

ABSTRACT
A major challenge to oncolytic virus therapy is that individual cancers vary in their sensitivity to oncolytic viruses, even when these cancers arise from the same tissue type. Variability in response may arise due to differences in the initial genetic lesions leading to cancer development. Alternatively, susceptibility to viral oncolysis may change during cancer progression. These hypotheses were tested using cells from a transgenic mouse model of prostate cancer infected with vesicular stomatitis virus (VSV). Primary cultures from murine cancers derived from prostate-specific Pten deletion contained a mixture of cells that were susceptible and resistant to VSV. Castration-resistant cancers contained a higher percentage of susceptible cells than cancers from noncastrated mice. These results indicate both susceptible and resistant cells can evolve within the same tumor. The role of Pten deletion was further investigated using clonal populations of murine prostate epithelial (MPE) progenitor cells and tumor-derived Pten−/− cells. Deletion of Pten in MPE progenitor cells using a lentivirus vector resulted in cells that responded poorly to interferon and were susceptible to VSV infection. In contrast, tumor-derived Pten−/− cells expressed higher levels of the antiviral transcription factor STAT1, activated STAT1 in response to VSV, and were resistant to VSV infection. These results suggest that early in tumor development following Pten deletion, cells are primarily sensitive to VSV, but subsequent evolution in tumors leads to development of cells that are resistant to VSV infection. Further evolution in castration-resistant tumors leads to tumors in which cells are primarily sensitive to VSV.
IMPORTANCE There has been a great deal of progress in the development of replication-competent viruses that kill cancer cells (oncolytic viruses). However, a major problem is that individual cancers vary in their sensitivity to oncolytic viruses, even when these cancers arise from the same tissue type. The experiments presented here were to determine whether both sensitive and resistant cells are present in prostate cancers originating from a single genetic lesion in transgenic mice, prostate-specific deletion of the gene for the tumor suppressor Pten. The results indicate that murine prostate cancers are composed of both cells that are sensitive and cells that are resistant to oncolytic vesicular stomatitis virus (VSV). Furthermore, androgen deprivation led to castration-resistant prostate cancers that were composed primarily of cells that were sensitive to VSV. These results are encouraging for the use of VSV for the treatment of prostate cancers that are resistant to androgen deprivation therapy.
INTRODUCTION
There has been a great deal of progress in the development of new replication-competent viruses that kill cancer cells (oncolytic viruses), in understanding their mechanisms of oncolysis, and in their advancement to clinical trials (1–3). The key biological underpinning of oncolytic virus therapy is that activation of proliferative signaling pathways in cancer cells often leads to downregulation of antiviral pathways, making cancer cells more susceptible to virus infection than normal cells (4–7). Vesicular stomatitis virus (VSV) is a well-established example of a highly cytolytic virus with a tropism for cancers that have downregulated their antiviral responses (5). Our laboratory and others have made a variety of genetic modifications to enhance the selectivity of VSV for cancers versus normal tissues (7–14). For example, viruses with mutations in the viral M protein, which is responsible for suppressing host antiviral responses, are defective in their ability to invade normal tissues (15, 16) but effectively infect cancers that are defective in their antiviral responses (3). Genetically engineered VSV is currently in a phase I clinical trial for localized treatment of hepatocellular carcinoma (2). However, one of the major challenges to oncolytic virus therapy is that individual cancer cell lines vary dramatically in their sensitivity to oncolytic viruses, even when these cancers arise from the same tissue type (7, 13, 17–22). The experiments presented here address the origin of these differences in susceptibility to oncolytic VSV among prostate cancers.
Human prostate cancer develops as normal prostate epithelium acquires a series of mutations and epigenetic changes that lead to invasive adenocarcinoma of the prostate (23). Further mutations lead to development of metastatic prostate cancer that spreads to other organs. For patients with localized prostate cancer, radiation therapy and/or radical prostatectomy typically achieve >90% disease-free survival within 5 years (24, 25). However, treatment for metastatic disease is less effective (26). Since androgenic effects are important for growth of normal and malignant prostatic cells, androgen deprivation therapy was developed to control prostate cancer growth. Nonetheless, nearly all men with metastatic prostate cancer eventually develop castration-resistant disease after treatment with androgen deprivation, such that the cancer cells continue to proliferate in the presence of low levels or absence of androgen (26). Patients who present with castration-resistant disease typically have a poor prognosis, even with recent improvements in therapy for this disease (26). These patients would be candidates for oncolytic virus therapy.
Individual prostate cancer cell lines vary dramatically in their sensitivity to VSV. Some prostate cancer cell lines, such as LNCaP, have substantial defects in their antiviral responses and are highly susceptible to oncolysis by VSV (13, 17). Other cell lines, such as PC3, retain their ability to mount an antiviral response and have constitutively high levels of expression of antiviral genes that render them more resistant to VSV than normal prostate epithelial cells (17, 27). The variability in response of prostate cancer cells to VSV infection may arise due to differences in the initial genetic lesions leading to prostate cancer development. Alternatively, susceptibility to VSV oncolysis may change during cancer progression. Human prostate cancers are heterogeneous in histologic architecture, immunophenotype, and genetic diversity (28). This also raises the possibility that susceptible and resistant cells may coexist in the same tumor. These hypotheses were tested using primary prostate epithelial cell cultures from transgenic Pb-Cre4 × Ptenloxp/loxp mice, in which prostate tumors develop from a single genetic lesion, i.e., deletion of the Pten gene (29).
Results presented here show that primary cultures from murine cancers derived from prostate-specific Pten deletion contained a mixture of cells that were susceptible and resistant to VSV. Furthermore, castration-resistant cancers contained a higher percentage of susceptible cells than cancers from noncastrated mice. These results indicate that both susceptible and resistant cells can evolve within the same tumor due to heterogeneity in tumor evolution. The role of Pten in sensitivity to VSV infection was further investigated using clonal populations of murine prostate epithelial (MPE) progenitor cells and tumor-derived Pten−/− cells. Deletion of Pten in MPE progenitor cells using a self-deleting lentivirus vector resulted in cells that were poorly responsive to interferon (IFN) stimulation, and were susceptible to VSV infection. In comparison, tumor-derived Pten−/− cells constitutively expressed higher levels of the antiviral transcription factor STAT1, activated STAT1 in response to VSV infection, and were correspondingly resistant to VSV infection. These results suggest that in the early stages of tumor development following Pten deletion, cells destined to become prostate cancer are primarily sensitive to VSV, but subsequent evolution in tumors leads to the development of cells that are resistant to VSV infection. Further evolution in castration-resistant tumors leads to tumors composed primarily of cells that are sensitive to VSV oncolysis.
MATERIALS AND METHODS
Mice.
Prostate-specific Pten−/− mice were generated by crossing PtenLoxp/Loxp (PtenL/L) mice with mice of the ARR2Probasin-Cre transgenic line PB-cre4, in which expression of the Cre recombinase is under the control of a modified rat prostate-specific probasin promoter, as previously described (30, 31). PtenL/L mice backcrossed to C57BL/6 mice (32) were a generous gift from Yong Chen (Wake Forest School of Medicine). DNA was acquired via tail snipping for genotyping. Castration procedure was performed on prostate-specific Pten−/− mice at 3 months of age under isoflurane anesthesia. Recovery from the procedure was carefully monitored. All animals were maintained in an isolated environment. Animal care was conducted in compliance with the state and federal Animal Welfare Acts and the standards and policies from the U.S. Department of Health and Human Services. The protocol was approved by the Institutional Animal Care and Use Committee of Wake Forest School of Medicine.
