

ABSTRACT
Japanese encephalitis is an acute zoonotic, mosquito-borne disease caused by Japanese encephalitis virus (JEV). Japanese encephalitis is characterized by extensive inflammation in the central nervous system (CNS) and disruption of the blood-brain barrier (BBB). However, the pathogenic mechanisms contributing to the BBB disruption are not known. Here, using a mouse model of intravenous JEV infection, we show that virus titers increased exponentially in the brain from 2 to 5 days postinfection. This was accompanied by an early, dramatic increase in the level of inflammatory cytokines and chemokines in the brain. Enhancement of BBB permeability, however, was not observed until day 4, suggesting that viral entry and the onset of inflammation in the CNS occurred prior to BBB damage. In vitro studies revealed that direct infection with JEV could not induce changes in the permeability of brain microvascular endothelial cell monolayers. However, brain extracts derived from symptomatic JEV-infected mice, but not from mock-infected mice, induced significant permeability of the endothelial monolayer. Consistent with a role for inflammatory mediators in BBB disruption, the administration of gamma interferon-neutralizing antibody ameliorated the enhancement of BBB permeability in JEV-infected mice. Taken together, our data suggest that JEV enters the CNS, propagates in neurons, and induces the production of inflammatory cytokines and chemokines, which result in the disruption of the BBB.
IMPORTANCE Japanese encephalitis (JE) is the leading cause of viral encephalitis in Asia, resulting in 70,000 cases each year, in which approximately 20 to 30% of cases are fatal, and a high proportion of patients survive with serious neurological and psychiatric sequelae. Pathologically, JEV infection causes an acute encephalopathy accompanied by BBB dysfunction; however, the mechanism is not clear. Thus, understanding the mechanisms of BBB disruption in JEV infection is important. Our data demonstrate that JEV gains entry into the CNS prior to BBB disruption. Furthermore, it is not JEV infection per se, but the inflammatory cytokines/chemokines induced by JEV infection that inhibit the expression of TJ proteins and ultimately result in the enhancement of BBB permeability. Neutralization of gamma interferon (IFN-γ) ameliorated the enhancement of BBB permeability in JEV-infected mice, suggesting that IFN-γ could be a potential therapeutic target. This study would lead to identification of potential therapeutic avenues for the treatment of JEV infection.
INTRODUCTION
Japanese encephalitis (JE) is an acute zoonotic, mosquito-borne infectious disease caused by JE virus (JEV) infection. JEV is a single-stranded, positive-sense RNA virus, belonging to the genus Flavivirus of the family Flaviviridae (1, 2). JEV is a neurotropic virus and infection causes an acute encephalopathy. JE commonly affects children in the South Pacific regions of Asia (3, 4). Of nearly 70,000 cases of JE reported each year, ca. 20 to 30% of cases are fatal, and a high proportion of patients that survive have serious neurological and psychiatric sequelae (5). Pathologically, JE is positively associated with severe neuroinflammation in the central nervous system (CNS) and the disruption of the BBB (6). However, it is not clear whether blood-brain barrier (BBB) disruption is a prerequisite for or a consequence of JEV infection in the CNS.
The BBB is a physical and physiological barrier composed of cerebral microvascular endothelium together with astrocytes, pericytes, neurons, and the extracellular matrix (7). Brain microvascular endothelial cells (BMECs) comprise the major component of the BBB, and the tight junctions (TJ) between BMECs determine and limit the paracellular permeability. The cytoplasmic TJ proteins, zonula occludens (ZO), interact with each other and connect the transmembrane TJ proteins occludin and claudins to the actin cytoskeleton. Occludin plays a key role in the formation of TJ complex and is sensitive to the modification in inflammation associated with oxidative stress, as recently reviewed (8). Claudins are another important family of transmembrane proteins to form the TJ backbone and maintain the integrity of the BBB. Brain endothelial cells predominantly express claudin-3 and claudin-5, and the depletion of claudin-5 induces the disruption of the BBB in mice (8). Together, these proteins and cells form an elaborate network that selectively regulates the transport of the compounds into and out of the brain (7, 9–11). Cell adhesion molecules (AMs) are cell surface molecules that facilitate intercellular binding and communication (12, 13). Intercellular cell adhesion molecule 1 (ICAM-1), vascular endothelial cell adhesion molecule 1 (VCAM-1), and platelet endothelial cell adhesion molecule 1 (PECAM-1) are responsible for recruiting leukocytes onto the vascular endothelium before extravasating to the injured tissues.
Many CNS diseases alter the function of the BBB (14, 15). Most neurotropic pathogens can cause changes to BBB permeability, such as Nipah virus, JEV, rabies virus (RABV), West Nile virus (WNV), and mouse adenovirus type 1 (MAV-1) (16–20). Some of these viruses (for example, Nipah virus and MAV-1) enhance BBB permeability by disrupting the TJ complex during infection of BMECs (16, 21), while others (such as HIV) disrupt the TJ complex and enhance BBB permeability via induction of inflammatory cytokines or chemokines such as gamma interferon (IFN-γ), interleukin-8 (IL-8), tumor necrosis factor alpha (TNF-α), and IL-1β, which indirectly contribute to BBB breakdown (20, 22). Human T-cell lymphotropic virus can infect endothelial cells and release proinflammatory mediators, facilitating the entry of virus into the CNS (23). WNV, which also belongs to the genus Flavivirus of the family Flaviviridae and shares some common biological feature with JEV, may degrade the TJ proteins and increase multiple matrix metalloproteinases (MMPs), contributing to BBB disruption (19).
In JEV-infected mice, clinical symptoms and mortality are associated with high virus titers in the brain, elevated production of inflammatory mediators, and BBB disruption (24, 25). However, it is not known whether BBB disruption is a prerequisite or a consequence of JEV infection. In the present study, the C57BL/6 mice were infected with JEV via the tail vein. It was found that the virus actively propagated in the brain before BBB permeability was found to be enhanced. BBB permeability was accompanied by a dramatic increase in the level of inflammatory cytokines and chemokines. Brain extracts from mice showing clinical signs of JE could enhance BBB permeability in vitro and administration of IFN-γ-neutralizing antibody ameliorated the enhancement of BBB permeability in JEV-infected mice. These data suggest that inflammatory cytokines and chemokines produced subsequent to CNS infection contribute to BBB disruption during JE pathogenesis.
MATERIALS AND METHODS
Virus and cells.
JEV P3 strain was used for both in vitro and in vivo studies. Virus was propagated in the brain of 1-day-old suckling mice intracerebrally with 15 μl containing 5 × 104 PFU of viral inoculum. When moribund, mice were euthanized and brains removed. A 10% (wt/vol) suspension was prepared by homogenizing the brain in Dulbecco modified Eagle medium (DMEM). The homogenate was centrifuged to remove debris, and the supernatant was collected and stored at −80°C (26). A baby hamster kidney fibroblast cell line (BHK-21) was used for viral titration. Mouse BMEC (bEnd.3) was obtained from American Type Culture Collection (Manassas, VA) and maintained in DMEM supplemented with fetal bovine serum (Gibco, Grand Island, NY).
Inoculation of mice with JEV.
Adult C57BL/6 mice (female, 6 to 8 weeks old) were obtained from the animal housing facility of the Chinese Academy of Sciences, Changsha, China, and maintained according to Committee for Protection, Supervision, and Control of Experiments on Animals guidelines, Huazhong Agricultural University. Mice were injected intravenously with 100 μl containing 105 PFU of JEV (strain P3) diluted with phosphate-buffered saline (PBS; pH 7.4). Control animals received sterile PBS by the same route. The time before intravenous injection is referred to as day 0. JEV-infected mice (six to seven mice) were sacrificed every day after JEV infection and continued for 7 days either for tissue sectioning or protein/RNA extraction.
Virus titration.
At the indicated times, mice were anesthetized and perfused with cold PBS through the left ventricle of the heart. The brain was then homogenized in 2 ml of cold DMEM, and virus in the supernatant was titrated by plaque assay on BHK-21 cells. BHK-21 cell monolayers were grown in 12-well plates and inoculated with serial dilutions of the virus preparation. After absorption for 60 min, virus inoculum was aspirated, and the cells were washed twice with PBS. An overlay consisting of DMEM containing 1.5% carboxy-methylcellulose (CMC; Wako) and 2% fetal calf serum (CMC-DMEM) was added to the cells, and the plates were incubated at 37°C in a CO2 incubator. After incubation for 5 days, the CMC-DMEM was removed, the cells were washed twice with PBS and then fixed and stained with a solution of 0.1% crystal violet and 10% formalin in PBS under UV light. After staining for 2 h, the cells were washed with water, and the plaques were counted. The virus titer was calculated from a virus dilution that produced 10 to 100 plaques per well and is expressed as PFU/ml. Virus titer in the serum was also determined by the same plaque procedure.
