

ABSTRACT
The interaction between hepatitis C virus (HCV) and cellular immune responses during very early infection is critical for disease outcome. To date, the impact of antigen-specific cellular immune responses on the evolution of the viral population establishing infection and on potential escape has not been studied. Understanding these early host-virus dynamics is important for the development of a preventative vaccine. Three subjects who were followed longitudinally from the detection of viremia preseroconversion until disease outcome were analyzed. The evolution of transmitted/founder (T/F) viruses was undertaken using deep sequencing. CD8+ T cell responses were measured via enzyme-linked immunosorbent spot (ELISpot) assay using HLA class I-restricted T/F epitopes. T/F viruses were rapidly extinguished in all subjects associated with either viral clearance (n = 1) or replacement with viral variants leading to establishment of chronic infection (n = 2). CD8+ T cell responses against 11 T/F epitopes were detectable by 33 to 44 days postinfection, and 5 of these epitopes had not previously been reported. These responses declined rapidly in those who became chronically infected and were maintained in the subject who cleared infection. Higher-magnitude CD8+ T cell responses were associated with rapid development of immune escape variants at a rate of up to 0.1 per day. Rapid escape from CD8+ T cell responses has been quantified for the first time in the early phase of primary HCV infection. These rapid escape dynamics were associated with higher-magnitude CD8+ T cell responses. These findings raise questions regarding optimal selection of immunogens for HCV vaccine development and suggest that detailed analysis of individual epitopes may be required.
IMPORTANCE A major limitation in our detailed understanding of the role of immune response in HCV clearance has been the lack of data on very early primary infection when the transmitted viral variants successfully establish the acute infection. This study was made possible through the availability of specimens from a unique cohort of asymptomatic primary infection cases in whom the first available viremic samples were collected approximately 3 weeks postinfection and at regular intervals thereafter. The study included detailed examination of both the evolution of the viral population and the host cellular immune responses against the T/F viruses. The findings here provide the first evidence of host cellular responses targeting T/F variants and imposing a strong selective force toward viral escape. The results of this study provide useful insight on how virus escapes the host response and consequently on future analysis of vaccine-induced immunity.
INTRODUCTION
Following primary hepatitis C virus (HCV) infection, approximately 75% of individuals fail to clear the virus (1), resulting in chronic hepatitis, progressive fibrosis, and increased risk of liver failure and hepatocellular carcinoma (2). Due to the high mutation rate, HCV exists within each infected host as a diverse, rapidly evolving population. However, the majority of new infections are initiated by only a few (1 to 3) unique transmitted/founder (T/F) variants (3, 4). To date, there has been no study of the selective pressures exerted by adaptive immune responses against HCV T/F viruses. Previous studies have investigated the immune response using reference consensus viral antigens in primary HCV infection and documented both HCV-specific CD4+ and CD8+ responses (5, 6), but these studies are limited by their focus on late stages of infection (i.e., after seroconversion) (7–10). It therefore remains unresolved whether the host T cell response targets viral populations that successfully establish a new infection and provides early selection pressure for viral evolution and immune escape. Understanding these mechanisms is important for the selection of immunogens for T cell-based vaccines that confer protection (3, 8, 10–12).
HCV can escape CD8+ T cell responses in several ways. For instance, amino acid mutations within the epitope or its flanking region can abrogate epitope processing and presentation, or continued antigen presentation can impair the antiviral activity of the T cell, resulting in T cell exhaustion (13). Current estimates suggest that 20 to 32% of the viral mutations observed over time at the consensus level in primary infection are driven by CD8+ T cell responses (5, 14–16). In contrast, studies on a small number of chimpanzees have reported that 65% of the early nonsynonymous mutations are located within HLA class I epitopes (17). Although viral escape from CD8+ T cell responses has been documented in acute HCV infection (6, 18–20), the lack of well-characterized longitudinal samples from the very early acute phase of primary HCV infections has prevented investigation of the dynamics of the immune response against the T/F viruses and the varied propensity of T/F viruses to escape.
We have previously identified T/F variants from the very early phase of primary HCV infection employing a mathematical model that describes the exponential growth of a viral population under neutral evolution, and with this we estimated the sequence and viral evolution of T/F viruses (3). Our study revealed that viral diversity initially increased following a strong genetic bottleneck after transmission of only 1 to 3 T/F variants (3, 4). In addition, after an initial increase in viral diversity, a second genetic bottleneck occurred at approximately 100 days postinfection (p.i.), which was then followed by clearance of the T/F viruses. In the case of the chronic progressors, a new viral population replaced the T/F virus. Based on this knowledge, we hypothesized that immune responses contributed to the selective forces driving a rapid viral evolution during acute HCV infection.
The rate at which immune escape variants arise in acute HCV infection is currently unknown. For human immunodeficiency virus (HIV) infection, studies of the rate of escape from CD8+ T cell responses against T/F viruses clearly show that escape can arise as early as a few weeks postinfection at a rate of 0.1 per day, suggesting that escape variants can rapidly replace T/F virus within a few days (21). Unlike HIV, HCV can be naturally cleared by the host immune response; hence, it is reasonable to hypothesize that a T/F-specific immune response exists in HCV infection and that this may be reproduced with a T cell-based vaccine with T/F-specific immunogens. However, understanding the rate of emergence of immune escape variants is key to immunogen selection. A T cell-based preventative vaccine against HCV which included multiple HLA class I-restricted genotype 1 epitopes has been tested in phase I and II studies (22, 23) and induced both CD4+ and CD8+ T cell responses, with some evidence of cross-genotype immunity. However, there are no data on the propensity for the development of escape variants from this vaccine, nor any direct evidence of protection.
This study examined the magnitude and timing of CD8+ T cell responses against HCV T/F viruses in very early HCV infection, along with the kinetics of escape mutations and their temporal association with sustained HCV-specific cellular immune responses.
MATERIALS AND METHODS
Subjects and viral sequences.
Incident cases prior to seroconversion were recruited from the Hepatitis C Incidence and Transmission Study in prisons (HITS-p), which is a prospective cohort of high-risk injecting drug users in Australia (24). Samples from these subjects were previously deep sequenced for analysis of viral evolution (3). Briefly, following detection of initial viremia, blood samples were collected frequently over a 24-week period until spontaneous clearance or chronic infection was established. HCV antibody (Ab) and HCV RNA testing was performed as described previously (25). The date of infection for each subject was estimated by subtracting the recognized mean preseroconversion window period of 51 days from the midpoint between the last HCV RNA-positive/HCV Ab-negative time point and the first seropositive time point (26). HLA class I typing was performed as previously described (27). Next-generation sequencing (NGS) data of the full-length viral population were previously reported and generated with Roche 454 FLX technology at multiple time points for each subject (3). The previous data were supplemented by an additional NGS data set from a viremic sample in subject Ch_240 at 140 days p.i. covering only the NS3 region from nucleotides (nt) 4742 to 6024 of the consensus reference genome of the T/F virus.
Bioinformatics analyses.
An optimized version of the previously published pipeline for 454 FLX data cleaning and alignment was utilized as follows (3): 454 FLX reads were cleaned with an average quality score of 15; read ends carrying low quality scores were trimmed using a sliding window of 8 nucleotides (nt), before alignment using the Burrows-Wheeler aligner–maximum entropy method (BWA-MEM) algorithm (28) with default parameters; on a subset of the data sets, the quality of the alignment was also judged based on the occurrence of indels causing frameshifts, which were mostly present in homopolymeric regions; and a visual check was performed on Tablet (29) before specific reads were eliminated. The alignment with BWA was successful for 95% to 99% of the reads. Aligned reads were then passed to Samtools (30) for generation of a BAM file.