Cells and viruses.
Primary cultures of epithelial cells were derived from prostates of normal PtenL/L, tumor-bearing Pten−/− and castration-resistant tumor-bearing Pten−/− mice at 3 and 6 months of age. The culture method was a modification of that described by Nandi et al. for primary cultures of mouse mammary epithelium (33) modified for prostate monolayer cultures (33, 34). Briefly, prostate lobes were microdissected and washed with HEPES-buffered saline containing 5% penicillin-streptomycin. The tissues were minced using a sterile razor blade and digested for 2 to 3 h using 225 U of collagenase (type IV; Worthington)/ml. Digestion was considered complete when stroma was released from the organoids. The digested tissue was layered onto a Percoll gradient, and the epithelial organoids were separated from the stromal cells and debris by gradient centrifugation (2,000 rpm, 20 min). The fraction containing epithelial organoids was isolated and washed repeatedly. The organoids were cultured in Dulbecco modified Eagle medium/F-12 (1:1) containing 1% fetal bovine serum, 10% bovine serum albumin (Sigma), 10 ng of cholera toxin (BD biosciences)/ml, 28 μg of bovine pituitary extract (Hammond Cell Tech)/ml, 80 μg of gentamicin/ml, 8 μg of insulin (Gibco)/ml, alpha-tocopherol (2.3 × 10−6 M), 5 μg of transferrin (Sigma)/ml, and trace elements. The organoids were cultured in dishes coated with rat-tail collagen. Cell outgrowths from the organoids formed a confluent monolayer by day 6 of harvest and were used for experiments at passage 2 and passage 3.
Generation and culture of MPE progenitor cells from PtenL/L mice, acutely deleted Pten−/− mice, and tumor-derived Pten−/− mice were previously described (35) and maintained in the medium described above (34–36). Briefly, PtenL/L cells were spontaneously immortalized from prostate epithelial cells of 8-week-old PtenL/L mice. Acutely deleted Pten−/− cells were derived by transducing PtenL/L cells with a self-deleting lentivirus expressing cre recombinase to achieve Pten deletion in vitro (37). Tumor-derived Pten−/− cells were obtained by spontaneously immortalized prostate cells from 16-week-old Pten−/− mice. The recombinant viruses, rwt and rM51R, as well as viruses encoding green fluorescent protein (GFP) and a palmitoylated form of dsRed were prepared in BHK cells as described previously (38).
Fluorescence microscopy and image analysis.
Primary cells were seeded onto imaging plates coated with rat-tail collagen. Cells were infected with viruses that express GFP at a multiplicity of infection (MOI) of 10 for 16 h, washed three times with phosphate-buffered saline (PBS), and fixed with 4% paraformaldehyde at room temperature for 15 min. The cells were permeabilized with PBS containing 0.25% Triton X-100 (PBST) for 15 min. The cells were washed with PBS and blocked with 1% bovine serum albumin in PBST for 30 min. The cells were then incubated with anti-pancytokeratin conjugated to phycoerythrin (PE; Sigma) for 1 h with constant agitation. The cells were then washed and incubated with 1 μg of DAPI (4′,6′-diamidino-2-phenylindole)/ml to label cellular DNA.
High-content images were acquired using a BD Pathway 855 instrument. Image analysis was performed using BD Attovision software. Briefly, a region of interest was defined using the area occupied by DAPI fluorescence to denote single cells. For each region of interest, the fluorescence intensity in GFP and PE channels were determined. The number of cells that expressed both viral protein (GFP) and cytokeratin (PE) were expressed as a percentage of the total number of cells that expressed cytokeratin (PE). The images shown are merged images of the GFP, PE, and DAPI channels.
Long-term culture cells were seeded on six-well tissue culture dishes. Cells were infected with VSV-GFP or VSV-dsRed at the multiplicities indicated in the figures. At 24 h postinfection, fluorescence and bright-field images were captured using a Retiga EX 1350 digital camera (QImaging Corp.) attached to a Nikon Eclipse TE 300 inverted microscope.
Cell viability assay.
Cells were grown in 96-well tissue culture dishes until 70% confluent and then infected with viruses at the MOIs indicated in the figures. At various times postinfection, live cells were measured by using an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (cell proliferation kit 1; Roche Diagnostics).
Metabolic labeling.
Cells were grown in six-well tissue culture dishes until 70 to 90% confluent and then infected with viruses at an MOI of 10. At the times postinfection indicated in the figures, cells were pulse-labeled for 20 min using [35S]methionine at 100 μCi/ml. Cells lysates were harvested in radioimmunoprecipitation assay (RIPA) buffer and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Radioactivity was analyzed by phosphorescence imaging (Typhoon FLA 9500; GE Healthcare Life Sciences).
Immunoblotting analysis.
Cells were seeded in six-well tissue culture dishes until 70 to 90% confluent and then mock or virus infected at the indicated MOIs. At the indicated times postinfection, cell lysates were prepared in RIPA buffer containing 1 μM aprotinin, 1 μM leupeptin, 1 μM E-64 protease inhibitor, 500 μM AEBSF [4-(2-aminoethyl)-benzenesulfonyl fluoride], and 1× PhosSTOP (Roche Applied Sciences). Proteins were resolved by SDS-PAGE and transferred onto 0.45-μm-pore-size polyvinyl difluoride membranes. The membranes were blocked for 1 h in PBS containing 0.1% Tween 20 and 5% nonfat dried milk. Immunoblots were then probed with antibodies against the viral matrix (M) protein, phospho-STAT1 (tyrosine 701; Cell Signaling), or STAT1 (Cell Signaling). Phospho-STAT1 was probed before total STAT1. Proteins were detected using enhanced chemiluminescence substrate (Thermo Scientific). The intensity of the bands was quantified by using ImageJ software.
IFN responsiveness assay.
Cells were pretreated with the concentrations of universal type I IFN (PBL interferon source) indicated in the figures. The cells were infected with virus at the indicated MOIs. At 72 h postinfection, the cells were treated with MTT reagent, and cell viability was determined as described above.
Statistical analysis.
Statistical analysis of two groups was performed using a Student t test. One-way analysis of variance (ANOVA) was performed on data with two or more groups. Two-way ANOVA was performed for all pairwise multiple comparisons. The Bonferroni post-test was performed for all multiple comparisons. Statistical analyses were performed with GraphPad Prism software.
RESULTS
Infection of primary prostate tumor cells with VSV.
Variability in the response of prostate cancer cells to VSV infection may arise due to differences in the initial genetic lesions leading to prostate cancer development. Alternatively, susceptibility to VSV oncolysis may change during cancer progression. Furthermore, prostate cancers are heterogeneous in cellular phenotypes (28). This raises the possibility that susceptible and resistant cells may coexist in the same tumor. These hypotheses were tested using primary prostate epithelial cell cultures from transgenic Pb-Cre4 × Ptenloxp/loxp mice, in which prostate tumors develop from a single genetic lesion, deletion of the Pten gene (29).
