wild type (HIV-GFP) and the CA mutant virus 5Mut was tested using the CsA washout assay. (A) 5Mut virus uncoats with kinetics similar to that of wild type. Shown are results from a representative assay, with error bars denoting standard error of the mean. (B) 5Mut virus has a half-life of uncoating similar to that of the wild type. Shown is the average half-life of uncoating, calculated from three independent experiments, with error bars denoting standard error of the mean.
Drug addition assay.
OMK or HeLa P4R5 cells were plated in 96-well dishes and spinoculated with HIV-GFP or CA mutant virus as in the CsA washout assay. Each experimental condition or control reaction was performed on triplicate wells. When inoculation medium was exchanged for warm medium, medium containing drug (and CsA for the experiment with OMK cells) was added to the time zero reaction. Drug addition continued in the same manner at various times postinfection. Controls included carrier (DMSO)-only treatment and continuous treatment with drug. Cells were harvested as in the CsA washout assay. The percentage of GFP-positive cells was determined by flow cytometry using the Accuri C6 flow cytometer (BD Biosciences) and averaged for each triplicate reaction, and standard error was calculated. For each experimental condition or control reaction, the percentage of GFP-positive cells was averaged from the triplicate wells. For each virus, the data were then normalized by setting the percentage of GFP-positive cells in the DMSO (no-drug) control to 100%. A dilution of each virus was used to achieve ∼30% infectivity in the DMSO control. This experiment was repeated three or four times, as noted in the figure legends. These repeat experiments were done within the same week to avoid variability in cell sensitivity to infection. The data presented in Fig. 1B and C and 5 are averages from these independent experiments, with error bars representing standard error of the mean. The data presented in Fig. 1A are from a representative experiment of three independent experiments, with error bars denoting standard error of the mean.
FIG 5.

Effect of CA mutations on drug susceptibility. The drug addition assay with PF74 (A and B), BI2 (C and D), or nevirapine (E and F) was performed over an extended time course with CA mutant viruses N74D, E45A, Q63/67A, A92E, and 5Mut. For each virus, data were normalized to the no-drug control. Shown are results from an average of three experiments, with error bars representing standard error of the mean.
Real-time PCR.
HeLa P4R5 cells were infected with HIV-GFP for 16 h in the presence of various concentrations of PF74, BI2, nevirapine, or DMSO carrier control. DNA was harvested using the DNeasy blood and tissue kit (Qiagen) and digested with DpnI to remove residual plasmid DNA. Real-time PCR was performed on 0.5 μg of each DNA sample using the iCycler iQ real-time PCR detection system (Bio-Rad) with early and late reverse transcription (RT) primers (25–27). The number of cells in each sample was determined by real-time PCR analysis of genomic β-actin DNA (26). Products were quantified using a standard curve generated by serial dilution of HIV-GFP or β-actin plasmid. The numbers of early and late RT products per cell were calculated and normalized using the DMSO control. This experiment was repeated three times. The data presented in Fig. 3 are from a representative experiment, with error bars representing standard error of the mean.
FIG 3.
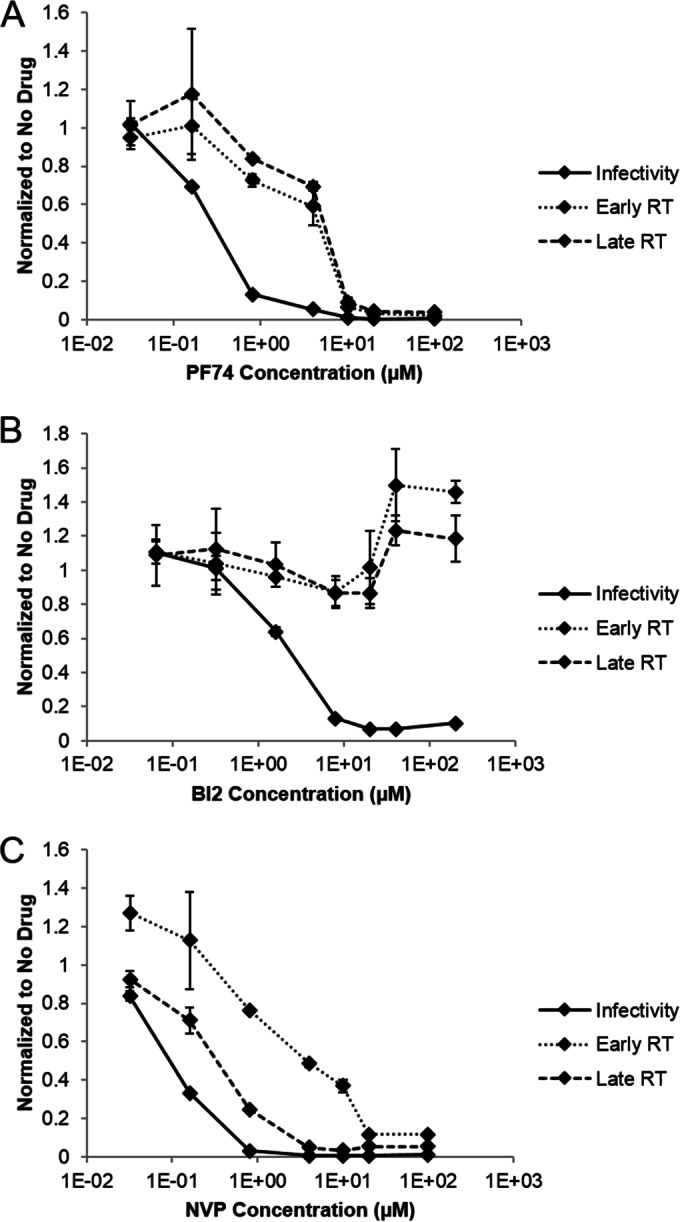
Effect of PF74 and BI2 on reverse transcription and infectivity. Real-time PCR was used to examine the effect of different concentrations of PF74 (A), BI2 (B), or nevirapine (C) on the process of reverse transcription in HELA P4R5 cells. An infectivity assay was also performed with the same drug concentrations. Data were normalized to the no-drug control then plotted on the same axis. Shown are results from a representative experiment from three independent experiments, with error bars representing standard error of the mean.