To accurately detect single nucleotide polymorphisms (SNPs) from the aligned and cleaned 454 FLX sequences, two different SNP calling algorithms were utilized: a per site quality score-based statistical test, LoFreq (31), and a Bayesian approach for error correction using the software package ShoRAH (32). Both algorithms implement a statistical error correction model (ShoRAH via probabilistic clustering and LoFreq via discriminating SNPs individually by inferring an error probability based on the quality score assigned to individual nucleotides) and a strand bias test, where a particular error is more likely to occur in reads traversing the genome in one direction as opposed to the other. LoFreq implements a Fisher exact test of strand bias, while ShoRAH has a beta-binomial model of forward read distribution and more details; comparison between these two algorithms can be found in McElroy et al. (32). The results from these two analyses were also manually curated to exclude the following SNPs: those that were called within, or adjacent to, homopolymer regions that presented a frameshift (indels); those not detected by both algorithms; and those present only in fewer than three forward reads and three reverse reads. With this approach the lowest frequency of occurrence detected was 0.05%, which is consistent with accurate benchmark tests of these algorithms with control data sets (32).
Analysis of T/F virus.
Analysis of T/F virus from NGS data was performed as previously described (3). Briefly, haplotypes of 1,500 nt were reconstructed from NGS data across the full genome using the updated software package ShoRAH (32, 33). Each of these haplotype distributions covering the full genome of HCV was tested with Poisson Fitter (34) for evidence of star-like phylogeny for T/F virus (3). Haplotype sequences from the first viremic time point of the HCV genome of each subject were used for phylogenetic analyses. This analysis was performed with MEGA, version 5 (35). Phylogenetic trees were constructed using PhyML, performing a maximum-likelihood estimate of phylogenies from nucleotide viral sequences. A generalized time-reversible (GTR) substitution model with a gamma-distributed rate variation among sites was chosen after a model selection test with JModelTEST (36).
CD8+ T cell epitope selection.
The set of HLA-restricted peptide epitopes tested in enzyme-linked immunosorbent spot (ELISpot) assays for each subject were determined by selecting a subset of the potential autologous epitopes for each infection. For each subject, this subset was selected from three major pools of potential epitopes, defined as follows.
(i) Pool 1 consisted of the set of autologous T/F virus HLA class I-restricted epitopes (9 to 11 amino acids) that are at least 90% homologous to previous experimentally confirmed epitopes from the Immune Epitope Data Base (IEDB [www.iedb.org]). The IEDB database of HLA class I-restricted epitopes consisted of 2,965 epitopes, of which 1,098 were reported with positive T cell assay results.
(ii) Pool 2 consisted of the set of epitopes identified by IEDB epitope prediction tools (using the IEDB-recommended procedure for 9 to 11 amino acids) from the T/F autologous sequence with a high predicted binding score (defined as a half-maximal inhibitory concentration [IC50] of <500 nmol). This set consisted of approximately 500 to 1,000 epitopes per subject. Approximately 30 to 50% of these epitopes were at least 90% homologous to the experimentally confirmed epitopes available in the IEDB database.
(iii) Pool 3 consisted of the set of epitopes from the longitudinal set of autologous T/F sequences associated with a nonsynonymous substitution that subsequently reached fixation over the observed course of the infection and with an increased IC50 of >250 nmol (suggesting an immune escape variant).
A maximum of 95 epitopes were selected for each subject to account for the total peripheral blood mononuclear cells (PBMC) available for each time point in the ELISpot matrix testing approach (see below). In particular, 40 epitopes were selected from pool 1, 40 were selected from pool 2, and the remaining 15 were from the potential escape variants (pool 3). Higher preference was given to the following categories: (i) predicted epitopes (pool 2) that were restricted against two or more HLA class I alleles present in the subject and (ii) the set of epitopes overlapping pools 1 and 2, as experimentally confirmed epitopes represented in the autologous virus. A post hoc decision process was also undertaken in cases where the maximum number of planned epitopes could not be tested due to limitations in the availability of PBMC following the thawing of frozen cells. In these cases, a subset of epitopes from the selected 95 was chosen by random sampling across the three pools (see above), prioritizing epitopes in preference categories i and ii.
Autologous CD8+ T cell ELISpot assay.
Synthesized peptides (Mimotopes, Australia) were utilized in autologous CD8+ T cell ELISpot assays in pools of ≤5 peptides per well in a matrix format using the robotic Biomek FX system (Beckman Coulter, USA) as previously described (37). Peptides were pooled such that any two wells shared only one peptide, and each peptide was tested in at least two separate wells. The protocol was adjusted to optimize the detection of HCV-specific T-cell responses as follows: PBMC were added to a 96-well ELISpot plate (MAIPS; Millipore, USA) precoated with gamma interferon (IFN-γ) (Mabtech, Sweden) at a concentration of 150,000 cells per well and incubated overnight with peptides (final concentration of 10 μg/ml). Plates were read and analyzed using an AID plate reader (Autoimmun Diagnostika GmbH, Germany). A peptide/well was considered positive if any corresponding peptide elicited an IFN-γ response at a magnitude of 20 spot-forming units (SFU)/million cells and a negative-control count of zero. To confirm a response to a particular peptide, the single peptide was tested in a confirmatory IFN-γ ELISpot assay, using one peptide per well at a concentration of 200,000 cells per well. At least one well per study subject was allocated as a positive control (anti-CD3 antibody; Mabtech, Sweden), and three wells were used as negative controls. The background was defined as the mean plus three times the standard deviation of the number of spots counted in the three negative-control wells.
For subject Cl_360, 93 peptides were tested per time point, while for subjects Ch_23 and Ch_240 the number of epitopes tested varied at each time point due to heterogeneous recovery of the cells after thawing. This number ranged from 3 to 95 (the median number of peptides tested was 45 and 11 for subjects Ch_23 and Ch_240, respectively). All subjects had at least one sample tested with >70 epitopes.
Estimation of the rate of CD8+ T cell escape.
The kinetics of CD8+ T cell escape was estimated with a simple population dynamics model as previously reported in HIV infection (38). The changes in the frequency of the escape mutant in the population were fitted by the following model: f(t) = f0/(f0 + (1 − f0)e−kt), where f(t) is the frequency of the escape variant in the population, k is the rate of escape, and it is assumed that the escape variant is present at time (t) zero at the frequency f0. The escape variant was defined as any genomic variant carrying the mutation that consequently becomes fixed in the population. Therefore, the frequency of the escape variant at each time point was the sum of the frequencies of each epitope carrying the escape mutation of interest, regardless of the other amino acid positions. For epitopes where there was no evidence of the escape variant, the zero value was replaced with 1/(n + 1), where n is the coverage of the deep-sequencing data. This model was used to estimate the rate of escape only in those cases with experimental evidence (ELISpot data generated in this study) of CD8+ T cell-driven immune escape. Estimates were performed in R using the nonlinear least-squares method for nonlinear models (nls) (39).
Test for selection pressure from CD8+ T cell responses.
Normalized Shannon entropy (S) scores across the HCV genome for each time point were calculated as a measure of overall viral diversity. S scores were then obtained for epitope regions, defined as the sum of genomic regions corresponding to HLA class I epitopes predicted with IEDB tools (see above), and the nonepitope region was defined as the remaining complementary part of the genome. This analysis excluded the time point at 140 days p.i. for subject Ch_240 as only a limited region of the genome was sequenced.
