

ABSTRACT
While geographic distance often restricts the spread of pathogens via hosts, this barrier may be compromised when host species are mobile. Migratory waterfowl in the order Anseriformes are important reservoir hosts for diverse populations of avian-origin influenza A viruses (AIVs) and are assumed to spread AIVs during their annual continental-scale migrations. However, support for this hypothesis is limited, and it is rarely tested using data from comprehensive surveillance efforts incorporating both the temporal and spatial aspects of host migratory patterns. We conducted intensive AIV surveillance of waterfowl using the North American Mississippi Migratory Flyway (MMF) over three autumn migratory seasons. Viral isolates (n = 297) from multiple host species were sequenced and analyzed for patterns of gene dispersal between northern staging and southern wintering locations. Using a phylogenetic and nucleotide identity framework, we observed a larger amount of gene dispersal within this flyway rather than between the other three longitudinally identified North American flyways. Across seasons, we observed patterns of regional persistence of diversity for each genomic segment, along with limited survival of dispersed AIV gene lineages. Reassortment increased with both time and distance, resulting in transient AIV constellations. This study shows that within the MMF, AIV gene flow favors spread along the migratory corridor within a season, and also that intensive surveillance during bird migration is important for identifying virus dispersal on time scales relevant to pandemic responsiveness. In addition, this study indicates that comprehensive monitoring programs to capture AIV diversity are critical for providing insight into AIV evolution and ecology in a major natural reservoir.
IMPORTANCE Migratory birds are a reservoir for antigenic and genetic diversity of influenza A viruses (AIVs) and are implicated in the spread of virus diversity that has contributed to previous pandemic events. Evidence for dispersal of avian-origin AIVs by migratory birds is rarely examined on temporal scales relevant to pandemic or panzootic threats. Therefore, characterizing AIV movement by hosts within a migratory season is important for implementing effective surveillance strategies. We conducted surveillance following birds along a major North American migratory route and observed that within a migratory season, AIVs rapidly reassorted and gene lineages were dispersed primarily within the migratory corridor. Patterns of regional persistence were observed across seasons for each gene segment. We show that dispersal of AIV gene lineages by migratory birds occurs quickly along migratory routes and that surveillance for AIVs threatening human and animal health should focus attention on these routes.
INTRODUCTION
Geographic distance often limits the spread of pathogens between susceptible host populations (1). However, highly mobile hosts can transfer pathogens quickly across space (2). An example is how the migratory behaviors of waterfowl in the order Anseriformes, a major reservoir host for influenza A virus (AIV) diversity, can spread these viruses across broad geographic distances (3–5). Much of the genetic diversity giving rise to AIVs which infect poultry, swine, and humans (4) is found in migratory ducks and geese. Each of the four human pandemic strains emerging in the last 100 years has contained genetic segments derived from avian-origin AIVs (6). Therefore, understanding the genomic diversity of AIVs circulating in the Anseriformes, along with other natural reservoirs, is important for preparing for future pandemic threats (7).
Influenza A virus is a single-stranded RNA virus of the order Orthomyxoviridae and contains eight separate RNA genomic segments that readily reassort with each other during coinfections to form ever-changing genomic constellations (8). In waterfowl, AIV infections are typically caused by low pathogenic (LP) avian-origin influenza A viruses (5, 9; but see reference 10). This absence of observable clinical signs suggests a limited effect on host species behavior (11), which presumably permits virus spread over varied distances via infected hosts during migration. Many studies implicate birds in the movement of AIVs (12–14) and have speculated on the potential for movement of highly pathogenic (HP) viruses out of regions of Asia where they are endemic (15, 16). However, there is limited evidence for the spread of diverse AIV strains by wild birds, especially over shorter periods; thus, the significance of host waterfowl in spreading AIV is still debated (17, 18). Recent studies described the movement of AIV genetic diversity in North America over decade-long time frames (19, 20). To better understand influenza A virus evolution in the natural host and to aid in our ability to effectively respond to viral threats to public and animal health, the movement of AIVs must be understood for shorter time frames that are relevant to disease events. These events can occur quickly, as witnessed in 2013 in China, where a novel H7N9 virus of avian origin was detected in humans and within months had caused hundreds of infections (21), as well as in recent outbreaks of Asian-origin, highly pathogenic H5 AIVs in wild and domestic birds in multiple locations in western North America (22).