CA visualization by high-resolution SIM.
Two days prior to infection, CHO A745 cells were seeded on glass coverslips in a 24-well dish at an initial density of 6 × 104 cells per well. Cells were infected with integrase-GFP-labeled virus with 10 μg/ml DEAE-dextran for 6 h, or infection was semisynchronized by incubation with virus for 2 h followed by medium change and then additional incubation for an additional 2 or 4 h. Cells incubated with virus for 2 h were reserved for either bafilomycin A- or secondary-antibody-only controls, while an additional coverslip was not infected. Diluted virus stocks were also spinoculated on glass at 1,200 × g for 90 min. All experimental and control samples were fixed with 3.7% formaldehyde in PIPES buffer for 5 min. Coverslips were then stained for 1 h using rabbit lamin B1 antibody at 1:1,000 (Abcam) and human antibody 71-31 for CA at 1:2,000 (AIDS Reagent Repository). Coverslips were then exposed to sequential secondary antibody staining for 30 min each with Alexa Fluor 405-labeled anti-rabbit (Abcam) and Alexa Fluor 647 anti-human (Jackson ImmunoResearch) antibodies. Coverslips were then mounted on slides using ProLong Gold antifade reagent (Invitrogen). Samples were imaged on a GE OMX superresolution structured illumination microscope (SIM), and images were reconstructed using the GE-API SoftWorx package. Background levels for each channel were chosen based on maximum values observed in the secondary-antibody-only (lamin and 71-31) and no-virus (integrase-GFP) controls. Images were examined with the SoftWorx imaging suite to identify integrase-GFP-labeled viral complexes localized to the nuclear membrane. The position of integrase-GFP-labeled viral complexes relative to the nuclear membrane was confirmed by volume projection. Particles were identified as nuclear if they were located completely inside the nucleus or overlapped by less than 25% with the interior side of the nuclear lamin stain. Particles were identified as localizing to the nuclear membrane if they overlapped with the lamin stain. In an image, either all cytoplasmic particles were counted or a minimum of 20 particles per image were randomly chosen with the CA channel turned off. Once a particle's location was categorized, the CA stain channel was made visible, and the intensity of CA staining was analyzed using the Data Inspector tool in a 4- by 4-pixel square specified to contain the entirety of 71-31 signal. The total CA intensity was recorded for the brightest z section associated with the integrase-GFP-labeled viral complexes. In each experiment, a minimum of 10 images were examined for each experimental condition. In the five images for the bafilomycin control, 20 cytoplasmic particles were randomly chosen with the CA channel turned off. The CA channel was then made visible, and each viral complex was scored for CA intensity. For the virus-on-glass control, 20 viral particles were randomly chosen and scored for CA intensity from each of 5 images, similar to the procedure for the bafilomycin control. To determine the background intensity in the CA channel, 20 virions were chosen randomly from each of 5 images in the secondary-antibody-only control, and the total CA intensity of a 4- by 4-pixel square was recorded. This imaging experiment was repeated 3 times. To compare the CA intensities from these 3 experiments, the CA intensity of each viral complex was normalized by dividing by the median CA intensity of the appropriate bafilomycin A control.
CA visualization by deconvolution microscopy.
Two days prior to infection, HeLa cells stably expressing HA-CPSF6 were seeded on glass coverslips in a 24-well dish at an initial density of 6 × 104 cells per well. Cells were infected with integrase-GFP-labeled virus with 10 μg/ml DEAE-dextran for 7.5 h. After fixation, cells were stained for CA and lamin as described above for the high-resolution structural imaging experiments. Control samples included a secondary-antibody-only control and a sample not exposed to virus. Coverslips were imaged on a DeltaVision wide-field microscope and analyzed using the SoftWorx imaging suite to identify integrase-GFP-labeled viral complexes localized to the nucleus.
RESULTS
Drug susceptibility in relation to uncoating.
To further characterize the role of CA in HIV-1 replication, we used two capsid binding drugs, PF74 and BI2. Both drugs have been shown to inhibit HIV-1 replication, and drug resistance mutations map to CA (28–30). Interestingly, these drugs have been suggested to have opposing effects on the viral capsid despite binding to a similar site on CA (28–32). PF74 has been proposed to induce rapid capsid disassembly, as treatment with this drug blocks the accumulation of viral cDNAs, increases the rate of uncoating in vitro, and decreases pelletable CA from infected cells (28, 29, 32). Contradicting these data, PF74 has also been shown to stabilize CA-NC assemblies in vitro (32, 33). BI2 does not affect reverse transcription and also stabilizes CA-NC complexes in vitro (30, 33).
To determine the relative timing of PF74 and BI2 action in relation to uncoating, we performed a drug addition assay in parallel with a CsA washout assay in OMK cells, as described above. In this particular CsA washout assay, approximately 50% of viral complexes uncoated by 1.5 h postinfection (Fig. 1A). In the parallel drug addition assay, OMK cells were synchronously infected with HIV-GFP under the same experimental conditions as in the CsA washout assay. However, CsA was maintained in the medium, and 10 μM PF74 or 20 μM BI2 was added at times corresponding to those used in the CsA washout assay. Cells were harvested at 2 days postinfection, and the percentage infected cells was determined at each time point of drug addition. These data were normalized by setting the percentage of GFP-positive cells in the no-drug control reaction to 100%. At 1.5 h postinfection, where 50% of the virus had uncoated in the CsA washout assay, PF74 and BI2 were able to inhibit the infectivity of 95% of the virus (Fig. 1A). Even at 4 to 5 h, where the uncoating curve in the CsA washout assay leveled off, indicating that uncoating was complete, 65 to 85% of the virus was still sensitive to inhibition by the capsid-targeting drugs (Fig. 1A). Therefore, virus is susceptible to the inhibitory action of PF74 and BI2 for hours after uncoating is completed as determined by the CsA washout assay.