In order to test the hypothesis that CD8+ T cell responses drive viral evolution, three statistical methods were utilized, as previously described (21). These analyses were based on viral sequences obtained via haplotype reconstruction using ShoRAH (see above) covering the surrounding genomic region (approximately 800 nucleotides) of the epitopes experimentally confirmed in this study. The first two methods were based on phylogenetic analyses that are implemented in the web-based package DataMonkey (www.datamonkey.org). The first method, the fixed-effect likelihood model (FEL), analyzed the ratio of nonsynonymous to synonymous evolutionary changes (dN/dS) at each codon position (a dN/dS of >1 is evidence of positive/diversifying selection), while the second method used a mixed-effects model of evolution (MEME) which is capable of identifying instances of both episodic and pervasive positive selection at the level of an individual site. For both of these methods, a phylogenetic tree constructed with PhyML was utilized along with the sequence files. A model test was performed on codon substitution models, implemented in DataMonkey (CodonTest) (40). Finally, the third method, enriched clustering statistics (ECS), is a clustering-based approach already utilized in HIV studies (21). This method statistically addresses the probability that clusters of mutations occur in a sequence region in excess of what would be expected with a random neutral model of selection. The false-discovery rate (FDR; denoted as q) was computed to correct for multiple testing (41) with a q of <0.1 to identify windows significantly enriched for mutated positions relative to the frequency of mutations outside the window.
Ethics statement.
Ethical approvals were obtained from the Human Research Ethics Committees of Justice Health (reference number GEN 31/05), the New South Wales Department of Corrective Services (reference number 05/0884), and the University of New South Wales (reference numbers 05094, 08081, 13237, 09075, 14170). Written informed consent was obtained from the participants.
Nucleotide sequence accession number.
The new NGS data are available at the European Nucleotide Archive under accession number PRJEB8405.
RESULTS
Evolution of T/F virus and viral populations during acute infection.
Three subjects with primary HCV infection followed from preseroconversion until infection outcome were analyzed (Table 1). The first available viremic sample ranged from 4 to 50 days p.i. One subject naturally cleared the infection (Cl_360), and two developed chronic infection (chronic progressors; Ch_240 and Ch_23) (Fig. 1). NGS data were available from 13 viremic time points from these subjects to study in-depth HCV RNA populations (see Table S1 in the supplemental material). The NGS data set had a median coverage of 3,814 reads per base (maximum coverage, 17,228 reads per base). An average of 187 single nucleotide polymorphisms (SNPs) per time point (n = 13) were detected, with a median frequency within the viral population of 3% (5 to 95 percentile range, 0.05% to 52%). Of these SNPs, an average of 106 (62%) were found at a frequency of >1% per time point (3). The sequence of the T/F virus was estimated from the distribution of variants at the earliest time point (HCV RNA positive and HCV Ab negative) using a mathematical model previously applied in both HIV and HCV which utilizes exponential viral growth and the distribution of viral variants to test the hypothesis that the observed population is consistent with a neutral evolution of the viral population (3).
TABLE 1.
Clinical and laboratory characteristics of the subjects
| Subjecta | Age at infection (yr) | Symptomatic | HLA type |
HCV genotype | No. of days p.i.b | Date of collection (day-mo-yr) | No. of T/F viruses | HCV Ab | HCV RNA | Viral load (IU/ml) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | ||||||||||
| Ch_240 | 21 | No | 0201 | 1501 | 0304 | 3a | −61 | 3-12-08 | 1 | − | − | 0 |
| 0201 | 5701 | 0602 | 44 | 6-26-08 | − | + | 54,887 | |||||
| 57 | 7-9-08 | + | + | 85,473 | ||||||||
| 71 | 7-23-08 | + | + | 64,063 | ||||||||
| 85 | 8-6-08 | + | + | 6,051 | ||||||||
| 99 | 8-20-08 | + | + | 497 | ||||||||
| 113 | 9-3-08 | + | + | 4,862 | ||||||||
| 140c | 9-30-08 | + | + | 1,034 | ||||||||
| 220 | 12-19-08 | + | + | 44,449 | ||||||||
| 310 | 3-19-09 | + | + | 29,552 | ||||||||
| 538 | 11-2-09 | + | + | 62,174 | ||||||||
| Ch_23 | 22 | Yes | 0201 | 4402 | 0501 | 1a | −165 | 11-6-08 | 2 | − | − | 0 |
| 0201 | 5701 | 0602 | 36 | 5-25-09 | − | + | 19,234,348 | |||||
| 44 | 6-2-09 | − | + | 17,907,338 | ||||||||
| 60 | 6-18-09 | + | + | 8,121,396 | ||||||||
| 74 | 7-2-09 | + | + | 397,185 | ||||||||
| 85 | 7-13-09 | + | + | 3,218 | ||||||||
| 101 | 7-29-09 | + | + | 398 | ||||||||
| 135 | 9-1-09 | + | + | 2,843,176 | ||||||||
| 197 | 11-2-09 | + | + | 5,896,155 | ||||||||
| 365 | 4-19-10 | + | + | 516,945 | ||||||||
| 3a | 635 | 1-14-11 | + | + | 14,738 | |||||||
| 781 | 6-9-11 | + | + | 344,388 | ||||||||
| Cl_360 | 28 | No | 3201 | 1402 | 0501 | 3a | −101 | 1-15-10 | 1 | − | − | 0 |
| 6802 | 4402 | 0802 | 30 | 5-27-10 | − | + | 5,648,631 | |||||
| 44 | 6-10-10 | − | + | 4,617,483 | ||||||||
| 58 | 6-24-10 | + | + | 14,170 | ||||||||
| 71 | 7-7-10 | + | + | 15,938 | ||||||||
| 83 | 7-19-10 | + | + | 1,060 | ||||||||
| 97 | 8-2-10 | + | + | <15 | ||||||||
| 132 | 9-6-10 | + | + | 57 | ||||||||
| 223 | 12-6-10 | + | − | 0 | ||||||||
| 422 | 6-23-11 | + | − | 0 | ||||||||
All subjects were male.
The number of days p.i. was estimated from the time to seroconversion. Boldface denotes time points that were deep sequenced for HCV variant analysis.
This data set covers only the NS3 region of the HCV genome.
FIG 1.