In North America, most waterfowl species undertake annual continental-scale migratory movements in one of four migratory flyways (Fig. 1) (23). The current hypothesis is that subclinically AIV-infected waterfowl move viruses over geographic space within short periods coinciding with mass migration from their breeding grounds in the North to their wintering sites in the South (17) and that viruses are potentially brought back north by migrating shorebirds (order Chardriiformes) the next spring (24). In the late summer and fall, large proportions of these flocks are comprised of immunologically naive juvenile birds that show a higher prevalence of AIV infections during this period (25). As these birds migrate, they encounter other naive individuals from different geographic areas, thus increasing the potential for continued transmission of specific strains of AIV originating from other geographic areas (26). However, the frequency of virus recovery is markedly lower at the wintering grounds and during the northern spring migration, and it is therefore difficult to establish the primary routes of AIV dispersal. However, if infected birds move AIVs between distinct breeding and wintering habitats within migratory pathways (i.e., along the flyway), then intensive AIV surveillance of these birds as they migrate should result in the detection of an increased number of AIV dispersal events annually within these corridors.
FIG 1.
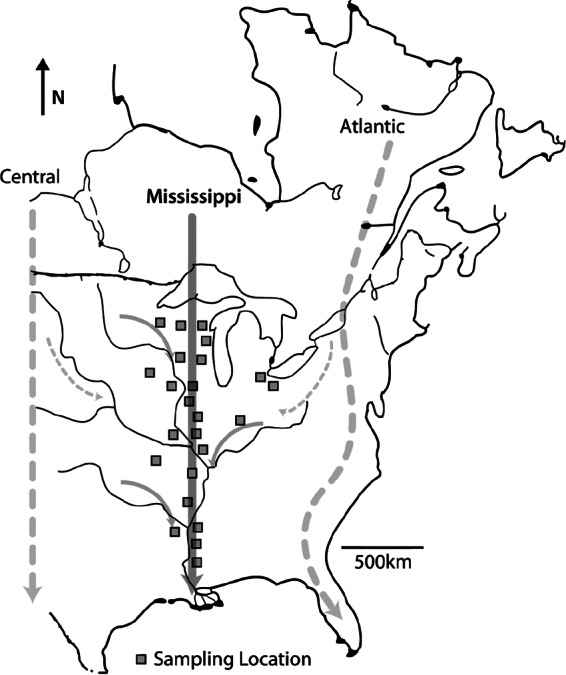
Map of the North American Mississippi Migratory Flyway and associated surveillance locations (black outlined squares) that were used in this study. Arrows are generalizations of bird movements during autumn migratory seasons. Three of the four major migratory bird flyways are represented (Central, Mississippi, and Atlantic).
The objective of this study was to determine the patterns and extents of AIV movement in the largest North American migratory bird flyway by volume, the Mississippi Migratory Flyway (MMF), by sampling birds migrating south on their autumn migration following peak AIV infectivity in these waterfowl (27). We strategically selected AIV surveillance sites along the migratory route to capture viruses at multiple locations as these viruses moved with their infected reservoir host populations. Here we describe the patterns of movement and the regional persistence of lineages of AIV gene segments within the MMF, providing insight into the dispersal of AIV by migratory birds over shorter time frames than have primarily been examined. In addition, we use our results from the MMF to make recommendations for the design of future wild bird surveillance efforts while further defining the natural history of AIV in an important natural reservoir.
MATERIALS AND METHODS
Sample collection, influenza A virus isolation, and sequencing.