Cyclophilin A (CypA) is a cellular protein that binds to the capsid and is required for optimal HIV-1 replication in a cell type-specific manner, although the mechanistic details of its function remain unclear (34–39). In addition, CypA has been reported to enhance the antiviral activity of PF74 in HeLa cells (29). In the drug addition assay in OMK cells, CsA was present to inhibit restriction by TRIM-CypA, which would also prevent the binding of cellular CypA to the viral capsid. Therefore, we repeated the drug addition assay in HeLa P4R5 cells in the absence of CsA treatment in order to separate the effects of CsA from drug susceptibility. Similar to the results in OMK cells, the PF74 and BI2 inhibited infectivity of 80 to 95% of the virus at 2 h postinfection, and by 5 h postinfection only 50% (BI2) or 20% (PF74) of the virus was resistant to inhibition (Fig. 1B). These data suggest that CypA binding or other CsA-sensitive effects do not change drug susceptibility in OMK cells. However, to better compare with previous experiments, we performed all subsequent experiments in HeLa P4R5 cells.
As at least 50% or more of the virus was susceptible to drug treatment at 5 h postinfection, we next performed drug addition experiments in HeLa P4R5 cells over a longer time scale, including a nevirapine control to determine when reverse transcription was complete. Virus became resistant to BI2 inhibition earlier than PF74 inhibition, with 50% of the virus susceptible to BI2 at 5 h postinfection, compared to 8 h for PF74. For both drugs, ∼80% of the virus was resistant to drug treatment by 12 h postinfection (Fig. 1C). A parallel drug addition assay with the nonnucleoside reverse transcriptase inhibitor nevirapine was used to reveal the progression of reverse transcription. Consistent with previous data, all virus became resistant to inhibition of reverse transcription by nevirapine by 8 h postinfection, indicating that reverse transcription was completed by 8 h postinfection (7, 9) (Fig. 1C). These experiments reveal that the virus can remain sensitive to PF74 and BI2 after reverse transcription is completed.
Virus was susceptible to the action of BI2 and PF74 after the completion of reverse transcription, hours past when uncoating was observed in the CsA washout assay. These data are not consistent with PF74 and BI2 affecting only uncoating of HIV-1 but suggest that these drugs may be targeting a downstream step in viral replication mediated by CA. Therefore, we next examined whether these compounds could affect capsid integrity in our system or could influence reverse transcription and whether the postuncoating effect we observed in the drug addition assay was dependent on an interaction with CA.
Effect of PF74 and BI2 on conical capsid integrity in HIV-infected cells.
PF74 has previously been suggested to induce rapid capsid disassembly in vitro and decrease pelletable capsid from CrFK cells (29). Therefore, we tested PF74 and BI2 for a similar effect in infected cells. To determine whether PF74 or BI2 can disrupt the conical capsid in vivo, we performed the recently developed capsid integrity assay (24). In this assay, virus is made using the iGFP proviral construct in which the fluorescent protein GFP is cloned into Gag between matrix and capsid and flanked by proteolytic cleavage sites (40). During viral maturation, GFP is cleaved out of Gag and can serve as a fluid-phase marker of the virus. Importantly, a subset of the GFP fluid-phase marker is trapped within the interior of the conical capsid and can be utilized to evaluate the integrity of the conical capsid via the ability of the capsid to retain this fluid-phase marker. This arrangement ensures that in the cytoplasm of the cell, virus with an intact capsid will retain the fluid-phase GFP label, whereas any virus with a conical capsid that is not an intact fullerene cone will lose the fluid-phase GFP protein.
For the capsid integrity assay, iGFP virus was colabeled with Cherry-Vpr to identify viral complexes. HeLa cells that express an HA-tagged version of rhTRIM5α were continuously infected with labeled virus in the presence of MG132 to capture the proteasome-independent intermediate of TRIM restriction (26, 41). After 6 h, cells were fixed, stained for HA, and imaged. The number of Cherry-Vpr-labeled viral complexes that localized to rhTRIM5α cytoplasmic bodies was counted, and each viral complex was characterized for the presence of GFP. In this experiment, localization to cytoplasmic bodies was used as a marker for viral fusion, as we have previously shown that only productively fused virus can localize to these structures (41).
We conducted the TRIM capture assay in the presence of PF74, BI2, or a DMSO carrier control. PF74 or BI2 did not alter the percentage of GFP-positive viral complexes captured in rhesus TRIM5a cytoplasmic bodies, suggesting that these compounds do not affect the integrity of the viral capsid (Fig. 2A). However, only viral complexes that localize to cytoplasmic bodies are counted in this assay, and TRIM can bind only to multimerized CA. A drug that induces very rapid capsid disassembly, as is proposed for PF74, may be able to facilitate disassembly before the viral complex could associate with TRIM protein. However, treatment with PF74 or BI2 did not decrease the number of viral complexes localizing to cytoplasmic bodies (Fig. 2B). The slight increase in the number of viral complexes that associated with TRIM bodies in the presence of PF74 and BI2 was not statistically significant. Therefore, PF74 and BI2 do not seem to affect capsid integrity in infected cells, suggesting that they do not induce rapid uncoating.
Effect of PF74 and BI2 on reverse transcription and infectivity.
In the drug addition assays, susceptibility of virus to PF74 and BI2 resembled reverse transcription kinetics more closely than uncoating kinetics (Fig. 1). Therefore, we examined the effect of different concentrations of PF74 and BI2 on the process of reverse transcription, using nevirapine as a negative control (Fig. 3C). Cells were infected in the presence of drug, and the extent of reverse transcription after 16 h was determined by quantitative PCR. Early and late RT products were inhibited to similar extents by different drug concentrations of PF74 (29) (Fig. 3A). Interestingly, at lower concentrations than what was used in the drug addition assays, PF74 was able to inhibit viral infectivity without an effect on reverse transcription. A similar observation of the effect of differing concentrations of PF74 on reverse transcription has been made in a recent study (31). In contrast, BI2 did not affect the production of early or late RT products at any concentration tested, which is consistent with other studies (30) (Fig. 3B). In addition, a concentration of BI2 which completely inhibited HIV replication without severe effects on cell viability could not be found, suggesting that a subset of viral complexes may not be susceptible to BI2. Given that PF74 and BI2 exhibit similar kinetics of activity, these data suggest that these drugs do not exclusively inhibit HIV infectivity in HeLa P4R5 cells by interfering with reverse transcription.
PF74 and BI2 target the HIV-1 capsid.