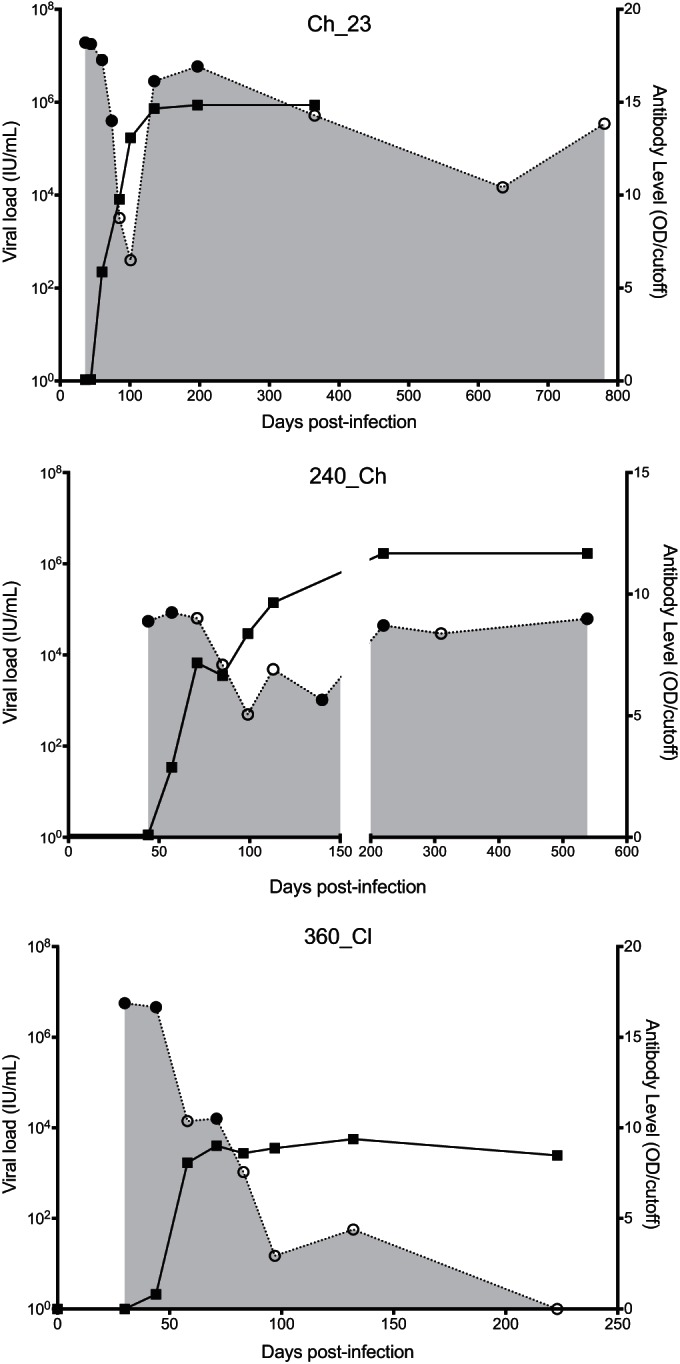
Longitudinal course of viral load and antibody levels. Two subjects were chronic progressors (Ch_23 and Ch_240), and one cleared the infection (Cl_360). Gray shading indicates viral load, with open circles indicating viremic time points and filled circles indicating time points from which viral samples were deep sequenced. Black lines indicate antibody level, with black squares indicating data points. OD, optical density.
In order to characterize the selective pressures that potentially contributed to the rapid emergence of new variants replacing the T/F virus, both algorithm-predicted and experimentally verified CD8+ T cell epitopes were identified (see Materials and Methods for details). Mutations within these epitopes were also assessed for reversion events, namely mutations towards common circulating variants that are assumed to be a fitter HCV strain (Table 2).
TABLE 2.
Fixation events identified in the two subjects with chronic infectionc
| Subject | Positiond | Genome regionf | Founder residue | Mutant residue | Time of fixation (days p.i.) | Max frequency (%)e | Reversion | CD8+ T cell epitopea |
|---|---|---|---|---|---|---|---|---|
| Ch_240 | 372 | E1 | A | V | 220 | 99.54 | No | No |
| 376 | E1 | V | I | 538 | 99.64 | No | No | |
| 400 | E2-HVR1 | T | A | 538 | 91.31 | No | No | |
| 405 | E2-HVR1 | I | V | 538 | 75.40 | No | No | |
| 408 | E2-HVR1 | K | N | 538 | 77.20 | No | No | |
| 443 | E2 | Y | H | 220 | 98.89 | No | No | |
| 483 | E2 | G | D | 220 | 99.69 | Yes | No | |
| 543 | E2 | T | A | 220 | 99.75 | No | No | |
| 583 | E2 | L | P | 538 | 80.52 | No | No | |
| 750 | NS2 | A | T | 220 | 99.34 | No | No | |
| 1509 | NS3 | T | N | 140 | 100.00 | No | Nob | |
| 1606 | NS3 | P | L | 140 | 92.60 | No | Yes | |
| 1641 | NS3 | V | I | 538 | 98.82 | Yes | Yes | |
| 1768 | NS4b | H | Y | 140 | 100.00 | No | No | |
| 1985 | NS5a | T | I | 220 | 99.79 | No | Nob | |
| 1999 | NS5a | T | A | 220 | 99.92 | No | Yes | |
| 2361 | NS5a | S | P | 538 | 78.43 | No | No | |
| 2497 | NS5b | T | A | 220 | 99.84 | Yes | No | |
| 2973 | NS5b | G | D | 220 | 99.78 | No | Yes | |
| Ch_23 | 402 | E2-HVR1 | L | F | 60 | 93.87 | No | No |
| 443 | E2 | Y | I | 60 | 96.80 | No | No | |
| 446 | E2 | N | R | 60 | 97.19 | No | No | |
| 542 | E2 | T | I | 135 | 99.57 | No | Yes | |
| 856 | NS2 | Q | H | 197 | 80.04 | No | No | |
| 921 | NS2 | F | S | 60 | 99.22 | No | No | |
| 1498 | NS3 | I | T | 135 | 91.34 | No | Yes | |
| 2629 | NS5b | K | N | 135 | 99.44 | No | Yes |
Mutation present in a CD8+ T cell epitope previously reported in literature.
Mutation present in a flanking region of HLA-I epitopes.
Boldface data represent experimentally validated responses in this study.
Polyprotein amino acid position with reference to genotype 1a strain H77 (GenBank accession number NC_004102) for Ch_23 and to genotype 3a strain CB (GenBank accession number AF046866) for Ch_240.
Max, maximum.
HVR1, hypervariable region 1.
A total of 27 high-frequency nonsynonymous nucleotide mutations (defined as mutations from the T/F that emerged at frequencies greater than 75% of the viral population) arose during primary infection, some of which reached fixation in the viral population as early as 66 days p.i. (Table 2). Of the 27 high-frequency mutations, 7 (26%) were associated with experimentally confirmed CD8+ T cell responses, suggestive of escape. Only three of these fixations were reversions toward the worldwide consensus (Table 2).
CD8+ T cell responses against T/F virus.
HLA class I-restricted epitopes were predicted from the sequences of T/F viruses, as well as from mutated variants dominating the infection at later time points. For each subject, autologous peptides were synthesized including both T/F and escape variants and further tested in IFN-γ ELISpot assays (see Materials and Methods for more details). Positive responses were identified for all three subjects (Table 3). The first detectable HCV-specific CD8+ T cell response was between 30 to 44 days p.i., after which all three subjects displayed a progressive increase in the breadth and magnitude of the responses. The increased breadth and magnitude of HCV-specific CD8+ T cell responses coincided with a decline in viral load (Fig. 2).
TABLE 3.
Summary of the HLA class I epitopes from T/F virus identified in this study with positive ELISpot responses
| Subject | HLA type | Epitope | Position (aa)a | Protein region | Previously reportedb |
|---|---|---|---|---|---|
| Ch_23 | HLA-B*44:02 | AEVIAPAVQT | 1743–1752 | NS4B | Yes |
| HLA-B*57:01 | FAWYLKGKW | 774–782 | E2 | No | |
| HLA-B*57:01 | KSKRTPMGF | 2629–2637 | NS5B | Yes | |
| HLA-B*57:01 | RAEAQLHAW | 852–860 | NS2 | No | |
| HLA-B*44:02 | AELIEANLLW | 2228–2237 | NS5A | Yes | |
| HLA A*02:01 | WLGNIIMFA | 2827–2835 | NS5B | No | |
| Ch_240 | HLA-B*57:01 | RAQAPPPSW | 1602–1610 | NS3 | Yes |
| HLA A*02:01 | RLGPVQNEV | 1633–1641 | NS3 | No | |
| Cl_360 | HLA-A*32:01 | YLTAYQATV | 1591–1599 | NS3 | No |
| HLA-A*68:02 | SVIDCNVAV | 1456–1464 | NS3 | Yes | |
| HLA-A*68:02 | ATDALMTGF | 1442–1500 | NS3 | Yes |
aa, amino acid.