Cells were cultured from mouse prostates according to the procedure described by Barclay et al. (34), which promotes epithelial cell growth. Cells cultured from normal mouse prostates or prostate tumors were analyzed for cytokeratin expression to identify cells of epithelial origin (Fig. 1). Cells from normal PtenL/L prostates, Pten−/− prostate tumors, and castration-resistant Pten−/− prostate tumors were seeded at passage 2 or 3 onto imaging plates, labeled with DAPI as a marker for the nucleus, and immunolabeled using a pan-cytokeratin antibody as a marker of epithelial cells. Mouse embryo fibroblasts were used as a negative control for cytokeratin labeling. Fluorescence of individual cells was quantified using a high-content cellular imaging system. A total of 85 to 95% of the cultured prostate cells were positive for cytokeratin, indicating their epithelial origin. The fluorescence intensity among individual cells varied considerably, a finding consistent with heterogeneity in cytokeratin composition among epithelial cells and differential reactivity of the pan-cytokeratin antibody. In previously published data, these cells also express markers of both the luminal and the basal epithelium (34).
FIG 1.

Primary cultures of mouse prostate epithelial cells express cytokeratins. Primary cultures of mouse prostate epithelial cells were cultured from normal PtenL/L prostates, Pten−/− prostate tumors, and castration-resistant Pten−/− prostate tumors. Cells were seeded into imaging plates and labeled with DAPI (blue) as a marker for cellular DNA and immunolabeled for cytokeratin (red) as a marker for epithelial cells. Using the DAPI label to define the region of interest to denote single cells, the percentages of cells immunolabeled with cytokeratin were determined and are shown in the graph. Murine embryonic fibroblasts (MEFs) were used as a negative control for cytokeratin immunolabeling.
In order to determine whether cells derived from Pten−/− prostate tumors are susceptible to VSV infection, primary prostate cells from 3-month-old prostate-specific Pten−/− mice and their littermate controls were infected with recombinant enhanced GFP (eGFP)-expressing virus containing either a wild-type (wt) M protein (rwt-GFP virus) or an isogenic virus containing mutant M protein (rM51R-GFP virus) at an MOI of 10 for 16 h. The cells were then fixed and stained with DAPI and anti-cytokeratin antibody. Fluorescence images of normal and tumor-derived cells infected with rwt-GFP are shown in Fig. 2A and B, respectively. The numbers of cells that express both GFP and cytokeratin were expressed as a percentage of total cytokeratin-positive cells, and data from multiple experiments are shown in Fig. 2C.
FIG 2.

Prostate tumors contain a mixture of resistant and susceptible cells. Primary cultures of prostate epithelial cells were established from Pten−/− prostate tumors of 3-month-old mice and their littermate normal PtenL/L controls. Cells were seeded into imaging plates and were infected with rwt-GFP or rM51R-GFP viruses (green). Cells were fixed, permeabilized, immunolabeled for cytokeratin (red) and stained with DAPI to label cellular DNA (blue), and then analyzed with a high-content imaging system. Representative fluorescence images are shown in panels A and B. The numbers of cells that were positive for GFP and cytokeratin were expressed as a percentage of total cytokeratin-positive cells (C and D). The data shown are averages of at least three experiments ± the standard errors of the mean (SEM).
Most of the normal PtenL/L prostate epithelial cells infected with rwt-GFP virus express GFP (ca. 75%). These results are similar to previous results indicating that normal human prostate epithelial cells are susceptible to VSV (13). In contrast, significantly fewer cells derived from Pten−/− prostate tumors (42%) expressed detectable GFP. The percentage of prostate cells that expressed detectable GFP from the M protein mutant virus (rM51R-GFP) was less than that of cells infected by rwt-GFP virus (Fig. 2D), likely due to the induction of antiviral responses in some of the cells by the M protein mutant virus. Nonetheless, the prostate epithelial cells infected with M protein mutant virus exhibited a similar trend in that fewer cells from Pten−/− tumors support GFP expression compared to normal PtenL/L prostate epithelial cells. These results indicate that even with a single defined initiating genetic lesion of Pten deletion, a heterogeneous response to VSV infection develops in prostate tumors during carcinogenesis.
In order to determine the susceptibility of castration-resistant Pten−/− prostate tumors to VSV infection, transgenic Pb-Cre4 × Ptenloxp/loxp mice were castrated at 3 months of age, and cells were cultured from castration-resistant Pten−/− prostate tumors at 6 months of age. Normal PtenL/L prostate cells and Pten−/− prostate tumor cells from 6-month-old noncastrated mice served as controls. The cells were infected, stained, and imaged, and subjected to the same analysis as in Fig. 2. In Fig. 3A and B are shown images of cells from castration-resistant Pten−/− and 6-month-old control Pten−/− tumors after infection with rwt-GFP virus, and quantification of multiple experiments with rwt-GFP and rM51R-GFP viruses is shown in Fig. 3C and D, respectively. Most of the castration-resistant Pten−/− prostate tumor cells expressed GFP similar to the results with normal PtenL/L cells. In contrast, the cells from control Pten−/− mice at 6 months still consisted of a mixture of cells that were primarily resistant to VSV and support very little viral gene expression, similar to the results with 3-month-old mice in Fig. 2. These results indicate that in the androgen-depleted environment of castrated mice, VSV-sensitive tumor cells are predominant over VSV-resistant tumor cells.
FIG 3.

Castration-resistant tumor cells are primarily susceptible to VSV infection. Pten−/− mice were castrated at 3 months of age and by 6 months of age had developed castration-resistant tumors. Primary cultures of prostate epithelial cells were established from Pten−/− prostate tumors of 6-month-old mice, castration-resistant tumors, and their littermate normal PtenL/L control prostates. Cells were seeded into imaging plates and infected with rwt-GFP and rM51R-GFP viruses (green). Cells were fixed, permeabilized, immunolabeled for cytokeratin (red) and stained with DAPI to label cellular DNA (blue), and then analyzed with a high-content imaging system. Representative fluorescence images are shown in panels A and B. The numbers of cells that were positive for GFP and cytokeratin were expressed as a percentage of total cytokeratin-positive cells (C and D). The data shown are averages of at least three experiments ± the SEM.
Susceptibility to VSV in clonal populations of murine prostate epithelial progenitor cells and tumor-derived Pten−/− cells.
The observation that Pten−/− prostate tumors contain a mixture of VSV-sensitive and VSV-resistant cells suggests that resistance does not result from Pten deletion alone but instead results from cellular changes during tumor progression. To further determine whether resistance to VSV develops from a single genetic lesion of Pten deletion or from tumor progression, clonal populations of mouse prostate epithelial cells in long-term culture were used. Murine prostate cells capable of long-term growth are derived from primary cultures such as those in Fig. 1 to 3 after passage 20 or higher (34) and will be referred to here as murine prostate epithelial (MPE) progenitor cells. MPE progenitor cells bear markers of both luminal and basal epithelial cells, as well as stem cell markers, such as Sca-1 and CD49f, and have the key stem cell properties of self-renewal and multilineage differentiation (39). Deletion of the Pten gene in MPE progenitor cells was achieved by transducing PtenL/L cells in vitro with lentivirus that expresses self-deleting Cre recombinase, and clonal populations of transduced and mock-transduced cells were derived as described previously (35). A clonal population of cells derived from tumors of 4-month-old Pten−/− mice was used to model tumor-derived Pten−/− cells (40). Pten−/− cells resulting from lentivirus transduction are referred to as acutely deleted to distinguish them from tumor-derived Pten−/− cells.