PF74 and BI2 have been shown to bind to CA in isothermal calorimetry and X-ray crystallography studies (28, 30–32). To determine whether the post-reverse transcription effect of these drugs on viral infectivity involves direct binding to CA, we next examined the effect of CA mutations on drug susceptibility kinetics, using nevirapine addition as a control for progression of reverse transcription (see Fig. 5E and F). The mutants N74D, E45A, and Q63/67A uncoat more slowly than wild type in the CsA washout assay to various degrees (6, 7). Q63/67A displays the greatest delay in uncoating compared to that of wild type, the N74D mutation yields the smallest effect on uncoating kinetics, and E45A is intermediate between these two mutants. The mutant A92E uncoats faster than the wild type in the CsA washout assay (6). Finally, we found that the PF74-resistant mutant 5Mut uncoated with kinetics similar to that of wild type in the CsA washout assay (Fig. 4). The 5Mut mutation has previously been found to increase the amount of pelletable capsid from infected cells, suggesting that this mutant uncoats more slowly than the wild type (29). This discrepancy may be due to the differences in the two assays, most notably that the CsA washout assay is based on viral infectivity.
After PF74 treatment, N74D and A92E showed drug susceptibility kinetics similar to that of wild type, whereas E45A and Q63/67A were more susceptible at later time points than wild type, to similar degrees (Fig. 5A and B). 5Mut displayed unique susceptibility kinetics, where approximately 50% of the virus was initially resistant to PF74 and this percentage increased rapidly over 2 h (Fig. 5B). Wild-type and N74D viruses had similar BI2 susceptibility kinetics. However, at early time points, E45A and Q63/67A had increased overall resistance to BI2 compared to wild type, and at later time points, they had a susceptibility similar to that of wild type (Fig. 5C and D). A92E and 5Mut displayed an increased overall resistance to BI2 compared to that of wild type at all time points but showed a similar decrease in drug susceptibility over time (Fig. 5D). Overall, a consistent change in drug susceptibility kinetics relative to uncoating kinetics was not observed across this panel of CA mutants. Therefore, these data suggest that PF74 and BI2 do not affect the process of uncoating in infected cells. However, we observed differences in overall BI2 susceptibility and the timing of PF74 susceptibility with different CA mutations. Therefore, these data also reveal that the ability of PF74 and BI2 to inhibit a post-reverse transcription step of HIV replication is a direct consequence of binding to the CA protein.
A subset of CA protein remains associated with nuclear HIV complexes.
PF74 and BI2 bind directly to a similar surface on CA (28, 30–32). However, we observed susceptibility to each drug for hours past when uncoating is observed in the CsA washout assay and after reverse transcription is complete. Collectively, these data support the hypothesis that some CA protein remains associated with the viral complex after the dissociation of the conical capsid structure. To test this hypothesis using a different methodology, we assayed for the presence of CA in viral complexes labeled with integrase-GFP using superresolution structural illumination microscopy (SIM), which allows for precise localization of viral complexes in the cytoplasm and nuclei of infected cells. For studies of viral complexes within infected cells, it is critical to identify the complexes that have been introduced into the cytoplasm after productive fusion. Because of the labeling approach utilized here, we cannot determine if cytoplasmic complexes are pre- or postfusion. Instead, we decided to utilize nuclear localization as the filter of viral complexes that have fused and are advancing toward integration. It is agreed in the field that only HIV complexes that have entered the cytoplasm after fusion and have shed their conical capsids (uncoated) enter the nucleus. Therefore, we specifically analyzed nuclear viral complexes for associated CA which would remain after uncoating.
In these imaging experiments, we used a virus labeled with integrase-GFP to ensure that the viral complex could be followed through the early steps of viral replication and into the nucleus (22, 42). This Gag-protease site-integrase-GFP was derived from a previous integrase-GFP construct from the Cereseto lab (22) by cloning into a Gag expression construct. The resultant Gag-integrase-GFP (GIG) fusion protein is cleaved by the viral protease during maturation to liberate the integrase-GFP protein from the Gag polyprotein. The previous integrase-GFP vector showed poor infectivity; however, virus made using a mixture of the GIG expression plasmid and an R9Δenv proviral plasmid had typical levels of viral infectivity, revealing that integrase-GFP could be incorporated without inhibiting infectivity. Because this virus was made using a mixed transfection, not all virions produced contained the same levels of the integrase-GFP label. To increase our ability to assay fused virions in the cytoplasm of cells, we used CHO A745 cells, which have been shown to allow for receptor-dependent binding and efficient fusion by VSV G-pseudotyped virus (15). We also focused our analysis on nuclear viral complexes which would have undergone productive viral fusion and uncoating.
For an imaging experiment, CHO A745 cells were plated on glass coverslips and infected with integrase-GFP-labeled virus in the presence of DEAE-dextran. In order to increase the chance of observing nuclear viral complexes, either virus was allowed to continuously infect cells for 6 h or infection was semisynchronized by removal of the inoculation medium after 2 h followed by an additional 2 or 4 h of incubation. After fixation, coverslips were stained for CA using the 71-31 monoclonal antibody, and the nuclear membrane was visualized using an antibody against lamin. The monoclonal antibody 71-31 was utilized because we empirically determined it had superior sensitivity to stain CA in viral intracellular complexes. Bafilomycin A was used as a control to prevent viral fusion and provide a range of initial antibody staining levels present in cells (Fig. 6C). Similarly, virus spun onto glass was used to determine the initial range of capsid staining levels of intact virions (Fig. 6D). Background levels for each channel were determined from a coverslip exposed only to secondary antibody for 71-31 and lamin and a coverslip that was not exposed to virus (Fig. 6E and F). All experimental and control coverslips were then imaged on a structured illumination superresolution OMX microscope. Although there were samples with continuous infection and semisynchronized infection, we did not observe any differences in the intensity of the CA signal between these samples, so the data were grouped together after analysis (Fig. 7).
FIG 6.