Epitopes identified with 90% blast homology in the IEDB. Only positive responses with a T cell-based assay are reported.
FIG 2.

CD8+ T cell ELISpot responses against T/F variants. Each panel shows the time course of viral load (shaded gray), and ELISpot responses against T/F epitopes (colored lines). Legends indicate the epitope sequence with corresponding color, and red letters indicate mutated residues that become fixed during the course of the infection. The earliest CD8+ responses were detected preseroconversion (antibody positive [Ab +ve], vertical dotted line). The decline in viremia preceded the peak of HCV-specific CD8+ T cell responses. In the two chronic progressors (Ch_23 and Ch_240), recrudescent viremia coincided with a decline and temporary loss of detectable HCV-specific CD8+ T cell responses around 100 to 150 days p.i. In a subject that cleared infection (Cl_360), the T/F-specific CD8+ T cell responses remained stable after viral clearance.
In Cl_360, three responses were identified against the T/F, all in NS3 (Table 3). In Ch_240, two responses were identified against two epitopes of the NS3 region, and in Ch_23 six responses were found in E2, NS2, NS4B, NS5A, and NS5B. In subject Cl_360, the earliest response was identified preseroconversion at 44 days p.i. (1442ATDALMTGF1450 which is a known HLA-A1-restricted immunodominant epitope but is also predicted as an HLA-A*68:02-restricted epitope) and remained high after viral clearance. The other two CD8+ T cell responses appeared at 97 days p.i. and were also sustained well after viral clearance (Fig. 2). The three epitopes targeted did not present amino acid mutations at frequencies of >0.1%. In both chronic progressors the earliest responses were similarly identified prior to seroconversion (1602RAQAPPPSW1610 in Ch_240 and 2838WLGNIIMFA2846 in Ch_23), with the peak of the response corresponding with the elimination of the T/F viruses. Recrudescent viremia and emergence of new viral variants coincided with a decline and temporary loss of detectable HCV-specific CD8+ T cell responses (Fig. 2). In both subjects, a rise in response magnitude after 300 days p.i. was observed for some of the epitopes. In subject Ch_23, this response coincided with a second infection with a GT3a virus detected at 635 days p.i. In subject Ch_240 this coincided with a rise in viremia without detection of a new infection but with the emergence of 19 high-frequency (>75% of the population) nonsynonymous substitutions between 220 and 538 days p.i. (Table 2).
Immune escape dynamics.
Longitudinal analysis of the 11 ELISpot-confirmed CD8+ T cell responses against T/F variants revealed that in six of the epitopes nonsynonymous mutations emerged at frequencies above 1% (see Fig. S1 in the supplemental material). Four of these epitopes were previously reported as being targeted by CD8+ T cell responses (Table 3). Four epitopes (two in Ch_23 and two in Ch_240) contained nonsynonymous substitutions that became fixed in the viral population, suggestive of CD8+ T cell-driven immune escape (Table 2). Three of these mutant epitopes were also synthesized and assayed for CD8+ T cell recognition via ELISpot assay. In subject Ch_23, both escape mutants, HLA-B57-restricted 852RAEA(Q/H)LHAW860 and 2629(K/N)SKRTPMGF2637, reached fixation at 197 days p.i. and 135 days p.i., respectively. Interestingly, the variant 2629NSKRTPMGF2637 was associated with a low-frequency positive response prior to fixation, i.e., at 60 and 74 days p.i. (Fig. 3), while the variant 852RAEAHLHAW860 was negative. A complex pattern of escape with substantial toggling before fixation was also observed in both epitopes.
FIG 3.

CD8+ T cell escape from T/F virus in chronic subject Ch_23. (A) Viral load (shaded gray) and CD8+ T cell response measured via ELISpot assay against epitope 852RAEAQLHAW860. No immune responses were detected against mutants 852RTEAQLHAW860 and 852RAEAHLHAW860 (only one time point was tested), with the latter reaching fixation at 197 days p.i. A detailed list of evolving epitope variants along with the time of detection and frequency of occurrence (%) in the viral population is reported. (B) Viral load (shaded gray) and CD8+ T cell responses measured via ELISpot assay against epitope 2629KSKRTPMGF2637 and escape variant 2629NSKRTPMGF2637. A detailed list of evolving epitope variants is reported below. The high-resolution analysis of these epitope regions indicated significant residue toggling across the epitope at low frequency before preferential selection of a variant that coincided with recrudescent viremia. The color coding of the residues in the text correlates with the epitope variant colors in the graphs. DPI, days p.i.
In subject Ch_240 the HLA-A02-restricted epitope 1633RLGPVQNEV1641 mutated at the C-terminal position V1641I (toward the predominant residue in global genotype 3a sequences). The escape variant also elicited a moderate response at 310 days p.i. (53 SFU/106) (Fig. 4) although this response was not tested at previous time points due to lack of samples. The HLA-B57-restricted NS3 epitope, 1602RAQAPPPSW1610, underwent significant evolution at residue 1606, before the 1606L mutation reached fixation at 140 days p.i. (Fig. 4). Due to limited availability of PBMC, this escape variant was not tested via ELISpot assay.
FIG 4.

CD8+ T cell escape from T/F virus in chronic subject Ch_240. (A) Viral load (shaded gray) and CD8+ T cell response measured via ELISpot assay against epitope 1633RLGPVQNEV1641. A moderate immune response was detected against escape variant 1633RLGPVQNEI1641 at 310 days p.i. (no further time points were tested), which reached fixation at 538 days p.i. A detailed list of evolving epitope variants along with the time of detection and frequency of occurrence (%) in the viral population is reported. (B) Viral load (shaded gray) and CD8+ T cell response against epitope 1602RAQAPPPSW1610. No immune responses were tested against escape variants. The variant 1602RAQALPPSW1610 rapidly reached fixation at 140 days p.i. A detailed list of evolving epitope variants is reported below. The high-resolution analysis of these epitope regions indicated significant residue toggling across the epitope at low frequency before preferential selection of a variant that coincided with recrudescent viremia. The color coding of the residues in the text correlates with the epitope variant colors in the graphs.
In three of the four epitopes with evidence of immune escape, the fixation events were mutations away from the consensus, and these mutations did not revert back to the T/F variant at a later time point. These data again suggest evidence of CD8+ T cell-driven immune pressure exerted on the T/F viruses (Table 2).
Rate of CD8+ T cell escape.
Mathematical modeling was employed to estimate the rate of immune escape in epitopes undergoing fixation events. The rate of escape from the variant 852RAEAHLHAW860 was estimated at 0.072 day−1 (standard error [SE], 0.0004; P value, 4.7 × 10−9), while for 2629NSKRTPMGF2637, the rate was estimated at 0.122 day−1 (SE, 0.037; P value, 0.03). The estimated rate of escape for the two escape variants in Ch_240, 1602RAQALPPSW1610 and 1633RLGPVQNEI1641, were 0.102 day−1 (SE, 0.016; P value, 0.008) and 0.026 day−1 (SE, 0.0005; P value, 0.000015), with a half-life of 95 and 370 days, respectively (Fig. 5).