We conducted AIV surveillance during the North American autumn migratory seasons annually at 18 to 28 study sites along the Mississippi Migratory Flyway (MMF) from July 2008 to February 2011 (Fig. 1). These dates span three complete autumn migratory cycles for Anseriformes species in the MMF (July 2008 to February 2009, July 2009 to February 2010, and July 2010 to February 2011). Specific study sites and dates for sampling along the MMF were chosen based on the migratory patterns of Anseriformes species within the Flyway (23), accessibility to sites, local consent and support of wildlife managers and hunters, and site-specific waterfowl species diversity and availability. Cloacal swabs were collected from live-captured and hunter-killed Anseriformes birds. The cloacal swabs were placed in a virus transport medium containing antibiotics (5). Live-captured birds were swabbed during the early portions of the migratory season (July and August of each year), while hunter-killed samples were sampled later in the season (September to February of each year). Samples from hunter-killed birds were collected on the opening day of the hunting season for each respective state hunting zone, with follow-up visits to the same locations 3 to 6 weeks later to obtain more comprehensive sampling of viral lineages present at each location. Hunting seasons occur earlier in the autumn (e.g., September) in northern states and continue later into the migratory season (e.g., February) in the southern states and are established based on the timing of regional migratory patterns of North American waterfowl.
Isolation of AIVs was carried out using standard viral isolation procedures after one passage through 10-day-old embryonating chicken eggs (28). The presence of AIV was confirmed by testing allantoic fluid with FluDetect kits (Synbiotics Corp., San Diego, CA). Antigenic subtyping of all isolates was done at the National Veterinary Service Laboratories, Ames, IA, by hemagglutinin and neuraminidase inhibition techniques (29) and for pathogenicity assessments of all H5 and H7 isolates.
Following isolation, the complete coding genomes of the influenza A viruses were sequenced as part of the NIH/NIAID-sponsored Influenza Genome Sequencing Project at the J. Craig Venter Institute. Viral RNA was isolated using a ZR 96 viral RNA kit (Zymo Research Corporation, Irvine, CA). The influenza A virus genomic RNA segments were simultaneously amplified from 3 μl of purified RNA by using a multisegment reverse transcription-PCR (M-RT-PCR) strategy (30, 31). Influenza virus M-RT-PCR products were randomly amplified and prepared for next-generation sequencing by using a sequence-independent single-primer amplification (SISPA) method (32). Two aliquots were then submitted for sequencing with 454 GS FLX+ (one plate) and Illumina HiSeq (one lane) sequencing technologies.
The sequence reads from the HiSeq or 454 GS FLX+ data were sorted by bar code and trimmed, and chimeric influenza virus sequences or non-influenza virus sequences were removed. The reads were then mapped to the best-matching reference virus by using the clc_ref_assemble_long program. At loci where platforms agreed on a variation (compared to the reference sequence), the reference sequence was updated to reflect the difference. A final mapping of all next-generation sequences to the updated reference sequences was then performed.
Influenza A virus sequence data preparation.
All nucleotide sequences for MMF AIV isolates were manually edited and aligned using Bioedit 7.2.0 (33). To address the potential for the AIVs isolated in this study to have originated from other MMF sampling sites, or possibly other flyways (i.e., Atlantic, Central, or Pacific), we supplemented our MMF AIV sequences with all available AIV sequences from avian species across North America obtained between June 2008 and March 2011 from the Influenza Research Database (IRD) (downloaded August 2014) to represent the existing and potentially contributing AIV genetic diversity observed in North America during our study (see Table S1 in the supplemental material) (34). All sequences supplemented from the IRD were trimmed to include only the open reading frame and were prepared and aligned similarly to the MMF AIV isolates. Any genomic segments with total nucleotides of <95% of the total length of the open reading frame were discarded (<1% of the total).
Phylogeographic analysis of AIV genetic structure.