Association of CA with viral complexes in cells. (A and B) High-resolution structural imaging was used to detect the presence of CA localizing to viral complexes in CHO cells. CHO cells were exposed to integrase-GFP-labeled virus (green), fixed, and stained for CA (red) and lamin to visualize the nuclear membrane (blue). Shown are examples of viral complexes localizing to the nucleus in a single z section (left), with a highlighted box presented as a volume projection in the z plane (right). The surface of the nucleus closest to the coverslip glass is labeled. (C to F) Control reactions for the high-resolution structural imaging experiment included an experimental sample exposed to bafilomycin A (C), integrase-GFP-labeled virus spun onto glass (D), an experimental sample stained only with secondary antibodies against human and rabbit (E), and a sample not infected with integrase-GFP-labeled virus (F). Shown is a single z section for each control. These images are the corresponding controls for the experiment presented in Fig. 7A and are displayed at the channel intensities used to analyze images for that experiment. (G and H). Deconvolution microscopy was used to detect the presence of CA localizing to viral complexes in HeLa cells. HeLa cells were exposed to integrase-GFP-labeled virus (green), fixed, and stained for CA (red) and lamin to visualize the nuclear membrane (blue). Shown is a single z section (left), with a highlighted box magnified to the right of the image.
FIG 7.
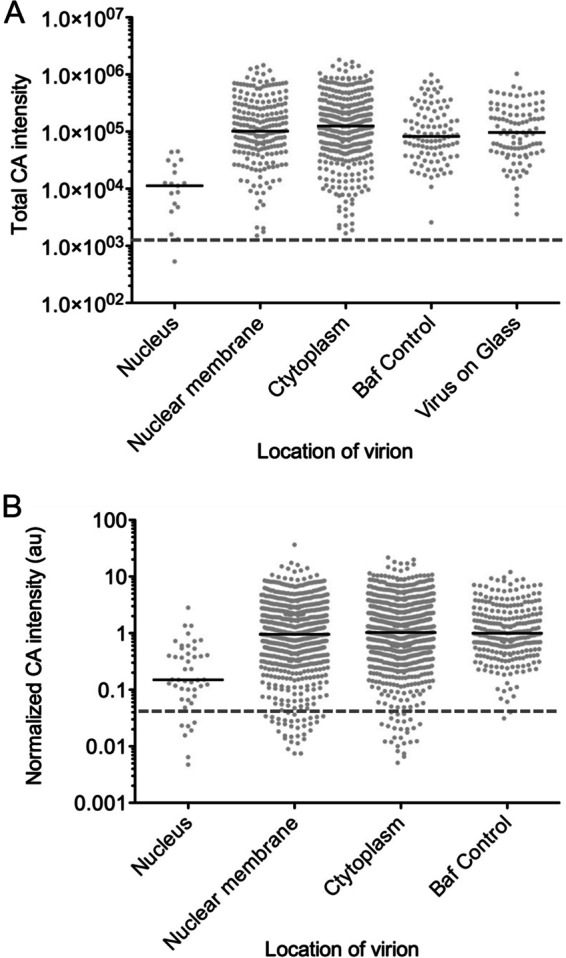
Quantitation of CA signal associated with viral complexes. High-resolution structural imaging was used to detect the presence of CA localizing to viral complexes in CHO cells, and the intensity of the CA signal associated with each viral complex was determined. (A) Scatter plot of total CA intensity from a representative experiment, with each dot representing one viral complex counted. CA intensity was determined across a 4- by 4-pixel region for each viral complex. Localization of the viral complex was determined relative to the nuclear membrane. Baf control and virus on glass are virions randomly chosen in the bafilomycin A and virus-on-glass controls, respectively. The black bar is the median of each column. The dashed line is the level of background in the CA staining channel as assayed from the secondary-antibody control. (B) Scatter plot of total CA intensity from 3 independent experiments after normalization using the median CA intensity of the bafilomycin control for each experiment. CA intensity was determined across a 4- by 4-pixel region from each viral complex. Localization of the viral complex was determined relative to the nuclear membrane. Baf control are cytoplasmic virions randomly chosen in the bafilomycin A control. The black bar is the median of each column. The dashed line is the level of background in the CA staining channel as assayed from the secondary-antibody control.
After SIM image reconstruction, we quantified the CA of viral complexes that localized inside the nucleus, at the nuclear membrane, and in the cytoplasm. Three-dimensional (3D) SIM reconstruction allowed for the precise localization of viral complexes relative to the nuclear membrane as delineated by the lamin stain (Fig. 6A and B). A viral complex was scored as inside the nucleus if localized entirely inside the lamin stain or if it overlapped less than 25% with lamin stain on the interior face of the nuclear membrane after examination in 3 dimensions. Although the distortion of signal in the z dimension is minimal with SIM imaging, there is still elongation of signal in the z plane which can make a nuclear complex appear to associate with the nuclear membrane as identified by lamin staining. Viral complexes were scored as localizing to the nuclear membrane if they overlapped with the lamin stain by more than 25%. Cytoplasmic viral complexes localized distinct from the lamin stain. Once localization was determined, viral complexes containing integrase-GFP were examined for CA based on 71-31 staining, and the total intensity of CA stain was recorded. In the bafilomycin control, individual cytoplasmic integrase-GFP-positive viral complexes were randomly chosen and scored for CA intensity. For the virus-on-glass control, integrase-GFP virions were randomly chosen and scored for CA intensity.
Utilizing this approach, we were able to identify integrase-GFP-positive viral complexes that localized inside the nucleus and stained for CA. The level of CA staining in these complexes was usually low but was detectable above background levels as determined by the secondary-antibody-only controls (Fig. 6). For reference, among the nuclear viral complexes that stained for CA, Fig. 6B depicts one of the brightest complexes, while Fig. 6A represents the more commonly observed complexes associated with lower CA intensity. Importantly, in the bafilomycin A control we did not observe any viral complexes localizing inside the nucleus (Fig. 6C). When comparing CA intensities for each category of virus localization, we observed that viral complexes localizing inside the nucleus had a lower median CA intensity than those complexes present at the nuclear membrane, in the cytoplasm, or in the bafilomycin A and virus-on-glass controls (Fig. 7A). The median CA intensities of viral complexes present at the nuclear membrane, in the cytoplasm, and in controls were similar, likely due the presence of a significant number of unfused virions in the infected cells. Therefore, we examined the range of CA intensities associated with each class of viral complex in the assay (Fig. 7A). From this comparison it was clear that the population of viral complexes with more intense CA staining was absent from nuclear complexes, compared to viral complexes localized to the nuclear membrane and cytoplasm and in controls. In addition, the population of viral complexes with the lowest associated CA stain was abundantly identified under conditions where individual viral complexes could fuse with the target cell but not in controls where virions remained intact. To provide an unbiased measure of the background level in the CA stain channel, we determined the average intensity of CA staining from 100 randomly chosen integrase-GFP viral complexes in the secondary-antibody control. When this background level of staining is represented as a dashed line (Fig. 7A), it appears that the vast majority of viral complexes contain some level of CA protein, including those localized to the nucleus.