FIG 5.

The rate of immune escape from T/F-specific CD8+ T cell responses. (A) In subject Ch_23, two T/F virus epitopes (in the legend) underwent escape mutations, the first (K2629N in black) at a rate of 0.122 per day and the second (Q856H in red) at a rate of 0.072 per day. (B) In subject Ch_240 two epitopes underwent escape mutations, the first (P1606L in black) at a rate of 0.102 per day and the second (V1641I in red) at a rate of 0.026 per day. Symbols represent the cumulative frequency of the escape variants within the epitope sequence. The lines represents the fit of the data with a mathematical model describing the escape dynamics under CD8+ T cell immune pressure (38).
ELISpot responses measured within 101 days p.i. were included to test the hypothesis that the magnitude of CD8+ T cell responses was associated with the likelihood of immune escape. The ELISpot responses against each T/F epitope that underwent immune escape were compared with the responses against each T/F epitope that did not escape within the time period of 101 days p.i. There were significantly higher immune responses in epitopes that developed escape variants during the early phase of infection (P < 0.05) (Fig. 6). The ELISpot responses against epitopes that did not undergo immune escape did not differ when analyzed by disease outcome (P > 0.05).
FIG 6.
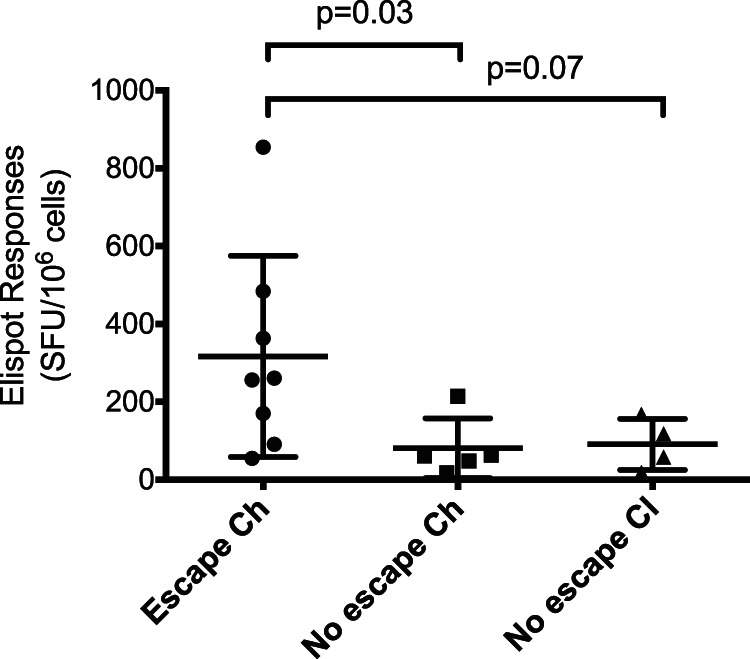
Magnitude of CD8+ T cell response associated with escape. The CD8+ T cell responses measured via ELISpot assay for each T/F epitope during the first 101 days p.i. was compared between epitopes that underwent escape (n = 2 in Ch_23 and n = 2 in Ch_240, for a total of 8 immune responses) and those that did not (n = 9). The conserved epitopes were also further divided based on disease outcome. The epitopes that elicited greater IFN-γ ELISpot responses were more likely to have escape variants emerge (P = 0.03). Statistical comparisons are Mann-Whitney tests. Scatter plots represent means ± 1 standard deviation.
Statistical validation of presence of immune pressure on experimentally validated CD8+ T cell responses.
To statistically validate the contribution of CD8+ T cell selection pressure on the evolution of T/F viruses, viral diversity was calculated within predicted major histocompatibility complex (MHC) class I epitopes. Normalized Shannon entropy (S) scores per genomic region across the HCV genome for each time point were calculated as a measure of overall viral diversity. Interestingly, epitope regions tended to have greater diversity than nonepitope regions from very early in infection although this increase was not statistically significant (Fig. 7).
FIG 7.
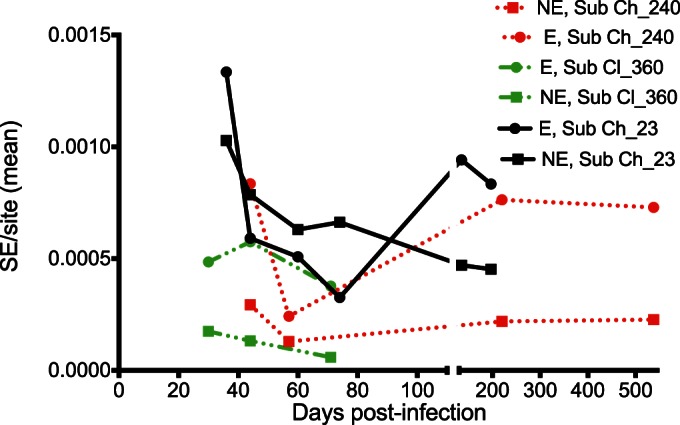
Mean Shannon entropy (SE) per site in epitope regions (E) and nonepitope regions (NE) calculated over time for the three subjects. Following a decline in the Shannon entropy per site within the first 100 days p.i. for all three subjects, the diversity index increased in the chronic phase of the infection. The mean Shannon entropy per site was generally higher within the epitope regions.
To validate the apparent immune pressure exerted by experimentally confirmed CD8+ T cell responses on viral evolution, evolutionary models and clustering analyses were utilized. There was evidence of selective pressure by CD8+ T cell responses provided first by a significant accumulation of SNPs in the targeted epitopes (P value of <0.05, q value of <0.2, using enriched clustering statistics [ECS]) (see Table S2 in the supplemental material). There was a positive correlation between the presence of diversifying selection within the epitope codons and the maximum value of the CD8+ T cell response during the first 100 days p.i. (P = 0.0004). There was also evidence of negative selection in codons in the flanking regions of two epitopes in subject Ch_240 and one epitope in subject Ch_23. Finally, for the subject who cleared the infection, as expected, there was no evidence of significant selective forces.
DISCUSSION
This study includes several novel observations that inform our understanding of the very early cellular immune responses against HCV T/F variants. First, we have shown that the rapid extinction of T/F viruses is strongly associated with the rapid onset of T/F-specific CD8+ T cell responses. Second, there was a significant association between the magnitude of CD8+ T cell responses and the likelihood of immune escape, suggesting a significant role of cellular immunity in driving T/F evolution. Third, in chronic infections T/F CD8+ T cell responses declined over the course of the infection. These results, along with evidence that there are very few T/F variants which represent a genetic bottleneck at transmission (3, 4), suggest that a potential route for design of T cell-based immunogens is to target T/F epitopes. However, this selection needs to be guided by a thorough investigation of the propensity of certain T/F epitopes to escape cellular responses.
The high-resolution analysis of viral diversity enabled by the deep-sequencing approach undertaken here suggests that CD8+ T cell responses play a role in driving extinction of the T/F viruses and that multiple novel variants evolved early in the infection from T/F viruses. However, the majority of these novel variants were eliminated within 100 days p.i., and only a few successfully initiated the later phase leading to chronic infection. It is evident that CD8+ T cell responses actively contribute to this selection process; whether this is the only force significantly contributing to the elimination of the T/F population remains to be resolved.