Measurement of BBB permeability.
BBB permeability was assessed either with Evans blue dye (EBD; 961 Da) or with sodium fluorescein dye (NaF; 376 Da). The EBD binds to serum albumin (69,000 Da) to become a high-molecular-weight protein tracer when injected into the circulation, whereas the NaF remains an unbound small molecule in the circulation. Mice were injected with 100 μl of EBD solution (2% in PBS) intravenously (27). After 1 h, all injected mice were sacrificed and transcardially perfused with 20 ml of normal saline. Whole brains were then removed and photographed.
NaF is also utilized as a tracer molecule to evaluate BBB permeability with a modified technique (28). The animals were injected with 10 mg of NaF in 0.1 ml of sterile saline, administered i.p. under anesthesia. After 10 min to allow circulation of the NaF, peripheral blood was collected. Serum (50 μl) was recovered and mixed with an equal volume of 15% trichloroacetic acid (TCA). After centrifugation for 10 min at 10,000 × g, the supernatant was recovered and made up to 150 μl by adding 30 μl of 5 M NaOH and 7.5% TCA. At the indicated times, mice were anesthetized and perfused with cold PBS through the left ventricle of the heart to flush out intravascular fluorescein. The brain tissues were homogenized in cold 7.5% TCA and centrifuged for 10 min at 10,000 × g to remove insoluble precipitates. After the addition of 30 μl of 5 M NaOH to 120 μl of supernatant, the fluorescence was determined using a BioTek Spectrophotometers (Bio-Tek Instruments, Wonooski, VT) with excitation at 485 nm and emission at 530 nm. Standards (125 to 4,000 μg/ml) were used to calculate the NaF content of the samples. NaF uptake into tissue is expressed as (μg of fluorescence spinal cord/mg of tissue)/(μg of fluorescence sera/ml of blood) to normalize values for blood levels of the dye at the time of tissue collection.
Quantitative real-time PCR analysis.
The left hemisphere of each mouse was collected and homogenized with DMEM. Total RNA from left hemisphere or cells was extracted with TRIzol (Invitrogen, Grand Island, NY). cDNA was synthesized by using ReverTra Ace qPCR RT kit (Toyobo, Japan) according to the manufacturer's instructions. Quantitative real-time PCR was performed using SYBR green (Invitrogen) on a StepOne Plus using StepOne software v2.2.2 (Applied Biosystems, Foster City, CA). The expression of mRNA was normalized with β-actin. A pair of primers, 5′-GGCTCTTATCACGTTCTTCAAGTTT-3′ and 5′-TGCTTTCCATCGGCCYAAAA-3′, was used for the JEV-C gene. To quantify the viral copy numbers, a standard curve was generated using the pcDNA3.0-HA/JEV-C gene plasmid as a template (the copy number ranged from 6.8 copies/μl to 6.8 × 107 copies/μl).
Luminex assay.
Blood was collected into serum separator tubes (Sarstedt, Nümbrecht, Germany) by retro-orbital bleeding. The serum was stored at −70°C until processing. The right hemisphere of each mouse was lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate) and protease inhibitor cocktail (Roche, South San Francisco, CA) and then centrifuged for 20 min at 12,000 rpm to remove debris.
Extracts from homogenized brains were analyzed simultaneously for the levels of preselected 13 mouse cytokines and chemokines using the Milliplex mouse cytokine magnetic kit (Millipore, Darmstadt, Germany) according to the manufacturer's instructions (29). The 13 cytokines and chemokines analyzed included TNF-α, IL-1β, IL-4, IL-6, IL-10, IL-12p70, IL-17, IFN-γ, C-C motif ligand 2 (CCL2), CCL3, CCL4, CCL5, and C-X-C motif ligand 10 (CXCL10). Premixed magnetic beads conjugated to antibodies for all 13 analytes were mixed with equal volumes of brain extracts in 96-well plates. Plates were protected from light and were incubated on an orbital shaker overnight at 4°C, followed by washing of magnetic beads with 200 μl of wash buffer three times. Detection antibodies were then added to each well, and the mixtures were incubated at room temperature for 1 h. Streptavidin phycoerythrin was added to each well, and the mixtures were incubated at room temperature for 30 min. The magnetic beads were resuspended in sheath fluid, and plates were assayed on a Luminex-3D system. The data of median fluorescence intensity were analyzed using a five-parameter logistic method for calculating cytokine and chemokine concentrations in brain homogenates.
Transendothelial permeability assay.
A transendothelial permeability assay was carried out as previously described with modifications (30). Mouse BMECs (b.End3) were cultured on 0.4-μm-pore-size Transwell filters until reaching 100% confluence. After treatments, FITC-dextran-10000 (10 kDa; Sigma-Aldrich, St. Louis, MO) was applied apically at 1 mg/ml for 30 min. Samples were removed from the lower chamber for fluorescence measurements with a fluorometer (excitation, 492 nm; emission, 520 nm).
Histopathology, IHC, and immunofluorescence.
Mice with signs of disease after infection were anesthetized with ketamine-xylazine (0.1 ml/10 g [body weight]) and perfused with PBS, followed by 4% paraformaldehyde as described previously (31). Brain tissues were removed and paraffin embedded for coronal sections (4 μm). The sections were stained with hematoxylin and eosin (H&E) for histopathology. For immunohistochemistry (IHC), the sections were deparaffinized and rehydrated in xylene and ethanol. Endogenous peroxidase was quenched by incubation in 3% hydrogen peroxide, and antigen retrieval was performed in 0.01 M citrate buffer. Sections were then blocked and incubated with primary rabbit anti-occludin polycolonal antibody (pAb; Santa Cruz, Santa Cruz, CA), rabbit anti-claudin-5 pAb (Invitrogen), and rabbit anti-ZO-1 pAb (Sigma) overnight at 4°C. After a washing step, biotinylated secondary antibodies were applied. The avidin-biotin-peroxidase complex (VectaStain standard ABC kit; Vector Laboratories, Burlingame, CA) was used to localize the biotinylated antibody. Diaminobenzidine (DAB; Vector Laboratories) was utilized for color development. Negative control was performed by substituting primary antibodies with PBS. For antigen quantification, sections were photographed and analyzed using an Olympus BX41 microscope (Olympus, Tokyo, Japan). The integrated optical density of DAB signals was determined by using an Image-Pro Plus (Media Cybernetics, Bethesda, MD).
In immunofluorescence, the following primary antibodies were used: rabbit anti-MAP2 pAb (1:1,000; Proteintech, China), rabbit anti-IBA1 pAb (1:500; Wako, Japan), and rabbit anti-glial fibrillary acidic protein (GFAP) pAb (1:800; Dako, Carpinteria, CA). Sections were incubated with either one primary antibody, followed by incubation with secondary antibody conjugated with either Alexa Fluor 488 or Alexa Fluor 555. The same sections were then incubated with another primary antibody, followed by incubation with the appropriate secondary antibody. Sections were imaged by using a laser confocal microscope (Leica, Germany). The data were obtained and processed using Adobe Photoshop 7.0 software (Adobe Systems, Pasadena, CA).
Western blotting.
BEnd.3 cells and mouse brains were lysed in RIPA buffer and protease inhibitor cocktail, homogenized, and centrifuged at 10,000 × g for 10 min at 4°C. After centrifugation, the insoluble material was removed, and the total protein concentration in supernatant was measured with a BCA protein assay kit (Beyotime, China). Brain extracts or cell lysates were electrophoretically separated by using sodium dodecyl sulfate–8 to 15% polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad, Richmond, CA) and blocked for 1 h at room temperature in 5% nonfat dry milk in Tris-buffered saline with Tween 20 (TBST). Membranes were then incubated overnight with either one of rabbit anti-ZO-1 (Sigma), rabbit anti-claudin-5 (Invitrogen), rabbit anti-occludin, rabbit anti-VCAM-1, rabbit anti-PECAM-1, rabbit anti-ICAM-1 (Santa Cruz), and mouse anti-β-actin (as loading control) antibodies. After extensive washing with TBST, the membranes were incubated with species-specific horseradish peroxidase-conjugated antibodies. Antibody binding was visualized using enhanced chemiluminescence reagents (Beyotime). All Western blots were quantified by using ImageJ, and the values represent the relative immunoreactivity of each protein normalized to the respective loading control. The data are presented as means ± the standard errors of the mean (SEM).