Control PtenL/L, acutely deleted, and tumor-derived Pten−/− cells were infected with rwt virus that encodes either GFP or a palmitoylated version of the fluorescent protein dsRed at increasing MOIs. Similar results were obtained with either virus. Figure 4 shows representative bright-field and fluorescence images of cells at 24 h postinfection with VSV-dsRed. Most control PtenL/L cells and acutely deleted Pten−/− cells expressed dsRed at MOI of 1, and nearly all expressed dsRed at an MOI of 10 or 50. However, tumor-derived Pten−/− cells expressed only low levels of dsRed independently of MOI. This result supports the conclusion that resistance does not result from Pten deletion alone but instead results from cellular changes during tumor progression.
FIG 4.

Tumor-derived Pten−/− cells support very little viral gene expression even at a high MOI. Vector control PtenL/L cells and acutely deleted Pten−/− cells (A) or nontransduced control PtenL/L cells and tumor-derived Pten−/− cells (B) were infected with rwt-dsRed virus at MOIs of 1, 10, and 50. Bright-field and fluorescence images were obtained at 24 h postinfection. The images shown are representative from two to three independent experiments.
The difference in viral gene expression in tumor-derived Pten−/− cells versus acutely deleted or normal prostate epithelial cells was confirmed by immunoblot analysis of viral M protein and by radiolabeling infected cells with [35S]methionine. Control PtenL/L, acutely deleted and tumor-derived Pten−/− cells were mock infected or infected with rwt virus. At 6 and 12 h postinfection, cell lysates were prepared and immunoblotted for VSV M protein (Fig. 5A). Individual band intensities were quantified and expressed as a percentage of band intensity of control PtenL/L cells at 12 h postinfection, and the results from multiple experiments are shown in Fig. 5B. There was no significant difference in M protein expression between control PtenL/L and acutely deleted Pten−/− cells, whereas M protein expression in tumor-derived Pten−/− cells was only detectable in longer exposures and was only ca. 10% of that of control cells.
FIG 5.
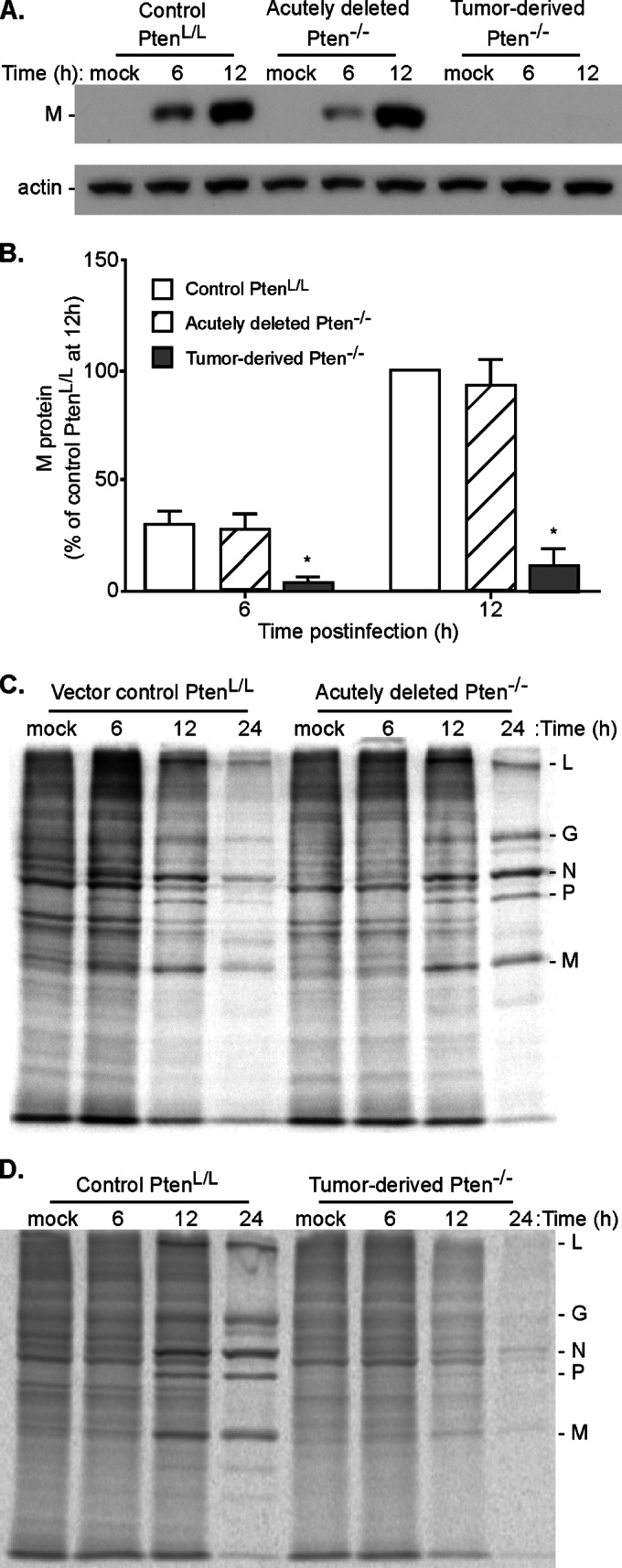
Viral protein expression in control PtenL/L cells and acutely deleted Pten−/− cells versus tumor-derived Pten−/− cells. (A) Control PtenL/L, acutely deleted Pten−/−, and tumor-derived Pten−/− cells were infected with rwt virus at an MOI of 10. Cell lysates were harvested at 6 and 12 h postinfection and probed for viral matrix protein (M) protein expression by immunoblotting. Representative immunoblots of at least three experiments are shown. (B) Viral matrix (M) protein expression was quantified and expressed as a percentage of M protein expression in control PtenL/L cells at 12 h postinfection. *, P < 0.05. (C) Vector control PtenL/L and acutely deleted Pten−/− cells were infected with rwt virus at an MOI of 10 or mock infected (M). At 6, 12, and 24 h postinfection, the cells were pulse-labeled with [35S]methionine. Cell lysates were harvested and resolved on SDS-PAGE gels. Representative phosphorimages are shown. Viral proteins L, G, N, P, and M are indicated on the right. (D) Nontransduced control PtenL/L and tumor-derived Pten−/− cells were infected with rwt virus at an MOI of 10 or mock infected (M). At 6, 12, and 24 h postinfection, cells were pulse-labeled with [35S]methionine and analyzed as in panel C.
The rates of viral protein synthesis were determined by pulse-labeling infected cells with [35S]methionine at 6, 12, and 24 h postinfection. Cell lysates were analyzed by SDS-PAGE and phosphorescence imaging. Figure 5C shows representative phosphorimages from vector control PtenL/L and acutely deleted Pten−/− cells, and in Fig. 5D, images from control PtenL/L and tumor-derived Pten−/− cells infected with rwt virus. Control PtenL/L and acutely deleted Pten−/− cells had a pattern of viral protein synthesis that is typical for permissive cell types, in which viral protein synthesis was detectable as early as 6 h after infection, and continued from 12 to 24 h after infection. However, the synthesis of viral proteins in tumor-derived Pten−/− cells was barely detectable above the background of host protein synthesis.