Given the relatively low number of viral complexes that we were able to detect unequivocally in the nucleus, we repeated the imaging experiment three times. The range of CA intensities did vary from experiment to experiment, likely due to differences in antibody staining efficiency and cell background. In order to compare the CA intensities from all three experiments, the CA intensity of each viral complex was normalized by dividing it by the median CA intensity of the appropriate bafilomycin A control (Fig. 7B). From these data, we found that viral complexes localizing to the nucleus had on average an ∼6-fold-lower level of CA signal than complexes at the nuclear membrane, in the cytoplasm, or in controls (Fig. 7B). When looking at the range of CA intensities, it is apparent that viral complexes which intensely stain for CA (those that have CA intensity equal to or greater than the median intensity in the bafilomycin control) can readily be found at the nuclear membrane, in the cytoplasm, and in the bafilomycin control but are not found in the nucleus. Therefore, it seems that a lower level of CA is a characteristic of viral complexes that have localized to the nucleus. We also normalized the level of background in the CA stain channel for each experiment using the secondary-antibody control. As seen by the dashed line in Fig. 7B, almost all of the viral complexes contain some CA signal. Finally, we tested whether we could detect low levels of CA associated with nuclear viral complexes in a different cell line. We repeated the experiment described above in HeLa cells infected continuously for 7.5 h and then imaged samples using deconvolution microscopy. Similar to our observations in CHO cells, we were able to detect decreased levels of CA associated with nuclear viral complexes in HeLa cells (Fig. 6G and H). Collectively, our imaging experiments suggest that viral complexes inside the nucleus contain a low but detectable level of CA.
DISCUSSION
Here we have attempted to reconcile our model of early uncoating with the observations that mutations in CA affect steps of viral replication that are nucleus related, including nuclear import pathway selection, infection of nondividing cells, and integration site selection (14, 16–21). First, we examined the effect of capsid-targeting drugs on HIV replication. In OMK and HeLa P4R5 cells, HIV was susceptible to PF74 and BI2 inhibition for hours past when the majority of uncoating was observed in the parallel CsA washout assay (Fig. 1 and 5) and even after the completion of reverse transcription. A similar timing of susceptibility to PF74 was recently reported by Price et al. using both high (30 μM) and low (1 μM) concentrations of the drug in HeLa cells (31). Importantly, HIV infection remained sensitive to inhibition by both drugs after reverse transcription was completed (Fig. 1C). The CsA washout assay is an indirect measure of uncoating, relying on the ability of the restriction factor TRIM-CypA to detect and restrict the infectivity of viral complexes (7, 11). However, we observed similar uncoating kinetics in a fluorescence microscopy-based uncoating assay in which CA is directly detected in infected cells by antibody staining (7, 14). A similar timing of uncoating and effect of reverse transcription on uncoating have also been observed in biochemical uncoating assays (8, 15). Therefore, the CsA washout assay provides a good monitor of uncoating in infected cells. These parallel CsA washout and drug susceptibility assays then reveal that PF74 and BI2 inhibit infection by targeting a CA-directed activity that takes place after uncoating as defined by the loss of the conical capsid structure.
To look for an effect of these drugs on uncoating, we performed a capsid integrity assay and examined the drug susceptibility of CA mutant viruses (24). PF74 has been proposed to induce rapid capsid disassembly due to its ability to increase the rate of uncoating in vitro and decrease pelletable CA from infected cells (28, 29, 32). However, BI2 and PF74 have also been shown to stabilize CA-NC assemblies in vitro, suggesting an opposite effect on the HIV capsid (30, 32, 33). We did not observe a change in capsid integrity with either drug in the capsid integrity assay (Fig. 2A). In these experiments, virus was exposed to drug before fusion with the cell, which should allow sufficient time for the drug to bind CA. A drug that induces very rapid capsid disassembly, as is proposed for PF74, may be able to facilitate disassembly before the viral complex could associate with TRIM protein. This situation would reduce the number of viral complexes localizing to TRIM cytoplasmic bodies. However, we did not observe a significant difference in the number of viral complexes that associated with TRIM bodies in the presence of PF74 and BI2 (Fig. 2B). Therefore, PF74 and BI2 do not seem to affect capsid integrity in infected cells, suggesting that they do not induce rapid uncoating. As TRIM can bind rapidly to incoming virus, it does remain possible that TRIM may associate with incoming virus before the effect of PF74 or BI2 (41). It is notable that published results obtained by utilizing the fate-of-capsid assay to indicate that PF74 causes disruption of the capsid are for assays conducted at very late time points (12 h) and so are impossible to compare to the results obtained at the early time points of the capsid integrity assay utilized here (32). We also examined the timing of drug susceptibility in a panel of CA mutants with various altered rates of uncoating (6, 7). Overall, the changes in drug susceptibility kinetics observed for each CA mutant were not consistent with its altered rate of uncoating (Fig. 5). E45A and Q63/67A were more susceptible to PF74 at later time points than the wild type. However, uncoating is dramatically delayed by the Q63/67A mutation compared to the E45A mutation, and this difference is not observed for the PF74 susceptibility kinetics (6, 7). Similarly, A92E and N74D uncoat faster and slower, respectively, than the wild type but have similar susceptibility kinetics in the drug addition assays. Therefore, from our imaging and cell-based studies, it seems unlikely that PF74 or BI2 induces rapid capsid disassembly in infected cells.