Neutralization of IFN-γ.
The time of intravenous injection with 105 PFU of JEV-P3 virus is referred to as day 0. Mice were injected intraperitoneally with 50 μg of neutralizing antibody (BioLegend, San Diego, CA) specific for IFN-γ or a rat IgG1 isotype control antibody (BioLegend) diluted in 200 μl of PBS at 0, 1, 2, and 3 days postinfection (dpi) with JEV. At 5 dpi, BBB permeability was determined based on the uptake of NaF. After animal euthanasia, brains were harvested, and the expression of TJ proteins in the brain was monitored by Western blotting.
Statistical analysis.
Data are expressed as means ± SEM, and the significance of differences between groups was evaluated by using a two-tailed Student t test or one-way analysis of variance, followed by Tukey's post hoc tests. Graphs were plotted and analyzed using GraphPad Prism (v5.0; GraphPad, La Jolla, CA).
RESULTS
JEV invades the brain by 2 dpi and replicates exponentially by 5 dpi.
To establish the C57BL/6 mouse model, mice were intravenously injected via the tail vein with JEV-P3 virus at 103 or 105 PFU per mouse. The clinical scores and body weight were monitored and recorded for 21 days. The mortalities were 50 and 90%, respectively, in mice infected with 103 and 105 PFU of virus (Fig. 1A). JEV-infected mice showed a progressive weight loss (Fig. 1B) and typical neurological signs, such as rigidity of the hindlimbs, abnormal gait, stiffness, and paralysis.
FIG 1.

Survival, body weight, viral burden, and BBB permeability analysis in mice after JEV-P3 infection. (A) Eight-week-old age-matched C57BL/6 mice were intravenously injected with 103 or 105 PFU of JEV-P3. Kaplan-Meier curves show the survival distribution for mice over the 21-day period following challenge (n = 20). (B) Average percent body weight changes were monitored. Body weights were determined every other day, beginning on day 0 until day 14. Mock-infected group, n = 18; infection group, n = 180. (C) The C gene copies of JEV in the brains, spleens, lymph nodes, and livers of JEV-infected mice were measured by qRT-PCR. (D) Evans blue dye was injected intravenously into mice. Representative brains are shown from mock-infected mice or from mice showing mild and severe symptoms. The data are expressed as means ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.005.
In the peripheral tissues (livers, spleens, and lymph nodes) of infected mice, the copy number of virus was significantly lower than in the brain at 5 dpi when the disease sign appeared (Fig. 1C). Changes in BBB permeability were monitored by perfusion with EBD (961 Da). Brains of mice with severe clinical signs absorbed more EBD than the brains of mice with mild clinical signs (Fig. 1D), an observation which indicates that the leakage of EBD into the brain parenchyma correlates with clinical severity. These results suggest that the titers of JEV are higher in the brain than in the peripheral organs, and enhancement in BBB permeability correlates with the clinical severity of JEV infection.
BBB permeability increased after virus invasion into the CNS.
To monitor the course of JEV infection in both the blood and the brain, virus titers were measured. Virus titers in the serum significantly declined from about 1,000 PFU/ml at 1 dpi to <100 PFU/ml at 2 dpi. After 2 dpi, the virus titer was difficult to detect with plaque assay since it variably dropped below the detection limit (Fig. 2A). In contrast, in the brain the virus became detectable at 2 dpi and reached a peak by 5 to 7 dpi (Fig. 2B).
FIG 2.

Viral loads and BBB permeability. Virus titers in the serum (A) and brain (B) were determined based on plaque formation in BHK-21 cells and are represented as geometric means (n = 5). The copy number of the JEV-C gene in the brain was monitored using qRT-PCR (C), and BBB permeability was determined with NaF uptake (D) for 7 consecutive days after injection with 105 PFU of JEV-P3. At 45 min after intraperitoneal injection with sodium fluorescein, the sera and brains were processed for the measurement of fluorescence, and the brain/serum fluorescence ratios were calculated. Each data point represents a single animal, and the lines represent the group medians. *, P < 0.05; **, P < 0.01.
To further determine the correlation of BBB disruption with viral replication, the virus copy number in the brain and the BBB permeability were monitored every day for 7 days (five to eight mice each day). NaF, a small molecule (376 Da) that is able to access and stain brain tissue only when the BBB is compromised, was used to monitor the alteration of BBB permeability. The copy number of the JEV-C gene increased significantly at 2 dpi, and the virus replicated exponentially in the brain between 2 and 4 dpi, when it reached its peak and remained steady (Fig. 2C). It may be because of the delayed protein synthesis and virus particle assembly that the virus titer did not increase pari passu with the copy number of JEV-C gene. Enhancement of the BBB permeability as detected by NaF uptake was not observed until 4 dpi and deteriorated further with progress of the disease (Fig. 2D). When the linear correlation between BBB permeability and C gene copies was analyzed, a good correlation (R2 = 0.9891) was found during viral exponential replication (1 to 4 dpi). However, BBB permeability was still deteriorating after viral replication reached its peak; thus, the correlation between BBB permeability and C gene copies was only 0.6299 from 1 to 6 dpi (data not shown). These results indicate that the virus gained entry into the CNS prior to BBB disruption and that enhanced BBB permeability correlates with the clinical severity of JEV infection.
Neurons are the major target to JEV infection in the CNS.
To determine the target for JEV infection in the CNS, C57BL/6 mice were infected with 105 PFU of JEV-P3 strain. When neurological signs became obvious, mice were sacrificed, and brain sections were stained with H&E. Perivascular and vascular infiltration of mononuclear cells formed perivascular cuffing in the cerebral parenchyma (Fig. 3A, black arrows in panels b and c). Activated microglia, which have large rod-shaped nuclei, encircled degenerating neurons (neuronophagia) and formed clusters around small foci of necrotic brain tissue (microglial nodules, Fig. 3A, red arrows in panels b and d). Immunohistochemistry of the cerebrum and cerebellar cortex showed that JEV envelope protein was mostly found in neurons as identified by MAP-2 staining (Fig. 3B, panels a to d). Astrocytes and microglia identified by GFAP and IBA-1 staining, respectively, were rarely costained by JEV protein (Fig. 3B, panels i to l and panels q to t). GFAP intensity showed a significant increase in the brains of JEV-infected mice compared to the brains of mock-infected mice, suggesting that astrocytes were activated (Fig. 3B, panels e to l). Likewise, IBA-1, a marker of activated microglia and macrophages, was also dramatically increased in the brains of JEV-infected mice (Fig. 3B, panels m to t). These data suggest that neurons are the major target for JEV infection and activation of astrocytes and microglia (or infiltrating macrophages) may further contribute to neuronal damage.
FIG 3.

Pathological features of JEV infection in mouse brain. (A) Brain histopathological changes of JEV-infected mice with disease signs were examined by H&E staining at 5 dpi. c, perivascular cuff (black arrows); d, microglia nodule (red arrows). (B) Immunofluorescent triple staining of neuron infection (a to d), astrocyte activation (e to i), and microglial activation (m to t). Brain sections were collected from mock-infected and JEV-infected mice at 5 dpi. E, JEV envelope protein (red). MAP-2, GFAP, and IBA-1 indicate markers for neurons, astrocytes, and microglia, respectively (green). DAPI stains for nuclei (blue). Subpanels e to h and m to p show brain sections from mock-infected mice.
The expression of TJs and AMs was altered in the JEV-infected mouse brain.
Tight junctions are important structures for maintaining BBB function, and the dysregulation of TJs is associated with BBB breakdown (32–34). To determine the mechanism by which JEV infection disrupts the BBB, the expression of occludin, claudin-5, and ZO-1 in the brains of JEV-infected mice was assessed by IHC and quantitative Western blotting. The expression of the transmembrane TJ proteins, occludin, and claudin-5 decreased on the walls of blood microvessels in the brains of JEV-infected mice compared to mock-infected mice at 5 dpi (Fig. 4Aa, b, d, and e). The immunoreactivity of the cytoplasmic TJ protein ZO-1 was partially lost in mice showing clinical signs (Fig. 4Ac and f). Tight junctions formed continuous seals around the inner side of the microvessels (black arrows). The continuity of occludin, claudin-5, and ZO-1 was disrupted (red arrows), and the aggregation of these proteins was observed (Fig. 4A) in JEV-infected mice. The IHC results were recapitulated by Western blotting (Fig. 4B). The expression levels of TJ proteins were significantly lower in mice infected with JEV than in mock-infected mice (Fig. 4C). Overall, the expression of occludin, claudin-5, and ZO-1 was reduced to 79.63, 56.65, and 65.35%, respectively, in the brains of JEV-infected mice (Fig. 4C) compared to those in mock-infected mice. These results suggest that the TJ complex is broken down in symptomatic JEV-infected mice.