To determine if gene migration events occurred nonrandomly and were indicative of the geographic structuring of genetic diversity (i.e., a lack of AIV movement) among MMF isolates, we used a method similar to that of Chen and Holmes (35). Specifically, we constructed maximum likelihood (ML) trees for the nucleic acid sequences of each of the MMF AIV internal segments that were isolated and sequenced in this study (polymerase PB2 [PB2], polymerase PB1 [PB1], polymerase PA [PA], nucleocapsid protein [NP], and matrix protein [MP]), using RAxML v. 7.2.8 (36). For this test, we used only AIVs collected in this study to specifically address the question of whether we could detect the movement of AIV diversity within the MMF by following bird populations through the flyway. The hemagglutinin (HA), neuraminidase (NA), and nonstructural (NS) gene segments were not included in this analysis due to the deep divergences observed between major clades. A GTR+Γ nucleotide substitution model was used based on results from Modeltest (37). We ran 1,000 bootstrap replicates to evaluate clade support for each segment tree. We then coded each isolate with the geographic location of sampling and used the Slatkin and Maddison test to count the number of gene migration events (s) on each gene segment tree (38). We reconstructed the geographic ancestral states at every internal node along the tree by using a parsimony method implemented in the Mesquite package (39). Any MMF AIV segment with identical nucleotide sequences was removed to avoid biasing the magnitude of gene migration. The observed number of state changes on each of the trees was counted and compared to the distribution of gene migration events (s) for a set of 10,000 randomized reassignments of isolates, using the same tree topology to calculate the expected s in a panmictic (i.e., no geographic barriers) population. The distribution of expected state changes from the randomized trees was then compared to the observed values to determine if any pattern of genetic structure existed within the tree. The P value for this test was calculated from the number of times that the observed s was larger than expected. Since study sites were not equidistant, we grouped sampling sites into two and four latitudinal distinctions. First, we used a broad classification between northern and southern sites (North/South). The second classification divided the study area into four latitudinal categories based on equal latitudinal divisions of isolates (North/mid-North/mid-South/South).
Estimation of viral migration rates.
As a geographically broader extension to the analysis with the Slatkin and Maddison test, we conducted a discrete phylogeography analysis to determine the rate and directionality of AIV gene migration within the Mississippi flyway and among other flyways (i.e., Atlantic, Central, and Pacific), using time-stamped sequence data with a relaxed Bayesian Markov chain Monte Carlo method as implemented in BEAST v1.8 (40, 41). Here we combined our smaller data set of MMF AIV isolates with all available AIV sequences from avian species across all North American flyways between June 2008 and March 2011 from the IRD, as discussed above, to represent the genetic diversity of AIVs in North America. State transition rate matrices (i.e., migration rates between discrete geographic categories) were measured separately for each of the five internal gene segments (PB2, PB1, PA, NP, and MP) to identify segment-specific migration rates independent of all other AIV segments within an isolate. Again, HA, NA, and NS were removed due to deep evolutionary divergences. Additionally, we ran a combined analysis where each of the five internal segment phylogenies and substitution rates was sampled and estimated independently but a single transition rate matrix was estimated using a joint migration process to identify the overall pattern of gene migration within the data set. A nonreversible, continuous-time Markov chain model was used to estimate rates between sites in the North and South (as defined above), as well as an additional, “other” category, which included all IRD AIVs outside the MMF (see Table S1 in the supplemental material). Due to the low frequency of AIV recovery in the southern latitudes, our AIV sample size in the South from 2008 to 2011 was limited (∼92 sequences for each segment) and likely does not represent the true phylogenetic diversity of viruses circulating in that region. Isolates from the “other” and “North” categories are more representative of the respective diversity of these categories. To avoid biasing migration estimates among geographic categories based on the sampling intensities and AIV prevalences in the “North” and “other” categories, we conducted 10 independent analyses whereby we randomly subsampled the IRD (with duplicates removed) and MMF sequences for the “other” and “North” data sets at sampling sizes equivalent to those collected in the South for each AIV segment (i.e., equal amounts from each of the three geographic categories). Each randomized data set represented the AIV diversity from 2008 to 2011 for North America and had approximately 280 sequences for each segment (i.e., 1,400 sequences per BEAST run for joint migration estimation). Additionally, subsampling of sequences made these joint analyses computationally feasible with the available computational resources.
For each BEAST run, we used an uncorrelated log-normal relaxed molecular clock and the HKY85 nucleotide substitution model. We used a time-aware Bayesian skyride coalescent prior for the segment-specific runs and a Bayesian skygrid coalescent for the runs estimating joint migration matrices. For every BEAST analysis, we performed 2 separate runs of 75 million generations each, with 25,000 generations sampled for each run to ensure convergence and effective sample size (ESS) values of >100. We used a burn-in of 10 to 15%, based on the exploration of the parameter space as measured in Tracer v1.5. For comparison of within- and between-flyway rates, we considered rate estimates between the North and South MMF locations to be within-flyway rates, while rate estimates that included other locations were considered between-flyway estimates. An edited BEAST v1.8 XML file implementing joint estimation is available for reference, as well as all R and Perl scripts used to conduct the analyses in this paper (https://github.com/friesac/Mississippi_Flyway_Material).