Also apparent in Fig. 5C and D is the inhibition of host protein synthesis by VSV, in which labeling of host proteins visible in mock-infected cells declined during the course of infection. The inhibition of host protein synthesis in tumor-derived Pten−/− cells was not notably different from that in control cells, suggesting that the low level of M protein expression in these cells was sufficient to inhibit host protein synthesis. For example, the amount of M protein required to inhibit host gene expression in permissive cells is 100- to 1,000-fold less than that produced during virus infection (41).
Susceptibility to VSV-induced cell death in MPE progenitor cells and tumor-derived Pten−/− cells.
VSV infection leads to cell death by multiple pathways, some of which are dependent on the ability of M protein to inhibit host gene expression and others of which are independent of M protein's inhibitory activity (42). These two classes of mechanisms can be distinguished by comparing cell death induced by virus with wt M protein (rwt virus) to that of M protein mutant virus (rM51R virus). Control PtenL/L, acutely deleted, and tumor-derived Pten−/− cells were infected with rwt or rM51R virus at an MOI of 10. At various times after infection, cells were incubated with MTT reagent. Cell viability was expressed as a percentage of mock-infected cells at that time point (Fig. 6). Most control PtenL/L and acutely deleted Pten−/− cells were killed by either virus by 36 to 48 h postinfection (Fig. 6A and B). At 12 and 24 h postinfection, the acutely deleted Pten−/− cells had significantly more viability than control PtenL/L cells. This may be due to more active survival signaling in Pten−/− cells. Tumor-derived Pten−/− cells were more resistant to virus-induced cell death than either PtenL/L or acutely deleted Pten−/− cells, with ca. 50% of tumor-derived Pten−/− cells remaining viable by 48 h postinfection with rwt virus (Fig. 6C) and ca. 75% remaining viable after infection with rM51R virus (Fig. 6D). Some of the resistant cells continue to divide after infection with VSV (unpublished data), which may account for the transient increase in viability observed at 36 h. Changes in the rate of MTT metabolism in individual cells could also contribute to this effect.
FIG 6.

In a single cycle infection, tumor-derived Pten−/− cells are more resistant to virus-induced cell death than control PtenL/L and acutely deleted Pten−/− cells. Vector control PtenL/L and acutely deleted (A and B) or nontransduced control PtenL/L and tumor-derived Pten−/− cells (C and D) were infected with rwt (A and C) or rM51R (B and D) viruses at an MOI of 10. Cell viability at the indicated times was determined by using an MTT assay and is expressed as a percentage of the mock-infected cells. The data show averages of at least three experiments ± the SEM. *, P < 0.05.
The difference in the responses of tumor-derived Pten−/− cells was even more striking in multiple cycle infections (MOI of 0.1, Fig. 7). PtenL/L or acutely deleted Pten−/− cells were largely nonviable by 48 h postinfection with either rwt or rM51R virus (Fig. 7A and B), indicating that virus had spread throughout the culture from the initially infected cells. In contrast, tumor-derived Pten−/− cells remained viable throughout the time course of infection with either virus.
FIG 7.

In a multiple cycle infection, tumor-derived Pten−/− cells are highly resistant to virus-induced cell death. Vector control PtenL/L and acutely deleted (A and B) or nontransduced control PtenL/L and tumor-derived Pten−/− cells (C and D) were infected with rwt (A and C) or rM51R (B and D) viruses at an MOI of 0.1. Cell viability was determined using MTT assay and is expressed as a percentage of the mock-infected cells. The data show averages of at least three experiments ± the SEM. *, P < 0.05.
Antiviral signaling through STAT1 in MPE progenitor cells and tumor-derived Pten−/− cells.
One of the hallmarks of human prostate cancer cells that are relatively resistant to VSV, such as PC3 cells, is that they have constitutively high levels of expression of antiviral gene products, such as the IFN-responsive transcription factor STAT1, and that STAT1 is activated by phosphorylation in cells infected with wild-type VSV, which normally suppresses IFN production and STAT1 activation in most cell types (17). To determine whether this is also the case for murine tumor-derived cells, control PtenL/L, acutely deleted, and tumor-derived Pten−/− cells were analyzed for STAT1 expression and activation by phosphorylation by immunoblots. Cells were mock infected or infected with rwt or rM51R virus at MOI of 10, and cell lysates were prepared at 6 and 12 h postinfection. Lysates were analyzed by immunoblots with antibodies against phospho-STAT1 or total STAT1 (Fig. 8). Mock-infected tumor-derived Pten−/− cells constitutively expressed much higher levels of STAT1 than PtenL/L or acutely deleted Pten−/− cells (Fig. 8A and C). Furthermore, STAT1 was phosphorylated in response to infection of tumor-derived cells with rwt virus (Fig. 8A and B), likely due to the production of type I IFNs by these cells. In contrast, there was little if any phosphorylation of STAT1 in PtenL/L or acutely deleted Pten−/− cells infected with rwt virus (Fig. 8A and B), due to the inhibitory activity of the wt M protein, which prevents IFN production in most cell types.
FIG 8.

Tumor-derived Pten−/− cells constitutively express high levels of STAT1, which is phosphorylated in response to infection with VSV with either wt or mutant M protein. Control PtenL/L, acutely deleted, and tumor-derived Pten−/− cells were infected with rwt (A) or rM51R (B) viruses at an MOI of 10. Cell lysates were harvested at the indicated times and probed for phospho-STAT1 and total STAT1 expression. Representative immunoblots for at least three experiments are shown. The levels of phospho-STAT1 and total STAT1 were quantified and are expressed as the percentage of levels in tumor-derived Pten−/− cells at 6 h postinfection. The data show averages of at least three experiments ± the SEM. *, P < 0.05.
All three cell types responded to infection with M protein mutant virus by phosphorylation of STAT1 (Fig. 8D and E), a finding consistent with the induction of type I IFN by this virus (43). The levels of phospho-STAT1 at 6 h postinfection differed among the three cell types in the following order: tumor-derived Pten−/− cells > PtenL/L cells > acutely deleted Pten−/− cells. Also notable in Fig. 8D and F was the increase in total STAT1 expression following infection with rM51R virus, which is consistent with the positive-feedback induction of STAT1 expression induced by type I IFNs. Again, the three cell types differed in the magnitude of this effect in the order tumor-derived Pten−/− cells > PtenL/L cells > acutely deleted Pten−/− cells.
IFN responsiveness of PtenL/L and Pten−/− MPE progenitor cells.
The differential response of phospho-STAT1 in PtenL/L versus acutely deleted Pten−/− cells infected with rM51R virus in Fig. 8D and E suggested that the acutely deleted cells might be less responsive to type I IFN than control PtenL/L cells. This hypothesis was tested in the experiments in Fig. 9 by pretreating the cells with the indicated doses of type I IFN for 16 h. The cells were then infected with rwt virus at MOI of 10, and cell viability was determined at 72 h. Control PtenL/L cells responded to IFN in a dose-dependent manner and were protected from rwt virus-induced cell death at the high dose. In contrast, acutely deleted Pten−/− cells were susceptible to rwt virus infection even at high doses of IFN, indicating that the deletion of Pten inhibits the IFN responsiveness of these cells.
FIG 9.