We next focused on the possibility of PF74 and BI2 targeting CA in postuncoating steps of viral replication. We observed differences in overall BI2 susceptibility and the timing of PF74 susceptibility with different CA mutants, confirming that this effect is dependent on an interaction with the CA protein (Fig. 5) (28–31). While we did observe an effect of PF74 on reverse transcription, this effect was not required to inhibit viral replication at lower drug concentrations (Fig. 3). A similar effect of different concentrations of PF74 on reverse transcription and infectivity has been observed in two recent studies (31, 42). In contrast to PF74, BI2 did not affect reverse transcription (Fig. 3). Finally, in drug addition studies, PF74 and BI2 inhibited infectivity at time points after reverse transcription was completed (Fig. 1). Therefore, our data suggest that the postuncoating effect of these drugs is not due to alterations in reverse transcription.
Since the timing of drug susceptibility is several hours past when uncoating is observed in the CsA washout assay, we hypothesized that some molecules of CA remain present in the viral complex after uncoating and completion of reverse transcription. In order to detect this predicted small amount of CA, we performed high-resolution structural imaging studies and analyzed the intensity of CA signal associated with integrase-GFP nuclear viral complexes compared to that associated with complexes localized at the nuclear membrane, in the cytoplasm, and in controls where virions remain intact. We were able to detect low levels of CA associated with integrase-GFP-positive viral complexes in the nuclei of CHO cells (Fig. 6A and B). We were also able to detect similar low levels of CA associated with integrase-GFP-labeled nuclear viral complexes in HeLa cells (Fig. 6G and H). This observation contrasts with a recent study by Peng et al. that utilized a related approach to determine if CA was associated with nuclear HIV complexes in TZM-bl cells (42). In these experiments, viral complexes were visualized using the related integrase-GFP construct from the Cereseto lab (22) and click labeling to stain the viral cDNA. Peng et al. observed that HIV nuclear complexes lacked CA as detected by a rabbit polyclonal antibody. We believe that this lack of CA staining was likely due to the staining conditions, as it is relatively easy to detect intact conical capsids, which contain thousands of copies of CA. We had to carefully titrate conditions with a very sensitive monoclonal antibody to CA (71-31) to efficiently detect the low-level CA staining in nuclear viral complexes. In contrast to what was observed in TZM-bl cells, in monocyte-derived macrophages Peng et al. found that the majority of viral complexes inside the nucleus contained CA signal. In our studies, the amount of CA signal associated with viral complexes inside the nucleus was reduced compared to that associated with viral complexes found at the nuclear membrane, in the cytoplasm, or in controls (Fig. 7). In fact, the brightly staining population of viral complexes that was readily detected in the cytoplasm and at the nuclear membrane was not found in the nucleus. These data therefore suggest that a subset of CA protein remains associated with the viral complex after uncoating.
Currently there are two primary models for the site of HIV-1 uncoating. In our model, HIV-1 sheds its conical core coat within a hour of fusion, as the reverse-transcribing viral complex is trafficked to the nucleus. In the second model, the intact capsid docks at the nuclear pore before uncoating. Although we set out to reconcile our model with existing data in the field, the data presented here cannot be used to distinguish between the models. We observed a lower intensity of CA staining associated with viral complexes inside the nucleus, indicating that the intact capsid is not imported into the nucleus (Fig. 7). However, in fixed-cell studies, we are unable to determine precisely where these nuclear viral complexes uncoat or if the loss of CA during uncoating is a progressive or rapid process. Viral complexes localizing to the cytoplasm and nuclear membrane are associated with a range of CA staining, including the low levels of CA protein found in nuclear viral complexes (Fig. 7). In support of model 1, it is possible that the capsid uncoats in the cytoplasm, accounting for the low level of capsid staining in viral complexes localized to the nucleus, nuclear membrane, and cytoplasm. In support of model 2, it is possible that the viral complexes in the nucleus originated from the brightly staining complexes in the cytoplasm that then docked and uncoated at the nuclear membrane. It is worth noting that a population of low-CA-staining viral complexes (similar to nuclear complexes) appears in the cytoplasm and at the nuclear membrane but is not present in the bafilomycin A and intact-virion (virus-on-glass) controls (Fig. 7). It is intriguing to speculate that this population may represent viral complexes that uncoated in the cytoplasm.
What is the nature of CA protein that localizes to the viral complex in the nucleus? CA monomers associate to form a hexameric lattice with 12 integrated pentamers to make the conical capsid of HIV-1 (43). Both PF74 and BI2 can associate with CA monomers (28, 30–32). PF74 binds with increased affinity to a site composed of the N-terminal domain of one CA monomer and the C-terminal domain of an adjacent monomer within or potentially between hexamers (31, 32). Therefore, the CA we detected in nuclear viral complexes could be monomeric, hexameric, or in another configuration, as similar effects on infectivity were observed for PF74 and BI2. Recently the high-affinity binding of PF74 to the hexameric form of CA has been used to support the model whereby HIV uncoats at the nuclear pore (31, 32). This conclusion seems premature given that the mechanism of the uncoating process is not known. It is possible that after uncoating of the conical capsid, a few CA hexamers remain associated with the viral complex, which then serve as targets for PF74 and BI2. It is also worth noting that there is ample CA present in the virion that is not part of the conical cone structure, so any CA remaining in the viral complex may or may not have originated from the conical capsid. Additionally, BI2 and PF74 inhibit HIV-1 with similar kinetics in our drug studies, suggesting that monomeric capsid may be sufficient to facilitate the effect of these drugs on infectivity.
Our cell-based assays, drug assays, and microscopy experiments support a model whereby a subset of CA remains associated with the viral complex after disassembly of the conical capsid. This residual CA protein could then facilitate downstream steps in viral replication, such as nuclear import and integration target site selection, while also providing a target for the antiviral action of BI2 and PF74. Additional investigation is needed to determine how BI2 and PF74 block infection, which will further elucidate the interplay between these critical steps of HIV-1 replication.
ACKNOWLEDGMENTS
We thank Paul Bieniasz and Theodora Hatziioannou for OMK and CHO A745 cells and Chris Aiken for providing PF74 and BI2. We also thank the NIH AIDS Research and Reference Reagent Program for the HeLa P4R5 cell line, nevirapine, and the 71-31 CA antibody. We thank the RHLCC flow cytometry core. We also thank Kelly Fahrbach and Joao Mamede for critical reviews of the manuscript.