Nucleotide identity analysis.
To characterize the movements of individual viruses rather than the generalized patterns identified in the phylogenetic analyses, in addition to accounting for the deep evolutionary splits confounding phylogenetic analyses of the HA, NA, and NS segments, we used pairwise percent nucleotide sequence identities (numbers of nucleotides shared between AIVs) to identify viruses moving between sampling localities (42). The percent identity for each genomic segment was measured between all MMF isolates from this study, in addition to the other North American AIV isolates found in the IRD database for the period between June 2008 and March 2011 (see Table S1 in the supplemental material). We used the blastn utility in BLAST+ to measure identity (43). The time between isolation events (days) and the geographic distance (kilometers) were recorded for each pairwise comparison. Time categories were used to group AIV comparisons among migratory seasons, as follows: within a season (≤200 days between isolations), across two seasons (>200 days but ≤600 days between isolations), and across three seasons (>600 days between isolations). Great-circle distances were calculated between the latitudes and longitudes of sampling sites for MMF isolates, and the latitude and longitude coordinates of the centroid of each state or province were used for IRD isolates.
For identity measures, we used a conservative criterion to infer movement of the same virus, which accounted for unequal mutation rates among AIV gene segments by using a variable nucleotide identity percentage for each AIV gene segment. The segment-specific cutoff was based on the percent nucleotide identity for observations in the upper 1% of a ranked distribution of every pairwise comparison for each segment (i.e., the nucleotide identity observed at the 100th pairwise comparison of 10,000 ranked pairwise comparisons = 1% cutoff). This value changes with each segment because of various levels of genetic diversity observed within a segment. The nucleotide identities of all eight genetic segments (i.e., genetic constellation) for each isolate were compared among isolates as a proxy measure of reassortment. Constellations shared between viruses collected on the same day and at the same geographic location were removed from the analysis. To assess the significance of high-identity pairwise comparisons, we used two-way contingency tables evaluated with Pearson's chi-square tests (44).
Nucleotide sequence accession numbers.
The consensus sequences obtained in this study were deposited in GenBank under the accession numbers listed in Table S1 in the supplemental material.
RESULTS
Virus isolates.
A total of 297 isolates were recovered from 9,261 cloacal swabs and environmental samples collected from July 2008 through February 2011 (Table 1). Samples were obtained from waterfowl sampled at 28 locations in nine U.S. states within the MMF (Fig. 1). Initial antigenic subtyping identified 46 unique HA and NA combinations that included 13 HA subtypes (H1 to H12 and H14) and 9 NA subtypes (N1 to N9). Analyses of sequencing data indicated 40 distinct HA-NA subtype combinations, with the most common being H4N6 (8.1% of isolates). An additional 34 isolates showed mixed isolations of multiple HA and NA subtype combinations. Nucleotide sequences were obtained for all eight genomic segments for 291 of the 297 AIV isolates (see Table S1 in the supplemental material). Variations in the number of sequenced segments for each were typically due to the sequencing of mixed infections or sequencing difficulties. After trimming of nucleotide sequences to the sizes of the open reading frames, the lengths of each of the segments were as follows: PB2, 2,277 bp; PB1, 2,271 bp; PA, 2,148 bp; HA, 1,638 to 1,719 bp; NP, 1,494 bp; NA 1,209 to 1,437 bp; MP, 979 bp; and NS, 835 bp. Sequences for an additional 1,468 AIVs were included from the IRD/GenBank, representing 13,457 nucleotide segments that were collected in North America between June 2008 and March 2011 (Table 1; see Table S1). Sequence information for all eight genomic segments was obtained for 1,184 of the 1,468 AIV isolates from the IRD, with 1,317 having at least five or more segments sequenced (see Table S1). Isolates from 39 different species were included in the total AIV sequence database.