Acutely deleted Pten−/− cells respond poorly to IFN stimulation. Control PtenL/L and acutely deleted Pten−/− cells were pretreated with the indicated concentrations of IFN and then infected with rwt virus at an MOI of 10. Cell viability was determined by using an MTT assay at 72 h postinfection and is expressed as a percentage of mock-infected cells. *, P < 0.05.
DISCUSSION
VSV is an attractive candidate oncolytic virus. To date, the virus has been tested as an oncolytic agent against a variety of cancer types, including glioblastomas, head and neck cancers, pancreatic adenocarcinomas, hepatocellular carcinomas, colorectal cancers, and breast and prostate cancers, to name a few (17, 20, 44–46). Even more excitingly, a recombinant VSV has recently moved to a phase I clinical trial for treatment of patients with hepatocellular carcinoma (http://clinicaltrials.gov/ct2/show/NCT01628640?term=vesicular+stomatitis+virus&rank=1). However, the common trend observed throughout the literature is that some cancer cell lines are susceptible to VSV oncolysis, whereas others are resistant, despite originating from the same tissue type. The basis for this diversity in response among cancers could be due to two general classes of mechanisms. One possibility is that different initiating mutations cause cancers to evolve through different paths such that one type of initiating mutation leads to sensitivity, whereas a different mutation leads to resistance. Alternatively, heterogeneity in the tumor microenvironment may lead to development of some cells that are sensitive to viral oncolysis and others that are resistant, even within the same tumor. The results presented here show that this second class of mechanism is responsible for the heterogeneity in responsiveness among prostate cancer cells originating from a single initiating mutation, deletion of the Pten gene.
The data presented here, together with earlier data, support a model for the progression of susceptibility versus resistance in prostate cancers shown in Fig. 10. In primary cultures of murine prostate epithelial cells, most of the cells derived from normal prostate epithelium were quite susceptible to VSV infection (Fig. 2, 3). Likewise, in murine clonal cell populations, normal MPE progenitor cells were sensitive to VSV infection (Fig. 4 to 7). VSV replication in normal murine prostates has been observed following intraprostatic inoculation in vivo (47, 48). However, the virus was quickly cleared in vivo, and there did not appear to be damage to the normal prostatic epithelium. This is likely due to the responsiveness of the normal epithelial cells to type I interferons (Fig. 9 and 10a) and other antiviral cytokines produced by immune cells. These results are similar to results with human cells, in which normal human prostate epithelial cells are also susceptible to VSV, but mount antiviral responses in response to type I IFN that can protect them from VSV cytolysis (13).
FIG 10.

Model for the development of VSV-susceptible and VSV-resistant cells in prostate cancers resulting from Pten deletion.
Similar to normal MPE progenitor cells, acutely deleted Pten−/− cells were susceptible to VSV infection. However, in contrast to normal MPE cells, acutely deleted Pten−/− cells were poorly responsive to IFN stimulation (Fig. 4 to 7 and 9). This suggests that in the early stages of tumor development, cells destined to become prostate cancer are primarily sensitive to VSV (Fig. 10b). Defects in the response to IFNs can also account for the observations of Moussavi et al. that virus replication in VSV-susceptible cells in Pten−/− tumors in vivo was greater than in normal prostates (47), suggesting that some of the cells in the tumors are defective in their antiviral responses. Together, these studies support the idea that the loss of Pten can decrease the protective effect of IFN against VSV infection. This is likely due to cross talk between the pathways activated by Pten deletion and the pathways activated by IFNs. For example, these pathways interact at the level of glycogen synthase kinase-3β (GSK-3β). GSK-3β is a serine/threonine kinase whose activity enhances STAT1 activation by inhibiting Src homology-2 domain-containing phosphatase 2 (SHP2) (49). GSK-3β is inhibited through phosphorylation by AKT following the loss of Pten. Further, activation of AKT and inhibition of GSK-3β activity following Pten loss leads to downregulation of STAT1 (50). This could be one mechanism through which loss of Pten may serve to downregulate antiviral responses.
Despite the suppressive effects on the antiviral response of acute deletion of Pten, by the time prostate cancers were well established in Pten−/− mice at 3 months of age, most of the prostate cancer cells were more resistant to VSV than normal prostate epithelial cells (Fig. 2, 3, and 10c). Similarly, a clonal population of tumor-derived Pten−/− cells was much more resistant to VSV infection than normal MPE progenitor cells (Fig. 4 to 7). These results are similar to results with human cells, in which normal human prostate epithelial cells are also susceptible to VSV, whereas some human prostate cancer cell lines such as PC3 are more resistant than normal cells (13). The resistance of PC3 cells is due to delays in several early steps in VSV replication that appear to be mediated by constitutive expression of antiviral genes, as well as robust responsiveness to type I IFNs (17, 27). Similarly, murine tumor-derived Pten−/− cells expressed high levels of the antiviral transcription factor STAT1, which was activated upon virus infection, even with wt VSV, which normally suppresses IFN synthesis in most cell types (Fig. 8).
Loss of Pten does not account for the difference in susceptibility between VSV-sensitive LNCaP and VSV-resistant PC-3 human prostate cancer cells, since neither of these two cell lines have functional Pten protein, and both have constitutively active AKT pathway (51). Similarly, as shown here, a clonal population of tumor-derived Pten−/− cells exhibited resistance to VSV infection (Fig. 4 to 7), whereas another cell line derived from murine Pten−/− tumors was shown to be susceptible to VSV (47). Together, these data are consistent with the idea that evolution within tumors can change the susceptibility to VSV infection in some of the cells by promoting antiviral gene expression.
What could be the cause of the changes that must have occurred during tumor evolution that enhance resistance to oncolytic VSV? Whereas activated AKT inhibits antiviral gene expression through inhibition of GSK-3β, activated AKT can also interact with the IFN response pathway at other levels to give a completely different phenotype, a phenotype that promotes IFN responsiveness and antiviral defense. For example, one downstream effector of AKT is protein kinase C-δ (PKC-δ). PKC-δ is activated by treatment with type I IFNs and associates with STAT1 to phosphorylate and activate STAT1 (reviewed in references 52 and 53). Another downstream substrate of AKT1 is EMSY, a transcriptional repressor that in conjunction with other binding partners represses transcription of IFN-stimulated genes (54). AKT1 phosphorylates EMSY and removes this repressive activity from ISREs, which allows transcription of antiviral genes (54). Thus, activation of the AKT pathway can also have antiviral effects.
A key result presented here is that castration-resistant Pten−/− tumor cells are primarily susceptible to virus infection (Fig. 3 and 10d). This is important because men with castration-resistant disease would be the target population for oncolytic virus therapy. At least two possibilities that are not mutually exclusive could contribute to this effect. One possibility is that cells that are IFN responsive and resistant to VSV infection are more sensitive to the effects of androgen withdrawal. As a result, the cells that remain and undergo expansion are castration resistant and susceptible to virus. Alternatively, androgen withdrawal induces death of tumor cells irrespective of their IFN responsiveness and sensitivity to virus infection. However, the progression of surviving cells to castration resistance is accompanied by the downregulation of antiviral genes. Both of these scenarios suggest a relationship between androgen signaling and antiviral response. Some castration-resistant tumors develop independence from androgen receptor (AR) signaling, such as appears to be the case of PC3 cells. However, in many castration-resistant prostate cancers, AR activation and signaling remains sustained through a variety of mechanisms (reviewed in reference 55), such as amplification of AR gene copy number, gain-of-function mutations in the AR gene, and alternative splice isoforms that encode constitutively active AR variants. Activation of AR inhibits IFN responsiveness and silencing AR stimulates IFN-induced gene expression (56), suggesting that androgen signaling and IFN signaling may be antagonistic. In a system in which AR signaling predominates, IFN responses may be attenuated, whereas cancers that have little or no AR signaling may retain IFN responsiveness.