This work was supported by NIH grants R01 AI47770 and P50 GM082545 to T.J.H., 5F32AI089359-03 to A.E.H., and R01 AI089401 to Chris Aiken. The RHLCC flow cytometry core is supported by a Northwestern University Flow Cytometry Core Facility and Cancer Center support grant (NCI CA060553).
REFERENCES
- 1.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol 77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzon T, Leschonsky B, Bieler K, Paulus C, Schroder J, Wolf H, Wagner R. 2000. Proline residues in the HIV-1 NH2-terminal capsid domain: structure determinants for proper core assembly and subsequent steps of early replication. Virology 268:294–307. doi: 10.1006/viro.1999.0178. [DOI] [PubMed] [Google Scholar]
- 4.Tang S, Murakami T, Agresta BE, Campbell S, Freed EO, Levin JG. 2001. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J Virol 75:9357–9366. doi: 10.1128/JVI.75.19.9357-9366.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang S, Murakami T, Cheng N, Steven AC, Freed EO, Levin JG. 2003. Human immunodeficiency virus type 1 N-terminal capsid mutants containing cores with abnormally high levels of capsid protein and virtually no reverse transcriptase. J Virol 77:12592–12602. doi: 10.1128/JVI.77.23.12592-12602.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hulme AE, Kelley Z, Okocha EA, Hope TJ. 2015. Identification of capsid mutations that alter the rate of HIV-1 uncoating in infected cells. J Virol 89:643–651. doi: 10.1128/JVI.03043-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci U S A 108:9975–9980. doi: 10.1073/pnas.1014522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Fricke T, Diaz-Griffero F. 2013. Inhibition of reverse transcriptase activity increases stability of the HIV-1 core. J Virol 87:683–687. doi: 10.1128/JVI.01228-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD. 2005. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J Virol 79:15567–15572. doi: 10.1128/JVI.79.24.15567-15572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hulme AE, Hope TJ. 2014. The cyclosporin A washout assay to detect HIV-1 uncoating in infected cells. Methods Mol Biol 1087:37–46. doi: 10.1007/978-1-62703-670-2_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sayah DM, Sokolskaja E, Berthoux L, Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 12.Sebastian S, Luban J. 2005. TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2:40. doi: 10.1186/1742-4690-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forshey BM, Shi J, Aiken C. 2005. Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J Virol 79:869–875. doi: 10.1128/JVI.79.2.869-875.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamashita M, Perez O, Hope TJ, Emerman M. 2007. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 3:1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kutluay SB, Perez-Caballero D, Bieniasz PD. 2013. Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog 9:e1003214. doi: 10.1371/journal.ppat.1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koh Y, Wu X, Ferris AL, Matreyek KA, Smith SJ, Lee K, KewalRamani VN, Hughes SH, Engelman A. 2013. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J Virol 87:648–658. doi: 10.1128/JVI.01148-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mamede JI, Sitbon M, Battini JL, Courgnaud V. 2013. Heterogeneous susceptibility of circulating SIV isolate capsids to HIV-interacting factors. Retrovirology 10:77. doi: 10.1186/1742-4690-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matreyek KA, Engelman A. 2011. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J Virol 85:7818–7827. doi: 10.1128/JVI.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qi M, Yang R, Aiken C. 2008. Cyclophilin A-dependent restriction of human immunodeficiency virus type 1 capsid mutants for infection of nondividing cells. J Virol 82:12001–12008. doi: 10.1128/JVI.01518-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albanese A, Arosio D, Terreni M, Cereseto A. 2008. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS One 3:e2413. doi: 10.1371/journal.pone.0002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shukair SA, Allen SA, Cianci GC, Stieh DJ, Anderson MR, Baig SM, Gioia CJ, Spongberg EJ, Kauffman SM, McRaven MD, Lakougna HY, Hammond C, Kiser PF, Hope TJ. 2013. Human cervicovaginal mucus contains an activity that hinders HIV-1 movement. Mucosal Immunol 6:427–434. doi: 10.1038/mi.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Z, Dobro MJ, Woodward CL, Levandovsky A, Danielson CM, Sandrin V, Shi J, Aiken C, Zandi R, Hope TJ, Jensen GJ. 2013. Unclosed HIV-1 capsids suggest a curled sheet model of assembly. J Mol Biol 425:112–123. doi: 10.1016/j.jmb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 26.Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ. 2006. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci U S A 103:7465–7470. doi: 10.1073/pnas.0510483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dismuke DJ, Aiken C. 2006. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J Virol 80:3712–3720. doi: 10.1128/JVI.80.8.3712-3720.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol 85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier JF, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C, Sundquist WI, Mason SW. 2013. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemother 57:4622–4631. doi: 10.1128/AAC.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, James LC. 2014. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 10:e1004459. doi: 10.1371/journal.ppat.1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, Ivanov DN, Yeager M. 2014. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A 111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fricke T, Brandariz-Nunez A, Wang X, Smith AB III, Diaz-Griffero F. 2013. Human cytosolic extracts stabilize the HIV-1 core. J Virol 87:10587–10597. doi: 10.1128/JVI.01705-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 35.Braaten D, Franke EK, Luban J. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J Virol 70:3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franke EK, Yuan HE, Luban J. 1994. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 37.Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG. 1994. Functional association of cyclophilin A with HIV-1 virions. Nature 372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- 38.Sokolskaja E, Sayah DM, Luban J. 2004. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol 78:12800–12808. doi: 10.1128/JVI.78.23.12800-12808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. 2005. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J Virol 79:176–183. doi: 10.1128/JVI.79.1.176-183.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huebner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. 2007. Sequence of human immunodeficiency virus type 1 (HIV-1) gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged EIV-1. J Virol 81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell EM, Perez O, Anderson JL, Hope TJ. 2008. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5 alpha. J Cell Biol 180:549–561. doi: 10.1083/jcb.200706154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, Cihlar T, Muller B, Krausslich HG. 2014. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 3:e04114. doi: 10.7554/eLife.04114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol 78:2545–2552. doi: 10.1128/JVI.78.5.2545-2552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]