FIG 4.

Expression of TJ proteins (occludin, claudin-5, and ZO-1) in the brains of JEV-infected mice. (A) Mice were infected intravenously with either 105 PFU of JEV-P3 or PBS. At 5 dpi, the animals were euthanized, and the brains were harvested, fixed, and sectioned to measure the expression of TJ proteins using IHC with antibodies to the respective TJ proteins. Black arrows indicate continuous seals that TJ formed around the inner sides of microvessels. Red arrows point to the continuity disruption of occludin, claudin-5, and ZO-1. Inset boxes in subpanels d, e, and f show high-magnification images of regions demarcated by dashed lines, respectively. (B) The expression of TJ proteins in JEV-infected mouse brains was detected by Western blotting. The infected group was divided into asymptomatic, mild, severe, or moribund. (C) Results of panel B were normalized with β-actin and densimetrically quantified as the fold change over that for mock-infected controls. The samples from asymptomatic, mild, severe, and moribund mice were quantified as the infected group. The data are means ± SEM of results from three independent experiments. The statistical significance in panel C was assessed using a two-tailed Student t test. Asterisks indicate statistical significance (*, P < 0.05).
Leukocyte adhesion and trafficking across the BBB depend on sequential activation and expression of AMs such as PECAM-1, VCAM-1, and ICAM-1 (35, 36). Therefore, the expression of PECAM-1, VCAM-1, and ICAM-1 was analyzed in brain tissues from JEV-infected mice. All AMs analyzed were significantly increased (ca. 1- to 2.5-fold) in JEV-infected mice compared to mock-infected mice, especially the expression of ICAM-1 (Fig. 5). Together, these data demonstrate that during JEV infection the enhancement of BBB permeability was associated with the reduction of TJs and the increase of cell AMs.
FIG 5.

Expression of AM proteins (PECAM-1, VCAM-1, and ICAM-1) in the brains of JEV-infected mice. (A) At 5 dpi, animals were euthanized, and the brains were then harvested and homogenized. After centrifugation, the supernatants from brain homogenates were analyzed by Western blotting for the expression of AM proteins. (B) The results of panel A were normalized with β-actin and densimetrically quantified as the fold change over that of mock-infected controls. The samples from asymptomatic, mild, severe, and moribund mice were quantified as the infected group. The data are means ± SEM of results from three independent experiments. Statistical significance was assessed by using a two-tailed Student t test. Asterisks indicate statistical significance (*, P < 0.05).
JEV infection induces the expression of inflammatory cytokines/chemokines in the CNS.
Neuroinflammation is a hallmark of JE. To determine whether the expression of inflammatory cytokines/chemokines in brain extracts might be responsible for the reduction of TJ protein expression in BMECs, simultaneous profiles of inflammatory cytokines/chemokines in serum and brain were analyzed with a Luminex assay. Overall, the level of these cytokines/chemokines in the serum was very low, while the level of these cytokines/chemokines in the brain extracts increased dramatically, and some levels were above the detection limit. The most significantly changed cytokines/chemokines are presented in Fig. 6. In serum, the expression of IFN-γ decreased in the first 4 dpi; the levels of chemokines CCL4 and CXCL10 increased briefly at 1 dpi, declined by 2 to 4 dpi, and increased again by 5 or 6 dpi. Notably, in the brain, the levels of CXCL10, IFN-γ, IL-6, and CCL4 significantly increased at 1 dpi, while the levels of CCL2 and CCL3 started to increase at 3 or 4 dpi. For the rest of the cytokines/chemokines (data not shown), IL-4 and IL-10 levels underwent no significant change, the levels of IL-1β, IL-17, and IL-12p70 had slight increase, and chemokine CCL5 increased by about 15- to 20-fold, less dramatically than the cytokines/chemokines shown in Fig. 6B. TNF-α also significantly increased at 4 or 5 dpi, with huge variations among individuals in the brain, but remained unchanged in serum (data not shown).
FIG 6.

Measurement of cytokines by using a Luminex assay in the brain extracts from mice infected with JEV-P3. C57BL/6 mice were infected intravenously with either 105 PFU of JEV-P3 or PBS. Each day from day 0 to 7 dpi, animals were euthanized, blood was collected, and the brains were harvested and homogenized. After centrifugation, the sera and supernatants from brain homogenates were used for measurement of the indicated cytokines using a Luminex assay. The expression levels of 6/13 cytokines/chemokines are shown in serum (A) and brain extracts (B). Pentagrams indicate the expression levels of cytokines beyond the upper detection limit. The data are shown as means ± SEM (n = 4) in each experimental condition. Statistical analyses were performed with two-tailed Student t test. Asterisks indicate statistical significance (*, P < 0.05; **, P < 0.01; ***, P < 0.005).
Inflammatory cytokines/chemokines disrupted the BBB.
Our studies described above indicate that the enhancement of BBB permeability occurred after virus replication in the CNS. To investigate whether JEV infection induces changes in BBB permeability, bEnd.3 cells were infected with JEV-P3 at multiplicities of infection (MOIs) of 1, 5, and 10. JEV antigen was found in only a few cells when infected at an MOI of 10 (data not shown). No significant changes in the expression of TJs were found in the infected cells as determined by quantitative reverse transcription-PCR (qRT-PCR; data not shown). These results indicate that JEV infection per se does not induce changes in BBB permeability. To determine whether inflammatory cytokines/chemokines are associated with alterations in BBB permeability, bEnd.3 cells were treated with brain extracts derived from JEV-infected or mock-infected mice. Significant increase in paracellular permeability was observed in bEnd.3 cells treated with brain extracts from JEV-infected mice. UV inactivation of the brain extracts did not alter the results (Fig. 7A). These results suggest that it is most likely the inflammatory cytokines/chemokines, not JEV infection per se, induced the enhancement of BBB permeability.
FIG 7.
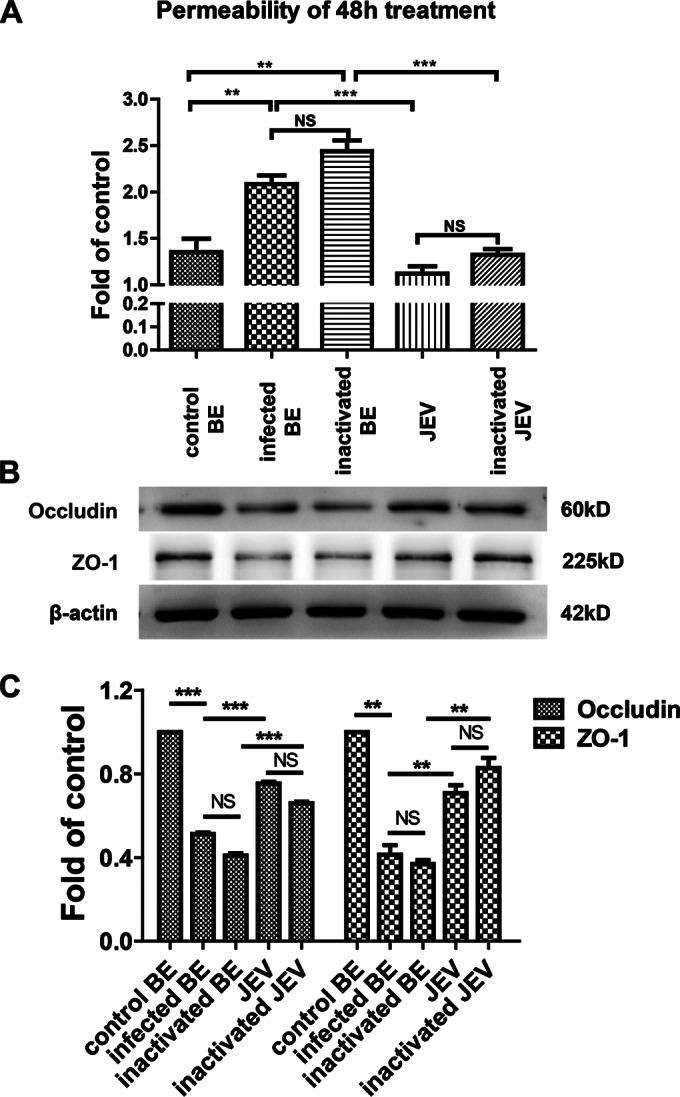
Effects of brain extracts on endothelial barrier permeability. (A) After b.End3 monolayers were grown to confluence on transwell membranes, the cells were treated with brain extracts, UV-inactivated brain extracts, JEV, or UV-inactivated JEV for 24 h or 48 h. FITC-dextran-10000 was applied apically at 1 mg/ml for 30 min, and then the permeability was measured from the samples of lower chamber with a fluorometer (excitation, 492 nm; emission, 520 nm). (B) Mouse BMECs were treated as described above, and then protein samples were collected and subjected to Western blotting for the TJ proteins occludin, claudin-5, and ZO-1. β-Actin was used as a loading control. (C) The results of panel B were normalized with β-actin and densimetrically quantified as the fold change versus control brain extracts (control BE). Means ± SEM of three independent experiments were shown. Asterisks indicate statistical significance (*, P < 0.05; **, P < 0.01; ***, P < 0.005).