This study strongly supports the hypothesis that T/F-specific CD8+ T cell responses drive evolution of escape variants in HCV infections. The rise in viral load after the genetic bottleneck at 100 days p.i. was coincident with the occurrence of dominant immune escape variants and a decline in the magnitude of T/F-specific responses. The results are consistent with the theory of vertical immunodominance proposed for HIV, whereby the probability of CD8+ T cell escape is proportional to the magnitude of the individual epitope-specific response relative to the total response within each subject at a given time point (42, 43). In support of this model of disease evolution is the fact that the decline in viral diversity observed within CD8+ T cell T/F epitopes was coincident with the first detectable CD8+ T cell responses, suggesting that CD8+ T cell responses were exerting purifying selection at identifiable epitopes.
T/F viruses rapidly escaped from CD8+ T cell responses at a rate of 0.02 to 0.1 day−1, suggesting that the presence of a CD8+ T cell response allows up to 10% of the viral population to escape each day. These remarkable viral dynamics are consistent with the range of escape found in HIV (42) although the estimated mean rate of escape in HIV is higher (0.24 day−1) (44). In contrast to data in HIV, rapid escape was observed here even at sites known to be associated with severe fitness costs. The highest rate of escape was in the well-described immunodominant HLA-B57 epitope, 2629KSKKTPMGF2637 (45), where the K2629N mutation has been shown in vitro to result in reduced replicative capacity (45). Although this mutation is known to cooccur with a Q2626K/R mutation, which restores replication to nearly wild-type levels, there was no evidence of compensatory mutations at residue 2626 or with any other linked mutations in NS5B in our analysis. Identification of such epitopes with rapid escape potential is important as they are likely to make poor vaccine candidates.
The limitations of this study include the limited sample availability during the first 4 weeks postinfection, which are needed to definitively validate that the very few variants observed in our earliest samples were indeed the T/F variants. In this regard it should be noted that in HIV these mathematically identified T/F viruses have been validated with experimental models of simian immunodeficiency virus (SIV) and with sexual transmission data (46). Although unlikely, it is theoretically possible that early selective pressure (innate responses or loss of reproductive fitness due to mutation events) may give rise to early fixation events with no survivors from an early lineage; hence, the detection of T/F virus may be inaccurate. In addition, it should be noted that the estimates of the rate of escape are based on infrequent sampling of viral populations around the second genetic bottleneck event (approximately 100 days p.i.) as low viral loads prevented amplification of the full HCV genome. The study is also limited by the lack of additional early PBMC samples, which precluded exhaustive testing of all potential epitopes for each subject as well as the experimental validation of the newly identified HLA class I epitopes that have not been previously reported.
That CD8+ T cell responses against conserved epitopes decline rapidly throughout infection indicates that escape is unlikely to be the only mechanism resulting in persistent viremia. CD8+ T cell exhaustion has been well documented in the chronic phase of HCV infection, with immune escape and exhaustion estimated to contribute equally to viral persistence (47). However, it remains unknown whether CD8+ T cell exhaustion occurs in early HCV infection and has a causative role in progression to chronic infection.
This study provides clear evidence for immune selection pressures as a driver for viral evolution and escape in the early phase of primary HCV infection. The findings raise significant concerns for HCV vaccine development, given that the general dogma has been to select epitopes which induce strong immune responses for inclusion in a prophylactic HCV vaccine and to prioritize those in which escape is associated with a significant fitness cost. In relation to the immunopathogenesis of primary HCV infection, the findings indicate that it is possible to mount a CD8+ T cell response against T/F viruses, but the strength of early responses against a poorly diversified viral population may exert strong selective pressure and lead to rapid immune escape. Further studies are needed to resolve the T/F epitopes targeted by immune responses and the propensity of T/F viruses to escape.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sukanya Raghuraman for her helpful suggestions on the manuscript.
The HITS-p investigators include Andrew R. Lloyd, Kate Dolan, Paul Haber, William Rawlinson, Carla Treloar, Greg Dore, Lisa Maher, and Fabio Luciani.
We declare that we have no conflicts of interest.
Author's contributions are as follows: R.A.B., A.R.L., and F.L. designed the research; R.A.B., F.L., P.D., B.C., S.G., P.L., M.W., A.C., and A.R.L. contributed samples and reagents; S.G., P.D., M.W., A.C., P.L., R.A.B., and F.L. performed the analyses; R.A.B., F.L., and A.R.L. wrote the paper.
This work was supported by funding from the National Health and Medical Council of Australia (NHMRC) (grant numbers 1042090 and 510488). A.R.L. and R.A.B. are supported by NHMRC Fellowships (510246 and 1060443).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03717-14.
REFERENCES
- 1.Grebely J, Page K, Sacks-Davis R, van der Loeff MS, Rice TM, Bruneau J, Morris MD, Hajarizadeh B, Amin J, Cox AL, Kim AY, McGovern BH, Schinkel J, George J, Shoukry NH, Lauer GM, Maher L, Lloyd AR, Hellard M, Dore GJ, Prins M, InC3 Study Group. 2014. The effects of female sex, viral genotype, and IL28B genotype on spontaneous clearance of acute hepatitis C virus infection. Hepatology 59:109–120. doi: 10.1002/hep.26639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seeff LB. 2002. Natural history of chronic hepatitis C. Hepatology 36:S35–46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 3.Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, Cameron B, Maher L, Dore GJ, White PA, Lloyd AR. 2011. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog 7:e1002243. doi: 10.1371/journal.ppat.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H, Stoddard MB, Wang S, Blair LM, Giorgi EE, Parrish EH, Learn GH, Hraber P, Goepfert PA, Saag MS, Denny TN, Haynes BF, Hahn BH, Ribeiro RM, Perelson AS, Korber BT, Bhattacharya T, Shaw GM. 2012. Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLoS Pathog 8:e1002880. doi: 10.1371/journal.ppat.1002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, Sidney J, Sette A, Pardoll D, Thomas DL, Ray SC. 2005. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med 201:1741–1752. doi: 10.1084/jem.20050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox AL, Mosbruger T, Lauer GM, Pardoll D, Thomas DL, Ray SC. 2005. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 42:104–112. doi: 10.1002/hep.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, Robbins G, Phillips R, Klenerman P, Walker BD. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med 191:1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urbani S, Amadei B, Fisicaro P, Tola D, Orlandini A, Sacchelli L, Mori C, Missale G, Ferrari C. 2006. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology 44:126–139. doi: 10.1002/hep.21242. [DOI] [PubMed] [Google Scholar]