TABLE 1.
Isolates included in the analyses in this study from each geographic location by migratory season (April to March)d
| Country | State or province | Categoryc | No. of isolates |
|||
|---|---|---|---|---|---|---|
| 2008–2009 | 2009–2010 | 2010–2011 | Total | |||
| USA | Alaska | O | 99 | 234 | 184 | 517 |
| Arkansas | S | 0 | 7 | 3 | 10b | |
| California | O | 97 | 27 | 39 | 163 | |
| Delaware | O | 0 | 58 | 2 | 60 | |
| Illinois | N | 7 | 23 | 34 | 64a | |
| Indiana | N | 3 | 4 | 2 | 9a | |
| Iowa | N | 0 | 4 | 14 | 18a | |
| Louisiana | S | 3 | 5 | 0 | 8 | |
| Michigan | N | 0 | 0 | 2 | 2a | |
| Minnesota | N | 130 | 36 | 2 | 168 | |
| Mississippi | S | 6 | 15 | 18 | 39a | |
| Missouri | S | 0 | 8 | 27 | 35a | |
| New Jersey | O | 27 | 71 | 0 | 98 | |
| North Dakota | O | 15 | 6 | 0 | 21 | |
| Ohio | N | 5 | 8 | 7 | 20a | |
| South Dakota | O | 2 | 0 | 0 | 2 | |
| Texas | O | 0 | 7 | 0 | 7 | |
| Wisconsin | N | 11 | 42 | 54 | 107a | |
| Canada | Alberta | O | 7 | 7 | 0 | 14 |
| Manitoba | O | 0 | 0 | 2 | 2 | |
| New Brunswick | O | 1 | 28 | 120 | 149 | |
| Newfoundland | O | 9 | 0 | 0 | 9 | |
| Nova Scotia | O | 0 | 4 | 48 | 52 | |
| Nunavet | O | 0 | 0 | 1 | 1 | |
| Prince Edward Island | O | 0 | 5 | 11 | 16 | |
| Quebec | O | 0 | 4 | 5 | 9 | |
| Guatemala | Unknown | O | 0 | 3 | 10 | 13 |
| Total | 422 | 606 | 585 | 1,613 | ||
Influenza A virus isolates collected and sequenced in this study.
Three of the 10 influenza A virus isolates were collected in this study.
Discrete categories were given to the isolates and used in the Slatkin and Maddison and discrete phylogeographic analyses. N, North; S, South; O, other.
The AIVs presented here represent isolates collected during this study or supplemented from the IRD/GenBank with at least five sequenced segments.
Phylogeographic analysis.
For the virus isolates collected in this study from birds as they migrated through the MMF, we observed fewer (P < 0.01) gene migration events than would be expected in a panmictic population, based on Slatkin and Maddison s values for five of the gene segments coding for internal AIV proteins (PB1, PB2, PA, NP, and MP) across each of the three migratory seasons (2008 to 2011) (see Fig. S1 to S5 in the supplemental material). This result was consistent for both geographic groupings (Table 2). No internal AIV segment appeared to be regionally structured during the 2008–2009 migratory season, potentially due to small sampling sizes causing incomplete representation of the existing genetic diversity in the tree topology. The 2009–2010 and 2010–2011 seasons each had segments that exhibited some level of reduced gene movement or lineage isolation. Note that regardless of the geographic grouping, the numbers of observed state changes (i.e., gene migration events) were consistent among segments within migratory years, with a maximum standard deviation (SD) of 5.0 events (s) for the observed state changes for the North/South groupings across the 2008–2011 seasons, suggesting no preferential movement of one internal gene over another within or among migratory seasons in the MMF (Table 2).
TABLE 2.