Furthermore, the expression of TJs in the bEnd.3 monolayer was monitored by Western blotting. The expression of occludin and ZO-1 was reduced significantly in cells treated with brain extracts from infected mice compared to those treated with virus alone (Fig. 7B and C). UV inactivation had no effect on the permeability of the monolayer. Thus, the increase in permeability and the reduction in TJs expression following JEV exposure were due to the soluble factors in brain extracts rather than to JEV infection in monolayer endothelial cultures. These results suggest that the inflammatory cytokines and chemokines enhance the BBB permeability through regulating the expression of TJ proteins.
Neutralization of IFN-γ ameliorated the disruption of BBB integrity and the downregulation of TJ proteins.
IFN-γ is the most important cytokine in the cytokine/chemokine production network directly promoting the expression of CXCL10, which could chemoattract peripheral blood lymphocytes into the brain (37). To determine whether IFN-γ contributes to the enhancement of BBB permeability, an anti-IFN-γ neutralizing antibody, or an isotype control antibody, was administered intraperitoneally at 0, 1, 2, and 3 dpi. BBB permeability was measured with NaF uptake at 5 dpi. As shown in Fig. 8A, treatment with an anti-IFN-γ antibody significantly decreased the uptake of NaF compared to isotype control, indicating that blocking IFN-γ can ameliorate the disruption of BBB integrity. To determine whether the anti-IFN-γ neutralizing antibody influences BBB permeability by regulating the expression of TJs, occludin, claudin-5, and ZO-1 were detected in the brain lysates of JEV-infected mice. It was found that the expression of occludin, claudin-5, and ZO-1 decreased less in the brains of mice treated with anti-IFN-γ antibody compared to those treated with the isotype control (Fig. 8B and C). In addition, virus titers were determined at 5 dpi after antibody administration. Compared to the isotype control, treatment with anti-IFN-γ neutralizing antibody did not significantly change the virus titer (Fig. 8D). These results suggest that IFN-γ contributes to BBB permeability enhancement and TJ protein expression in JEV infection.
FIG 8.
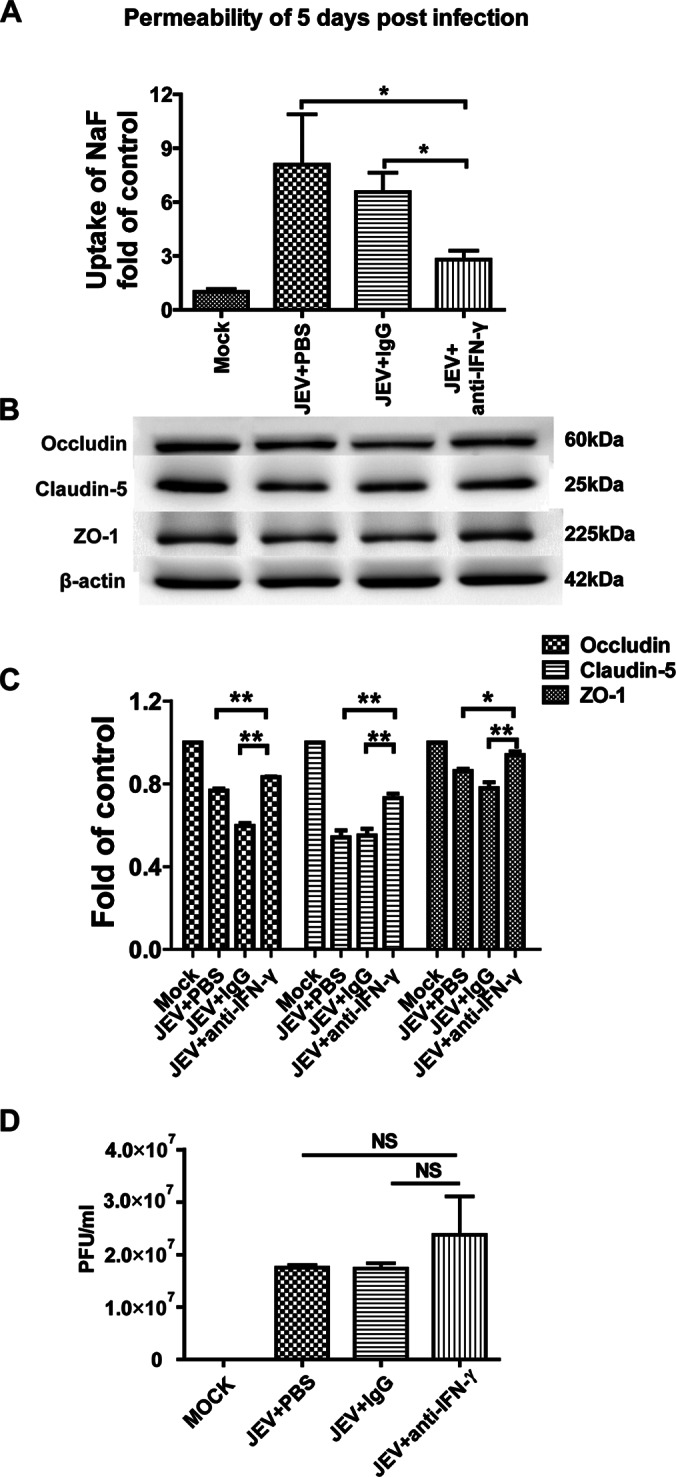
Effects of anti-IFN-γ neutralizing antibody on the BBB permeability of JEV-infected mice. (A) Eight-week-old age-matched C57BL/6 mice were intravenously injected with 105 PFU of JEV-P3. At 0, 1, 2, and 3 dpi, the mice were intraperitoneally injected with anti-IFN-γ neutralizing antibody or isotype control. At 5 dpi, the BBB permeability was determined with NaF uptake. (B) At 5 dpi, brain tissues were collected to measure the expression of TJ proteins by Western blotting. (C) The results of panel B were normalized with β-actin and densimetrically quantified as the fold change versus a mock-infected control. (D) At 5 dpi, viral loads in the brain were determined with a plaque assay after antibody administration. The data are means ± SEM of the results from three independent experiments. The statistical significance values in panel C were assessed by using two-tailed Student t test. Asterisks indicate the statistical significance (*, P < 0.05; **, P < 0.01).
DISCUSSION
Understanding the mechanisms of JEV pathogenesis in the CNS is critical to JE prevention and treatment. In the present study, we demonstrate that JEV entry into the CNS occurs prior to BBB dysfunction in a mouse model of JE. It is not the virus itself, but the inflammatory cytokines/chemokines that caused reduction of the expression of TJ proteins, resulting in the enhancement of BBB permeability. IFN-γ was found to be a key cytokine in this process. Indeed, neutralizing IFN-γ can ameliorate the disruption of BBB integrity in JE.