- 9.Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. 2001. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med 194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dustin LB, Rice CM. 2007. Flying under the radar: the immunobiology of hepatitis C. Annu Rev Immunol 25:71–99. doi: 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- 11.Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JC, Strazzera A, Chien DY, Munoz SJ, Balestrieri A, Purcell RH, Alter HJ. 2000. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 12.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. 2005. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med 201:1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann-Haefelin C, Spangenberg HC, Blum HE, Thimme R. 2007. Host and viral factors contributing to CD8+ T cell failure in hepatitis C virus infection. World J Gastroenterol 13:4839–4847. doi: 10.3748/wjg.v13.i36.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuntzen T, Timm J, Berical A, Lewis-Ximenez LL, Jones A, Nolan B, Schulze zur Wiesch J, Li B, Schneidewind A, Kim AY, Chung RT, Lauer GM, Allen TM. 2007. Viral sequence evolution in acute hepatitis C virus infection. J Virol 81:11658–11668. doi: 10.1128/JVI.00995-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfafferott K, Gaudieri S, Ulsenheimer A, James I, Heeg M, Nolan D, John M, Rauch A, Mallal S, Lucas A, Klenerman P, Diepolder HM, Lucas M. 2011. Constrained pattern of viral evolution in acute and early HCV infection limits viral plasticity. PLoS One 6:e16797. doi: 10.1371/journal.pone.0016797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merani S, Petrovic D, James I, Chopra A, Cooper D, Freitas E, Rauch A, di Iulio J, John M, Lucas M, Fitzmaurice K, McKiernan S, Norris S, Kelleher D, Klenerman P, Gaudieri S. 2011. Effect of immune pressure on hepatitis C virus evolution: insights from a single-source outbreak. Hepatology 53:396–405. doi: 10.1002/hep.24076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Callendret B, Bukh J, Eccleston HB, Heksch R, Hasselschwert DL, Purcell RH, Hughes AL, Walker CM. 2011. Transmission of clonal hepatitis C virus genomes reveals the dominant but transitory role of CD8+ T cells in early viral evolution. J Virol 85:11833–11845. doi: 10.1128/JVI.02654-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowen D, Walker C. 2005. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 19.Guglietta S, Garbuglia AR, Pacciani V, Scotta C, Perrone MP, Laurenti L, Spada E, Mele A, Capobianchi MR, Taliani G, Folgori A, Vitelli A, Ruggeri L, Nicosia A, Piccolella E, Del Porto P. 2005. Positive selection of cytotoxic T lymphocyte escape variants during acute hepatitis C virus infection. Eur J Immunol 35:2627–2637. doi: 10.1002/eji.200526067. [DOI] [PubMed] [Google Scholar]
- 20.Tester I, Smyk-Pearson S, Wang P, Wertheimer A, Yao E, Lewinsohn DM, Tavis JE, Rosen HR. 2005. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J Exp Med 201:1725–1731. doi: 10.1084/jem.20042284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med 206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, Halliday J, Kelly C, Bowen D, Fergusson J, Kurioka A, Ammendola V, Del Sorbo M, Grazioli F, Esposito ML, Siani L, Traboni C, Hill A, Colloca S, Davis M, Nicosia A, Cortese R, Folgori A, Klenerman P, Barnes E. 2014. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med 6:261ra153. doi: 10.1126/scitranslmed.3009185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A, Meyer J, Huddart R, Smith K, Townsend R, Brown A, Antrobus R, Ammendola V, Naddeo M, O'Hara G, Willberg C, Harrison A, Grazioli F, Esposito ML, Siani L, Traboni C, Oo Y, Adams D, Hill A, Colloca S, Nicosia A, Cortese R, Klenerman P. 2012. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med 4:115ra1. doi: 10.1126/scitranslmed.3003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leung P, Bull R, Lloyd A, Luciani F. 2014. A bioinformatics pipeline for the analyses of viral escape dynamics and host immune responses during an infection. Biomed Res Int 2014:264519. doi: 10.1155/2014/264519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teutsch S, Luciani F, Scheuer N, McCredie L, Hosseiny P, Rawlinson W, Kaldor J, Dore GJ, Dolan K, Ffrench R, Lloyd A, Haber P, Levy M. 2010. Incidence of primary hepatitis C infection and risk factors for transmission in an Australian prisoner cohort. BMC Public Health 10:633. doi: 10.1186/1471-2458-10-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Page-Shafer K, Pappalardo BL, Tobler LH, Phelps BH, Edlin BR, Moss AR, Wright TL, Wright DJ, O'Brien TR, Caglioti S, Busch MP. 2008. Testing strategy to identify cases of acute hepatitis C virus (HCV) infection and to project HCV incidence rates. J Clin Microbiol 46:499–506. doi: 10.1128/JCM.01229-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaudieri S, Rauch A, Park LP, Freitas E, Herrmann S, Jeffrey G, Cheng W, Pfafferott K, Naidoo K, Chapman R, Battegay M, Weber R, Telenti A, Furrer H, James I, Lucas M, Mallal SA. 2006. Evidence of viral adaptation to HLA class I-restricted immune pressure in chronic hepatitis C virus infection. J Virol 80:11094–11104. doi: 10.1128/JVI.00912-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McElroy K, Zagordi O, Bull R, Luciani F, Beerenwinkel N. 2013. Accurate single nucleotide variant detection in viral populations by combining probabilistic clustering with a statistical test of strand bias. BMC Genomics 14:501. doi: 10.1186/1471-2164-14-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zagordi O, Klein R, Daumer M, Beerenwinkel N. 2010. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res 38:7400–7409. doi: 10.1093/nar/gkq655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giorgi EE, Funkhouser B, Athreya G, Perelson AS, Korber BT, Bhattacharya T. 2010. Estimating time since infection in early homogeneous HIV-1 samples using a poisson model. BMC Bioinformatics 11:532. doi: 10.1186/1471-2105-11-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 36.Posada D, Buckley TR. 2004. Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53:793–808. doi: 10.1080/10635150490522304. [DOI] [PubMed] [Google Scholar]
- 37.Almeida CA, Roberts SG, Laird R, McKinnon E, Ahmed I, Pfafferott K, Turley J, Keane NM, Lucas A, Rushton B, Chopra A, Mallal S, John M. 2009. Automation of the ELISpot assay for high-throughput detection of antigen-specific T-cell responses. J Immunol Methods 344:1–5. doi: 10.1016/j.jim.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asquith B, Edwards CT, Lipsitch M, McLean AR. 2006. Inefficient cytotoxic T lymphocyte-mediated killing of HIV-1-infected cells in vivo. PLoS Biol 4:e90. doi: 10.1371/journal.pbio.0040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.R Core Development Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 40.Delport W, Scheffler K, Botha G, Gravenor MB, Muse SV, Kosakovsky Pond SL. 2010. CodonTest: modeling amino acid substitution preferences in coding sequences. PLoS Comput Biol 6:e1000885. doi: 10.1371/journal.pcbi.1000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog 8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu MK, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, Brackenridge S, Li H, Pavlicek JW, Cai F, Rose-Abrahams M, Treurnicht F, Hraber P, Riou C, Gray C, Ferrari G, Tanner R, Ping LH, Anderson JA, Swanstrom R, Cohen M, Karim SS, Haynes B, Borrow P, Perelson AS, Shaw GM, Hahn BH, Williamson C, Korber BT, Gao F, Self S, McMichael A, Goonetilleke N. 2013. Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest 123:380–393. doi: 10.1172/JCI65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganusov VV, Goonetilleke N, Liu MK, Ferrari G, Shaw GM, McMichael AJ, Borrow P, Korber BT, Perelson AS. 2011. Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. J Virol 85:10518–10528. doi: 10.1128/JVI.00655-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oniangue-Ndza C, Kuntzen T, Kemper M, Berical A, Wang YE, Neumann-Haefelin C, Foote PK, Hills-Evans K, Reyor LL, Kane K, Gladden AD, Bloom AK, Power KA, Thimme R, Lauer GM, Henn MR, Kim AY, Allen TM. 2011. Compensatory mutations restore the replication defects caused by cytotoxic T lymphocyte escape mutations in hepatitis C virus polymerase. J Virol 85:11883–11890. doi: 10.1128/JVI.00779-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw GM, Hunter E. 2012. HIV transmission. Cold Spring Harb Perspect Med 2:a006965. doi: 10.1101/cshperspect.a006965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, Pircher H, Thimme R. 2010. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog 6:e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.