Slatkin and Maddison tests of phylogeographic structure for five of the eight gene segments of influenza A viruses, using two-category (North/South) and four-category (North/mid-North/mid-South/South) latitudinal distinctionsc
| Gene segment | 2008–2009 |
2009–2010 |
2010–2011 |
2008–2011 |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n |
s valued |
n |
s valued |
n |
s valued |
n |
s valued |
|||||||||||||
| Two categories |
Four categories |
Two categories |
Four categories |
Two categories |
Four categories |
Two categories |
Four categories |
|||||||||||||
| O | E | O | E | O | E | O | E | O | E | O | E | O | E | O | E | |||||
| PB2 | 30 | 3.0 | 5.7 | 11.0 | 13.6 | 98 | 20.0 | 24.1 | 40.0 | 46.6a | 160 | 33.0 | 48.3b | 66.0 | 81.2b | 288 | 47.0 | 66.4b | 120.0 | 141.9b |
| PB1 | 28 | 5.0 | 4.8 | 9.0 | 12.0 | 95 | 19.0 | 23.9a | 36.0 | 45.5b | 152 | 36.0 | 44.3b | 67.0 | 75.0a | 275 | 60.0 | 74.2b | 117.0 | 135.8b |
| PA | 30 | 4.0 | 5.7 | 12.0 | 13.3 | 98 | 20.0 | 24.7a | 40.0 | 46.7a | 164 | 37.0 | 48.2b | 69.0 | 82.2b | 292 | 55.0 | 76.6b | 126.0 | 143.9b |
| NP | 27 | 5.0 | 5.6 | 12.0 | 12.4 | 97 | 17.0 | 24.7b | 38.0 | 46.1b | 147 | 40.0 | 44.8 | 70.0 | 74.4 | 271 | 61.0 | 76.3b | 124.0 | 135.5b |
| MP | 26 | 5.0 | 4.8 | 13.0 | 11.6 | 86 | 19.0 | 21.3 | 39.0 | 41.8 | 145 | 39.0 | 43.5 | 65.0 | 73.6a | 257 | 56.0 | 69.2b | 114.0 | 128.3b |
Significant difference (P < 0.05).
Significant difference (P < 0.01).
The AIV segments used in this analysis were from the MMF isolates collected in this study from birds as they moved in the Mississippi Migratory Flyway during migratory seasons.
O, observed; E, expected. The standard deviations of the observed s values were as follows: for the 2008–2009 season, 0.8 for two categories and 1.5 for four categories; for the 2009–2010 season, 1.1 for two categories and 1.7 for four categories; for the 2010–2011 season, 2.4 for two categories and 2.1 for four categories; and for the 2008–2011 seasons, 5.0 for two categories and 4.9 for four categories.
Estimation of viral migration rates.
For each of the 10 randomized BEAST data sets in which we estimated joint migration rate matrices across five of the internal segments, we found significantly higher migration rates estimated between North and South MMF sites than between the MMF sites and the “other” category (Fig. 2). While the 95% highest posterior density (HPD) intervals overlap, within-flyway migration rate estimates were higher than between-flyway rates (Fig. 2). North → South rates were equivalent to South → North rates (Fig. 2). For segment-specific runs (i.e., estimated individually), transition rates between MMF isolates and isolates from other flyways were skewed toward elevated within-flyway rates, but not to the magnitude observed with the joint estimation (data not shown).
FIG 2.
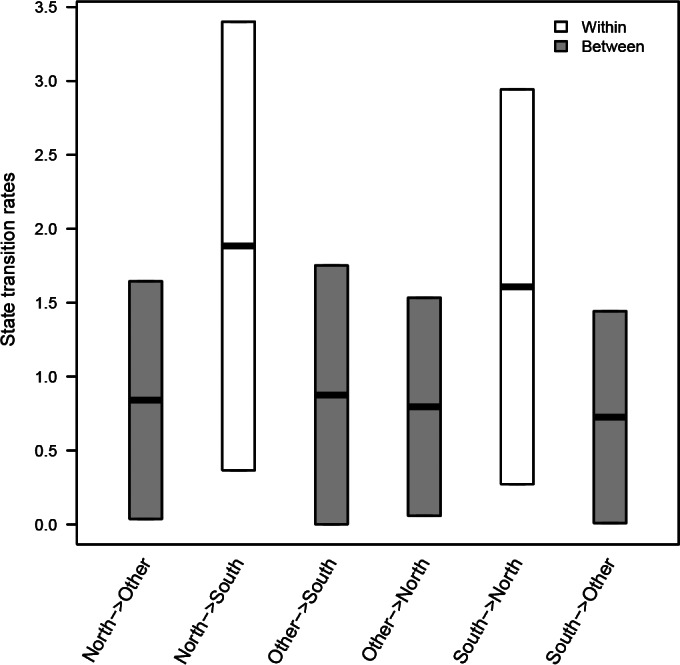
Box plots of the 95% HPD joint migration rate estimates combined across all BEAST discrete phylogeography randomized runs for avian-origin influenza A viruses sampled in the Mississippi Migratory Flyway (North and South) and isolates from the IRD (Other). Rate estimates included all PB2, PB1, PA, NP, and MP gene segments collected in this study and those publically available in the Influenza Research Database/GenBank.