Some viruses require disruption of the BBB to enter the CNS while others can enter the CNS and then cause BBB disruption. WNV can infect endothelial cells, facilitating the entry of cell-free virus into the CNS without disturbing the BBB, although BBB disruption can be observed later, accompanied by the degradation of TJ proteins and an increase in MMPs (38). In tick-borne encephalitis virus (TBEV) infection, the permeability of the BBB increased at later stages when high virus load was present in the brain, which means BBB breakdown was not necessary for TBEV entry into the brain (39). Conversely, infection of human microvascular endothelial cells (HMEC-1) with dengue virus (DENV) can induce cell apoptosis, suggesting that DENV might directly cause BBB breakdown in order to gain access to the CNS (40). HIV-1 and HIV-1 Tat protein have been shown to disrupt BBB integrity via modification of claudin-5, thereby allowing HIV-1 to enter the brain (41). Thus, depending on the virus, loss of BBB integrity can be the gateway for viral entry into the CNS (HIV-1 and DENV) or it can be the consequence of CNS infection (WNV and TBEV). To determine whether BBB disruption is a prerequisite for or a consequence of CNS infection in JE, BBB permeability and JEV viral loads in the CNS were monitored in mice after intravenous infection. Our data show that JEV-infected mice do not exhibit signs of BBB leakiness until 4 dpi, whereas virus is detectable in the brain as early as 2 dpi, indicating that CNS infection occurs prior to BBB dysfunction, similar to the other two flaviviruses, WNV and TBEV (39, 42, 43).
Different flaviviruses maneuver different pathways for neuroinvasion. Some viruses such as Murray Valley encephalitis virus and St. Louis encephalitis virus are speculated to enter the CNS via the olfactory pathway, which is independent of BBB dysfunction (44). Several routes were proposed for WNV entry into CNS. Hematogenous route via crossing the BBB was suggested to be one of the major routes by which WNV enters the CNS since high viremia correlated with early WNV entry into the CNS, although the associated mechanisms remain unclear (45). Retrograde axonal transport via peripheral nervous system was another route of WNV entering the CNS (19). It was also suggested that WNV virus enters the CNS by infected monocytes, dendritic cells, or macrophages (46). Moreover, the infection of endothelial cell in vitro gives another possibility that WNV might cross the BBB via brain endothelial cells (38). Similarly, the exact routes of JEV gaining entry into the CNS remain unclear. JEV may enter the CNS via a hematogenous route since the infection is diffuse throughout the brain (47). Both macrophages and dendritic cells are capable of being infected by JEV; thus, it remains possible that they contribute to the transmission of virus into the CNS (48–50). However, monocytes appear to have the ability to effectively control flavivirus infection, and in vivo experiments have failed to identify infected microglia or infected infiltrating macrophages in animal models of flavivirus encephalitis (25, 51). JEV could cross the BBB via endothelial transcytosis and/or endothelial infection, as reported previously (24). However, there is little to no evidence in vivo for endothelial infection by flaviviruses (52). In our in vitro model, JEV infection of the endothelial cell line b.End3 was extremely low, even at an MOI as high as 10. There was no significant change in the expression of proteins associated with tight-junction integrity (occludin, claudin-5, and ZO-1; data not shown). These observations are consistent with previous work showing that unequivocal endothelial infection was seen in only 1 of 167 mice following intraperitoneal infection with flavivirus (52). Therefore, it seems unlikely that the primary route of CNS entry is through JEV-infected microvascular endothelial cells.
In our study, it was found that inflammation induced by JEV infection triggered BBB disruption in the CNS. Large amounts of chemokines and cytokines were produced in the CNS, including CXCL10, CCL2, CCL3, CCL4, CCL5, TNF-α, IL-6, and IFN-γ in mice infected with JEV. The inflammatory mediators observed during the course of infection were more of a Th1-type immune response. Indeed, the Th2 cytokines IL-4 and IL-10 were unchanged in the brain of JEV-infected mice. Importantly, the upregulation of these inflammatory mediators reached a peak just before BBB breakdown. Furthermore, the level of CXCL10 was dramatically elevated in the CNS almost immediately after infection, similar to the observations in mice during WNV infection (53). Since neurons are the major target for JEV infection, it is possible that infected neurons are the early source of CXCL10 production as seen in other neurotropic virus infections (53, 54). Coincidently, IFN-γ, a protein that induces the expression of CXCL10, was also upregulated early after infection and could further promote CXCL10 production. We observed that IFN-γ increased in both mixed glial culture and BV-2 cells (a microglial cell line) after JEV infection. Both glia and neurons could be the source of IFN-γ early in infection (55). Although the ability of glial cells to be infected by flavivirus remains debatable, the studies by us and others clearly show that they are well activated during JE (25, 56). Activation of glial cells results in the production of multiple inflammatory chemokines and cytokines (25, 57–60). JEV-activated astrocytes have been shown to be a source of CCL5 and CXCL10 and important for the migration of NK cells and monocytes into the CNS (56, 57). The local immune responses initiated by activated microglial cells may provide protection against JEV infection of the CNS; however, they are also the major source of TNF-α, which can be deleterious to neurons (56). The elevated levels of the proinflammatory cytokines IL-6 and TNF-α and the chemokines CCL2 and CCL5 in the CNS could induce irreversible neuronal damage and correlates with an increased mortality rate (25, 56).
The functions of chemokines and cytokines in the CNS extend beyond their role as mediators of neuroinflammation. Our data showing that brain extracts from mice with JE, but not virus alone, can increase the permeability of an in vitro BBB model strongly suggest that cytokines and/or chemokines contribute to the BBB disruption in vivo. Indeed, we went on to show that blocking IFN-γ in vivo abolishes the increased BBB permeability associated with JEV infection. There are two pathways by which inflammatory cytokines can regulate BBB permeability. First, inflammatory cytokines can upregulate the endothelial AMs, which in concert with chemotactic chemokines, facilitate rolling and adhesion of leukocytes on the endothelial wall, and migration into the affected site (61, 62). In the brains of JEV-infected mice, the expression levels of ICAM-1, VCAM-1, and PECAM-1 are significantly elevated, indicating the potential for increased recruitment of mononuclear leukocytes from the periphery to the CNS. Second, inflammatory cytokines or chemokines (IL-6, IFN-γ, CXCL10, and CCL2 to CCL5) can deteriorate BBB permeability via downregulation of the TJ proteins claudin-5, occludin, and ZO-1 (19). Brain extracts from JEV-infected mice contain high levels of chemokines, cytokines, viruses, and soluble cellular proteins, and these extracts significantly enhanced BBB permeability in vitro even when the virus was inactivated with UV. It has been reported that a recombinant RABV expressing a chemokine (CXCL10, CCL5, or CCL3) could enhance BBB permeability in mice (63) and reduce the expression of TJ protein claudin-5, occludin, and ZO-1 in BMECs (37). This is consistent with our mouse model of JE, where we show increased expression of CXCL10, CCL3, and CCL5, along with decreased expression of claudin-5, occludin, and ZO-1 in the CNS in vivo. Furthermore, the enhancement of BBB permeability is chemokine and cytokine dependent in many other CNS diseases (64). For example, CCL2 has been demonstrated to reduce the expression of ZO-1, ZO-2, occludin, and claudin-5 in HIV-1-infected BMECs through Rho and Rho kinase signaling (65). In EAE, IL-17-induced reactive oxygen species activate myosin light-chain kinase and reduce expression levels of ZO-1 and occludin in BMECs (66). For some reason, experiments in vitro with transendothelial monolayers cannot represent the expression changes in claudin-5 that are observed in vivo. It should be understandable that the in vitro simplified system fails to replicate the precise cellular condition in vivo. It has been reported that IFN-γ, but not TNF-α, is associated with enhanced BBB permeability through peroxynitrite-dependent radicals in RABV infection (28). Consistent with these observations, neutralizing IFN-γ with antibodies ameliorated the enhancement of BBB permeability and partially restored the expression of TJ proteins in JEV-infected mice. Taken together, our data show that inflammatory mediators, particularly IFN-γ, play a central role in enhancing BBB permeability in JEV infection by downregulating TJ protein expression.
In summary, the evidence presented here demonstrates that inflammatory chemokines and/or cytokines in the CNS, but not JEV itself, mediate BBB breakdown during JE. As depicted in Fig. 9, JEV invades the CNS without prior disruption of the BBB permeability where it infects neurons. Infected neurons may produce chemokines that can also induce the activation of glial cells, which in turn produce a preponderance of inflammatory chemokines and cytokines. These inflammatory mediators can break down the BBB by reducing the expression of TJ proteins and thereby damage the integrity of the tight junctions between microvascular endothelial cells. Inflammatory mediators can further compromise the BBB barrier by inducing increased expression of adhesion molecules on BBB endothelial cells, allowing for increased infiltration of inflammatory cells from the periphery to the CNS. Increased inflammatory infiltrates can lead to further neuroinflammation and neuronal injury. Nevertheless, advanced investigations are warranted to determine the pathways by which JEV invades the CNS and induces inflammation. Understanding the mechanisms of inflammation-induced BBB disruption in JVE infection may provide a potential target for therapeutic intervention in CNS diseases that are driven or exacerbated by a compromised BBB.