The observation that prostate cancers resulting from Pten deletion consist of a mixture of cells that are susceptible to VSV and cells that have developed resistance is an important result that can account for the wide variation in susceptibility among prostate cancer cell lines. The presence of cells in prostate cancers that are resistant to virus infection does not imply that virus will be ineffective. It is well established that the antitumoral effects of oncolytic viruses in vivo involve additional mechanisms besides direct viral oncolysis (reviewed in reference 2). These mechanisms include disruption of the tumor vasculature and induction of antitumor immunity. Also, it may be possible to enhance the susceptibility of cancer cells that are initially resistant to oncolytic viruses by treatment with pharmacologic agents that suppress antiviral gene expression (27, 57). That being said, cancers with a large percentage of resistant cells are not as likely to be responsive to oncolytic virus therapy as those with a large percentage of susceptible cells. In this respect, the observation that castration increases the percentage of VSV-susceptible cells in Pten−/− prostate cancers is encouraging for the use of oncolytic VSV for the treatment of prostate cancers for which androgen ablation therapy has failed.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (NIH) grants R01 AI032983 and R01 AI105012 (D.S.L.). We also acknowledge the support of the Cellular Imaging Shared Resource and the Cell and Virus Vector Core Laboratory of the Comprehensive Cancer Center of Wake Forest University, supported by NIH grant P30 CA012197.
We thank Yong Q. Chen (Wake Forest School of Medicine) for the mouse strains used here. We thank Zachary D. Cary (Health Advances) for the VSV strains expressing dsRed.
REFERENCES
- 1.Bell J, McFadden G. 2014. Viruses for tumor therapy. Cell Host Microbe 15:260–265. doi: 10.1016/j.chom.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. 2014. Going viral with cancer immunotherapy. Nat Rev Cancer 14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 3.Parato KA, Senger D, Forsyth PA, Bell JC. 2005. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer 5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- 4.Balachandran S, Barber GN. 2000. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life 50:135–138. doi: 10.1080/713803696. [DOI] [PubMed] [Google Scholar]
- 5.Barber GN. 2004. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol 17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- 6.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med 6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 7.Stojdl DF, Lichty BD, ten Oever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275. doi: 10.1016/S1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 8.Ammayappan A, Nace R, Peng KW, Russell SJ. 2013. Neuroattenuation of vesicular stomatitis virus through picornaviral internal ribosome entry sites. J Virol 87:3217–3228. doi: 10.1128/JVI.02984-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayala-Breton C, Barber GN, Russell SJ, Peng KW. 2012. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum Gene Ther 23:484–491. doi: 10.1089/hum.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janelle V, Brassard F, Lapierre P, Lamarre A, Poliquin L. 2011. Mutations in the glycoprotein of vesicular stomatitis virus affect cytopathogenicity: potential for oncolytic virotherapy. J Virol 85:6513–6520. doi: 10.1128/JVI.02484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller JM, Bidula SM, Jensen TM, Reiss CS. 2010. Vesicular stomatitis virus modified with single chain IL-23 exhibits oncolytic activity against tumor cells in vitro and in vivo. Int J Interferon Cytokine Mediator Res 2010:63–72. doi: 10.2147/IJICMR.S9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muik A, Stubbert LJ, Jahedi RZ, Geibeta Y, Kimpel J, Dold C, Tober R, Volk A, Klein S, Dietrich U, Yadollahi B, Falls T, Miletic H, Stojdl D, Bell JC, von Laer D. 2014. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res 74:3567–3578. doi: 10.1158/0008-5472.CAN-13-3306. [DOI] [PubMed] [Google Scholar]
- 13.Ahmed M, Cramer SD, Lyles DS. 2004. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology 330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 14.Obuchi M, Fernandez M, Barber GN. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol 77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed M, Marino TR, Puckett S, Kock ND, Lyles DS. 2008. Immune response in the absence of neurovirulence in mice infected with m protein mutant vesicular stomatitis virus. J Virol 82:9273–9277. doi: 10.1128/JVI.00915-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trottier MD, Lyles DS, Reiss CS. 2007. Peripheral, but not central nervous system, type I interferon expression in mice in response to intranasal vesicular stomatitis virus infection. J Neurovirol 13:433–445. doi: 10.1080/13550280701460565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carey BL, Ahmed M, Puckett S, Lyles DS. 2008. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J Virol 82:12104–12115. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebert O, Harbaran S, Shinozaki K, Woo SL. 2005. Systemic therapy of experimental breast cancer metastases by mutant vesicular stomatitis virus in immune-competent mice. Cancer Gene Ther 12:350–358. doi: 10.1038/sj.cgt.7700794. [DOI] [PubMed] [Google Scholar]
- 19.Porosnicu M, Mian A, Barber GN. 2003. The oncolytic effect of recombinant vesicular stomatitis virus is enhanced by expression of the fusion cytosine deaminase/uracil phosphoribosyltransferase suicide gene. Cancer Res 63:8366–8376. [PubMed] [Google Scholar]
- 20.Ahmed M, Puckett S, Lyles DS. 2010. Susceptibility of breast cancer cells to an oncolytic matrix (M) protein mutant of vesicular stomatitis virus. Cancer Gene Ther 17:883–892. doi: 10.1038/cgt.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P, Grdzelishvili VZ. 2012. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J Virol 86:3073–3087. doi: 10.1128/JVI.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart JHt, Ahmed M, Northrup SA, Willingham M, Lyles DS. 2011. Vesicular stomatitis virus as a treatment for colorectal cancer. Cancer Gene Ther 18:837–849. doi: 10.1038/cgt.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tapia-Laliena MA, Korzeniewski N, Hohenfellner M, Duensing S. 2014. High-risk prostate cancer: a disease of genomic instability. Urol Oncol 32:1101–1107. doi: 10.1016/j.urolonc.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Boorjian SA, Karnes RJ, Rangel LJ, Bergstralh EJ, Blute ML. 2008. Mayo Clinic validation of the D'Amico risk group classification for predicting survival following radical prostatectomy. J Urol 179:1354–1360. doi: 10.1016/j.juro.2007.11.061. [DOI] [PubMed] [Google Scholar]