FIG 9.

Schematic representation of possible mechanisms regulating BBB permeability during JEV infection. During infection, JEV particles penetrate the BBB from the periphery into the CNS without changing BBB permeability. Neurons are vulnerable to JEV infection. Once neurons are infected, the virus begins multiplying and replicating in neurons, which causes the first round of neuronal injury accompanied by the production of cytokines or chemokines. These cytokines/chemokines activate microglia and astrocytes, which in turn stimulates more production of proinflammatory cytokines/chemokines and contributes to further neuronal injury. Cytokines and chemokines can activate immune cells inside the brain that initiate and/or potentiate BBB dysfunction and alter the architecture of tight junctions and adhesion molecules on BBB. Furthermore, transendothelial migration of leukocytes causes acute neuronal tissue damage. Consequently, BBB permeability increases.
ACKNOWLEDGMENTS
This study was supported by the National Natural Sciences Foundation of China (31172294 and 81261160325), fundamental research funds for the Central University (2011PY036), and a start-up research fund from Huazhong Agricultural University.
REFERENCES
- 1.Taylor JL, Schoenherr C, Grossberg SE. 1980. Protection against Japanese encephalitis virus in mice and hamsters by treatment with carboxymethylacridanone, a potent interferon inducer. J Infect Dis 142:394–399. doi: 10.1093/infdis/142.3.394. [DOI] [PubMed] [Google Scholar]
- 2.Weaver SC, Powers AM, Brault AC, Barrett AD. 1999. Molecular epidemiological studies of veterinary arboviral encephalitides. Vet J 157:123–138. doi: 10.1053/tvjl.1998.0289. [DOI] [PubMed] [Google Scholar]
- 3.Chen CJ, Raung SL, Kuo MD, Wang YM. 2002. Suppression of Japanese encephalitis virus infection by nonsteroidal anti-inflammatory drugs. J Gen Virol 83:1897–1905. [DOI] [PubMed] [Google Scholar]
- 4.Solomon T, Dung NM, Kneen R, Gainsborough M, Vaughn DW, Khanh VT. 2000. Japanese encephalitis. J Neurol Neurosurg Psychiatr 68:405–415. doi: 10.1136/jnnp.68.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell GL, Hills SL, Fischer M, Jacobson JA, Hoke CH, Hombach JM, Marfin AA, Solomon T, Tsai TF, Tsu VD, Ginsburg AS. 2011. Estimated global incidence of Japanese encephalitis: a systematic review. Bull World Health Organ 89:766–774, 774A-774E. doi: 10.2471/BLT.10.085233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diagana M, Preux PM, Dumas M. 2007. Japanese encephalitis revisited. J Neurol Sci 262:165–170. [DOI] [PubMed] [Google Scholar]
- 7.Pardridge WM. 1999. Blood-brain barrier biology and methodology. J Neurovirol 5:556–569. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- 8.Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. 2012. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 9:23. doi: 10.1186/2045-8118-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehner C, Gehwolf R, Tempfer H, Krizbai I, Hennig B, Bauer HC, Bauer H. 2011. Oxidative stress and blood-brain barrier dysfunction under particular consideration of matrix metalloproteinases. Antioxid Redox Signal 15:1305–1323. doi: 10.1089/ars.2011.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mariano C, Sasaki H, Brites D, Brito MA. 2011. A look at tricellulin and its role in tight junction formation and maintenance. Eur J Cell Biol 90:787–796. doi: 10.1016/j.ejcb.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Rubin LL, Staddon JM. 1999. The cell biology of the blood-brain barrier. Annu Rev Neurosci 22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi H, Boelte KC, Lin PC. 2007. Endothelial cell adhesion molecules and cancer progression. Curr Med Chem 14:377–386. doi: 10.2174/092986707779941032. [DOI] [PubMed] [Google Scholar]
- 13.Munro JM. 1993. Endothelial-leukocyte adhesive interactions in inflammatory diseases. Eur Heart J 14(Suppl K):72–77. [PubMed] [Google Scholar]
- 14.Andersen IH, Marker O, Thomsen AR. 1991. Breakdown of blood-brain barrier function in the murine lymphocytic choriomeningitis virus infection mediated by virus-specific CD8+ T cells. J Neuroimmunol 31:155–163. doi: 10.1016/0165-5728(91)90021-X. [DOI] [PubMed] [Google Scholar]
- 15.Morgan L, Shah B, Rivers LE, Barden L, Groom AJ, Chung R, Higazi D, Desmond H, Smith T, Staddon JM. 2007. Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience 147:664–673. doi: 10.1016/j.neuroscience.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 16.Gralinski LE, Ashley SL, Dixon SD, Spindler KR. 2009. Mouse adenovirus type 1-induced breakdown of the blood-brain barrier. J Virol 83:9398–9410. doi: 10.1128/JVI.00954-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuang Y, Lackay SN, Zhao L, Fu ZF. 2009. Role of chemokines in the enhancement of BBB permeability and inflammatory infiltration after rabies virus infection. Virus Res 144:18–26. doi: 10.1016/j.virusres.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathur A, Khanna N, Chaturvedi UC. 1992. Breakdown of blood-brain barrier by virus-induced cytokine during Japanese encephalitis virus infection. Int J Exp Pathol 73:603–611. [PMC free article] [PubMed] [Google Scholar]
- 19.Roe K, Kumar M, Lum S, Orillo B, Nerurkar VR, Verma S. 2012. West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J Gen Virol 93:1193–1203. doi: 10.1099/vir.0.040899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toborek M, Lee YW, Flora G, Pu H, Andras IE, Wylegala E, Hennig B, Nath A. 2005. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol 25:181–199. doi: 10.1007/s10571-004-1383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erbar S, Maisner A. 2010. Nipah virus infection and glycoprotein targeting in endothelial cells. Virol J 7:305. doi: 10.1186/1743-422X-7-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopez-Ramirez MA, Fischer R, Torres-Badillo CC, Davies HA, Logan K, Pfizenmaier K, Male DK, Sharrack B, Romero IA. 2012. Role of caspases in cytokine-induced barrier breakdown in human brain endothelial cells. J Immunol 189:3130–3139. doi: 10.4049/jimmunol.1103460. [DOI] [PubMed] [Google Scholar]
- 23.McGavern DB, Kang SS. 2011. Illuminating viral infections in the nervous system. Nat Rev Immunol 11:318–329. doi: 10.1038/nri2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.German AC, Myint KSA, Mai NTH, Pomeroy I, Phu NH, Tzartos J, Winter P, Collett J, Farrar J, Barrett A, Kipar A, Esiri MM, Solomon T. 2006. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model. Trans R Soc Trop Med Hyg 100:1135–1145. doi: 10.1016/j.trstmh.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P, Sen E, Basu A. 2007. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55:483–496. doi: 10.1002/glia.20474. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Hou L, Ye J, Liu X, Dan H, Jin M, Chen H, Cao S. 2010. Development of a convenient immunochromatographic strip for the diagnosis of infection with Japanese encephalitis virus in swine. J Virol Methods 168:51–56. doi: 10.1016/j.jviromet.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 27.Liu TH, Liang LC, Wang CC, Liu HC, Chen WJ. 2008. The blood-brain barrier in the cerebrum is the initial site for the Japanese encephalitis virus entering the central nervous system. J Neurovirol 14:514–521. doi: 10.1080/13550280802339643. [DOI] [PubMed] [Google Scholar]
- 28.Phares TW, Fabis MJ, Brimer CM, Kean RB, Hooper DC. 2007. A peroxynitrite-dependent pathway is responsible for blood-brain barrier permeability changes during a central nervous system inflammatory response: TNF-alpha is neither necessary nor sufficient. J Immunol 178:7334–7343. doi: 10.4049/jimmunol.178.11.7334. [DOI] [PubMed] [Google Scholar]
- 29.Famakin B, Mou Y, Spatz M, Lawal M, Hallenbeck J. 2012. Downstream Toll-like receptor signaling mediates adaptor-specific cytokine expression following focal cerebral ischemia. J Neuroinflamm 9:174. doi: 10.1186/1742-2094-9-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai CY, Ou YC, Chang CY, Pan HC, Chang CJ, Liao SL, Su HL, Chen CJ. 2012. Endothelial Japanese encephalitis virus infection enhances migration and adhesion of leukocytes to brain microvascular endothelia via MEK-dependent expression of ICAM1 and the CINC and RANTES chemokines. J Neurochem 123:250–261. doi: 10.1111/j.1471-4159.2012.07889.x. [DOI] [PubMed] [Google Scholar]