- 25.Zelefsky MJ, Yamada Y, Fuks Z, Zhang Z, Hunt M, Cahlon O, Park J, Shippy A. 2008. Long-term results of conformal radiotherapy for prostate cancer: impact of dose escalation on biochemical tumor control and distant metastases-free survival outcomes. Int J Radiat Oncol Biol Phys 71:1028–1033. doi: 10.1016/j.ijrobp.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 26.Suzman DL, Antonarakis ES. 2014. Castration-resistant prostate cancer: latest evidence and therapeutic implications. Ther Adv Med Oncol 6:167–179. doi: 10.1177/1758834014529176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben Yebdri F, Van Grevenynghe J, Tang VA, Goulet ML, Wu JH, Stojdl DF, Hiscott J, Lin R. 2013. Triptolide-mediated inhibition of interferon signaling enhances vesicular stomatitis virus-based oncolysis. Mol Ther 21:2043–2053. doi: 10.1038/mt.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, Kim R, Rubin MA, Pienta KJ. 2004. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res 64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 29.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. 2003. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4:209–221. doi: 10.1016/S1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 30.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. 2002. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, Sangiorgi FO, Maxson RE, Sucov HM, Roy-Burman P. 2001. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev 101:61–69. doi: 10.1016/S0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 32.Berquin IM, Min Y, Wu R, Wu J, Perry D, Cline JM, Thomas MJ, Thornburg T, Kulik G, Smith A, Edwards IJ, D'Agostino R, Zhang H, Wu H, Kang JX, Chen YQ. 2007. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J Clin Invest 117:1866–1875. doi: 10.1172/JCI31494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nandi S, Imagawa W, Tomooka Y, McGrath MF, Edery M. 1984. Collagen gel culture system and analysis of estrogen effects on mammary carcinogenesis. Arch Toxicol 55:91–96. doi: 10.1007/BF00346045. [DOI] [PubMed] [Google Scholar]
- 34.Barclay WW, Cramer SD. 2005. Culture of mouse prostatic epithelial cells from genetically engineered mice. Prostate 63:291–298. doi: 10.1002/pros.20193. [DOI] [PubMed] [Google Scholar]
- 35.Axanova LS, Chen YQ, McCoy T, Sui G, Cramer SD. 2010. 1,25-dihydroxyvitamin D(3) and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells. Prostate 70:1658–1671. doi: 10.1002/pros.21201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu M, Shi L, Cimic A, Romero L, Sui G, Lees CJ, Cline JM, Seals DF, Sirintrapun JS, McCoy TP, Liu W, Kim JW, Hawkins GA, Peehl DM, Xu J, Cramer SD. 2012. Suppression of Tak1 promotes prostate tumorigenesis. Cancer Res 72:2833–2843. doi: 10.1158/1538-7445.AM2012-2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM. 2001. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc Natl Acad Sci U S A 98:11450–11455. doi: 10.1073/pnas.201415498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitlow ZW, Connor JH, Lyles DS. 2006. Preferential translation of vesicular stomatitis virus mRNAs is conferred by transcription from the viral genome. J Virol 80:11733–11742. doi: 10.1128/JVI.00971-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barclay WW, Axanova LS, Chen W, Romero L, Maund SL, Soker S, Lees CJ, Cramer SD. 2008. Characterization of adult prostatic progenitor/stem cells exhibiting self-renewal and multilineage differentiation. Stem Cells 26:600–610. doi: 10.1634/stemcells.2007-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Wu J, Suburu J, Gu Z, Cai J, Axanova LS, Cramer SD, Thomas MJ, Perry DL, Edwards IJ, Mucci LA, Sinnott JA, Loda MF, Sui G, Berquin IM, Chen YQ. 2012. Effect of dietary polyunsaturated fatty acids on castration-resistant Pten-null prostate cancer. Carcinogenesis 33:404–412. doi: 10.1093/carcin/bgr290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyles DS, McKenzie MO, Ahmed M, Woolwine SC. 1996. Potency of wild-type and temperature-sensitive vesicular stomatitis virus matrix protein in the inhibition of host-directed gene expression. Virology 225:172–180. doi: 10.1006/viro.1996.0585. [DOI] [PubMed] [Google Scholar]
- 42.Kopecky SA, Willingham MC, Lyles DS. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J Virol 75:12169–12181. doi: 10.1128/JVI.75.24.12169-12181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol 77:4646–4657. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wollmann G, Tattersall P, van den Pol AN. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J Virol 79:6005–6022. doi: 10.1128/JVI.79.10.6005-6022.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cary ZD, Willingham MC, Lyles DS. 2011. Oncolytic vesicular stomatitis virus induces apoptosis in U87 glioblastoma cells by a type II death receptor mechanism and induces cell death and tumor clearance in vivo. J Virol 85:5708–5717. doi: 10.1128/JVI.02393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moerdyk-Schauwecker M, Shah NR, Murphy AM, Hastie E, Mukherjee P, Grdzelishvili VZ. 2013. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: role of type I interferon signaling. Virology 436:221–234. doi: 10.1016/j.virol.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moussavi M, Fazli L, Tearle H, Guo Y, Cox M, Bell J, Ong C, Jia W, Rennie PS. 2010. Oncolysis of prostate cancers induced by vesicular stomatitis virus in PTEN knockout mice. Cancer Res 70:1367–1376. doi: 10.1158/1538-7445.AM10-1367. [DOI] [PubMed] [Google Scholar]
- 48.Moussavi M, Tearle H, Fazli L, Bell JC, Jia W, Rennie PS. 2013. Targeting and killing of metastatic cells in the transgenic adenocarcinoma of mouse prostate model with vesicular stomatitis virus. Mol Ther 21:842–848. doi: 10.1038/mt.2012.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai CC, Kai JI, Huang WC, Wang CY, Wang Y, Chen CL, Fang YT, Lin YS, Anderson R, Chen SH, Tsao CW, Lin CF. 2009. Glycogen synthase kinase-3beta facilitates IFN-γ-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J Immunol 183:856–864. doi: 10.4049/jimmunol.0804033. [DOI] [PubMed] [Google Scholar]
- 50.Tseng PC, Huang WC, Chen CL, Sheu BS, Shan YS, Tsai CC, Wang CY, Chen SO, Hsieh CY, Lin CF. 2012. Regulation of SHP2 by PTEN/AKT/GSK-3β signaling facilitates IFN-γ resistance in hyperproliferating gastric cancer. Immunobiology 217:926–934. doi: 10.1016/j.imbio.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 51.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. 1998. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A 95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 53.Reich NC, Pfeffer LM. 1990. Evidence for involvement of protein kinase C in the cellular response to interferon alpha. Proc Natl Acad Sci U S A 87:8761–8765. doi: 10.1073/pnas.87.22.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ezell SA, Polytarchou C, Hatziapostolou M, Guo A, Sanidas I, Bihani T, Comb MJ, Sourvinos G, Tsichlis PN. 2012. The protein kinase Akt1 regulates the interferon response through phosphorylation of the transcriptional repressor EMSY. Proc Natl Acad Sci U S A 109:E613–E621. doi: 10.1073/pnas.1115029109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen MM, Abate-Shen C. 2010. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bettoun DJ, Scafonas A, Rutledge SJ, Hodor P, Chen O, Gambone C, Vogel R, McElwee-Witmer S, Bai C, Freedman L, Schmidt A. 2005. Interaction between the androgen receptor and RNase L mediates a cross-talk between the interferon and androgen signaling pathways. J Biol Chem 280:38898–38901. doi: 10.1074/jbc.C500324200. [DOI] [PubMed] [Google Scholar]
- 57.Shulak L, Beljanski V, Chiang C, Dutta SM, Van Grevenynghe J, Belgnaoui SM, Nguyen TL, Di Lenardo T, Semmes OJ, Lin R, Hiscott J. 2014. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J Virol 88:2927–2940. doi: 10.1128/JVI.03406-13. [DOI] [PMC free article] [PubMed] [Google Scholar]