- 31.Yan X, Prosniak M, Curtis MT, Weiss ML, Faber M, Dietzschold B, Fu ZF. 2001. Silver-haired bat rabies virus variant does not induce apoptosis in the brain of experimentally infected mice. J Neurovirol 7:518–527. doi: 10.1080/135502801753248105. [DOI] [PubMed] [Google Scholar]
- 32.Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. 2006. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci 26:1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liou ML, Hsu CY. 1998. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res 293:389–394. doi: 10.1007/s004410051130. [DOI] [PubMed] [Google Scholar]
- 34.Zhou J, Stohlman SA, Hinton DR, Marten NW. 2003. Neutrophils promote mononuclear cell infiltration during viral-induced encephalitis. J Immunol 170:3331–3336. doi: 10.4049/jimmunol.170.6.3331. [DOI] [PubMed] [Google Scholar]
- 35.MacLean AG, Rasmussen TA, Bieniemy D, Lackner AA. 2004. Activation of the blood-brain barrier by SIV (simian immunodeficiency virus) requires cell-associated virus and is not restricted to endothelial cell activation. Biochem Soc Trans 32:750–752. doi: 10.1042/BST0320750. [DOI] [PubMed] [Google Scholar]
- 36.Nottet HS, Persidsky Y, Sasseville VG, Nukuna AN, Bock P, Zhai QH, Sharer LR, McComb RD, Swindells S, Soderland C, Gendelman HE. 1996. Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain. J Immunol 156:1284–1295. [PubMed] [Google Scholar]
- 37.Chai Q, He WQ, Zhou M, Lu H, Fu ZF. 2014. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol 88:4698–4710. doi: 10.1128/JVI.03149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR. 2009. West Nile virus infection modulates human brain microvascular endothelial cell tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology 385:425–433. doi: 10.1016/j.virol.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruzek D, Salat J, Singh SK, Kopecky J. 2011. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T cells. PLoS One 6:e20472. doi: 10.1371/journal.pone.0020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasquez Ochoa M, Garcia Cordero J, Gutierrez Castaneda B, Santos Argumedo L, Villegas Sepulveda N, Cedillo Barron L. 2009. A clinical isolate of dengue virus and its proteins induce apoptosis in HMEC-1 cells: a possible implication in pathogenesis. Arch Virol 154:919–928. doi: 10.1007/s00705-009-0396-7. [DOI] [PubMed] [Google Scholar]
- 41.Andras IE, Toborek M. 2011. HIV-1-induced alterations of claudin-5 expression at the blood-brain barrier level. Methods Mol Biol 762:355–370. doi: 10.1007/978-1-61779-185-7_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrey JD, Olsen AL, Siddharthan V, Motter NE, Wang H, Taro BS, Chen D, Ruffner D, Hall JO. 2008. Increased blood-brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J Gen Virol 89:467–473. doi: 10.1099/vir.0.83345-0. [DOI] [PubMed] [Google Scholar]
- 43.Morrey JD, Siddharthan V, Olsen AL, Roper GY, Wang H, Baldwin TJ, Koenig S, Johnson S, Nordstrom JL, Diamond MS. 2006. Humanized monoclonal antibody against West Nile virus envelope protein administered after neuronal infection protects against lethal encephalitis in hamsters. J Infect Dis 194:1300–1308. doi: 10.1086/508293. [DOI] [PubMed] [Google Scholar]
- 44.Monath TP, Cropp CB, Harrison AK. 1983. Mode of entry of a neurotropic arbovirus into the central nervous system: reinvestigation of an old controversy. Lab Invest 48:399–410. [PubMed] [Google Scholar]
- 45.King NJ, Getts DR, Getts MT, Rana S, Shrestha B, Kesson AM. 2007. Immunopathology of flavivirus infections. Immunol Cell Biol 85:33–42. doi: 10.1038/sj.icb.7100012. [DOI] [PubMed] [Google Scholar]
- 46.Bai F, Kong KF, Dai J, Qian F, Zhang L, Brown CR, Fikrig E, Montgomery RR. 2010. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J Infect Dis 202:1804–1812. doi: 10.1086/657416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misra UK, Kalita J. 2010. Overview: Japanese encephalitis. Prog Neurobiol 91:108–120. doi: 10.1016/j.pneurobio.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 48.Aleyas AG, George JA, Han YW, Rahman MM, Kim SJ, Han SB, Kim BS, Kim K, Eo SK. 2009. Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J Immunol 183:2462–2474. doi: 10.4049/jimmunol.0801952. [DOI] [PubMed] [Google Scholar]
- 49.Cao S, Li Y, Ye J, Yang X, Chen L, Liu X, Chen H. 2011. Japanese encephalitis virus wild strain infection suppresses dendritic cells maturation and function, and causes the expansion of regulatory T cells. Virol J 8:39. doi: 10.1186/1743-422X-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen ST, Liu RS, Wu MF, Lin YL, Chen SY, Tan DT, Chou TY, Tsai IS, Li L, Hsieh SL. 2012. CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog 8:e1002655. doi: 10.1371/journal.ppat.1002655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. 2008. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med 205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King NJC. 2011. The immunopathogenesis of neurotropic flavivirus infection, p 25–53. In Ruzek D. (ed), Flavivirus encephalitis. InTech, Rijeka, Croatia. doi: 10.5772/22243. [DOI] [Google Scholar]
- 53.Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol 79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steain M, Gowrishankar K, Rodriguez M, Slobedman B, Abendroth A. 2011. Upregulation of CXCL10 in human dorsal root ganglia during experimental and natural varicella-zoster virus infection. J Virol 85:626–631. doi: 10.1128/JVI.01816-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Racz I, Nadal X, Alferink J, Banos JE, Rehnelt J, Martin M, Pintado B, Gutierrez-Adan A, Sanguino E, Bellora N, Manzanares J, Zimmer A, Maldonado R. 2008. Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. J Neurosci 28:12136–12145. doi: 10.1523/JNEUROSCI.3402-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen CJ, Ou YC, Lin SY, Raung SL, Liao SL, Lai CY, Chen SY, Chen JH. 2010. Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J Gen Virol 91:1028–1037. doi: 10.1099/vir.0.013565-0. [DOI] [PubMed] [Google Scholar]
- 57.Bhowmick S, Duseja R, Das S, Appaiahgiri MB, Vrati S, Basu A. 2007. Induction of IP-10 (CXCL10) in astrocytes following Japanese encephalitis. Neurosci Lett 414:45–50. doi: 10.1016/j.neulet.2006.11.070. [DOI] [PubMed] [Google Scholar]
- 58.Giulian D. 1993. Reactive glia as rivals in regulating neuronal survival. Glia 7:102–110. doi: 10.1002/glia.440070116. [DOI] [PubMed] [Google Scholar]
- 59.Kong LY, Wilson BC, McMillian MK, Bing G, Hudson PM, Hong JS. 1996. The effects of the HIV-1 envelope protein gp120 on the production of nitric oxide and proinflammatory cytokines in mixed glial cell cultures. Cell Immunol 172:77–83. doi: 10.1006/cimm.1996.0217. [DOI] [PubMed] [Google Scholar]
- 60.Mishra MK, Kumawat KL, Basu A. 2008. Japanese encephalitis virus differentially modulates the induction of multiple proinflammatory mediators in human astrocytoma and astroglioma cell-lines. Cell Biol Int 32:1506–1513. doi: 10.1016/j.cellbi.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 61.Man S, Ubogu EE, Ransohoff RM. 2007. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol 17:243–250. doi: 10.1111/j.1750-3639.2007.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roe K, Orillo B, Verma S. 2014. West Nile virus-induced cell adhesion molecules on human brain microvascular endothelial cells regulate leukocyte adhesion and modulate permeability of the in vitro blood-brain barrier model. PLoS One 9:e102598. doi: 10.1371/journal.pone.0102598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao L, Toriumi H, Kuang Y, Chen H, Fu ZF. 2009. The roles of chemokines in rabies virus infection: overexpression may not always be beneficial. J Virol 83:11808–11818. doi: 10.1128/JVI.01346-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spindler KR, Hsu TH. 2012. Viral disruption of the blood-brain barrier. Trends Microbiol 20:282–290. doi: 10.1016/j.tim.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. 2003. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci 116:4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- 66.Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR. 2010. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J 24:1023–1034. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]