Pairwise comparisons of sequence identity.
On average, 515,088 pairwise comparisons were examined for each AIV segment to determine the relationships of AIVs within the MMF and between other North American flyways (Table 3). Based on a 1% cutoff value, there were μ = 492.75 (σ = 382.73) “matching”/similar segments identified per gene segment. The percent identity for the 1% cutoff criterion was adjusted for each segment and resulted in percent identity scores ranging from 99.65% (PB2, PB1, HA, and NA) to 99.80% (MP) (Table 3). The numbers of high-identity observations ranged from 176 (PA) to 1,349 (NS), demonstrating the range of independent evolutionary constraints operating on each segment (i.e., NS is more conserved).
TABLE 3.
Summary statistics on pairwise comparisons between avian-origin influenza A virus isolates from the Mississippi Migratory Flyway and the Influenza Research Database/GenBank at 99% nucleotide identity and 1% cutoff values
| Gene segment | Length (nucleotides [nt]) | No. of MMF sequences | No. of IRD sequencesa | No. of pairwise comparisonsb | Maximum no. of differing nt for 99% identity | No. of high-identity matches | Maximum no. of differing nt for 1% cutoff | Resulting % identity | No. of high-identity matches |
|---|---|---|---|---|---|---|---|---|---|
| PB2 | 2,277 | 311 | 1,666 | 518,126 | 23 | 3,965 | 8 | 99.65 | 336 |
| PB1 | 2,271 | 298 | 1,652 | 492,296 | 23 | 4,279 | 8 | 99.65 | 325 |
| PA | 2,148 | 314 | 1,727 | 542,278 | 21 | 5,861 | 5 | 99.77 | 176 |
| HA | 1,719 | 301 | 1,749 | 526,449 | 17 | 3,368 | 6 | 99.65 | 237 |
| NP | 1,494 | 305 | 1,573 | 479,765 | 15 | 6,162 | 4 | 99.73 | 299 |
| NA | 1,437 | 321 | 1,812 | 581,652 | 14 | 4,054 | 5 | 99.65 | 676 |
| MP | 979 | 298 | 1,650 | 491,700 | 10 | 17,628 | 2 | 99.80 | 544 |
| NS | 835 | 303 | 1,612 | 488,436 | 8 | 15,074 | 2 | 99.76 | 1,349 |
Viruses include all available GenBank AIVs from avian species obtained from June 2008 to March 2011. These include the MMF sequences.
Every MMF sequence was compared to each IRD sequence.
Similar nucleotide lineages were concentrated more often at surveillance locations in close geographic proximity and for samples collected close together in time (χ2 = 587.66; P < 0.01) (Table 4; Fig. 3). The maximum distance between any two high-identity matches was observed within a migratory season for seven of the eight segments (PA excluded). The mean distance between high-identity matches increased marginally across seasons (NS excluded), but observations of these matches decreased significantly (χ2 = 271.27; P < 0.01) (Table 4). In addition, during the three migratory seasons in which this study took place, we observed significantly more matching lineages within the MMF than within the other flyways (χ2 = 302.42; P < 0.01) (Table 5). Note that while the NS segment was overrepresented in the high-identity matches (Table 3), the removal of this segment from the tests did not change the pattern or significance of the result.
TABLE 4.
Counts of all high-identity segment matches at a 1% cutoff for avian-origin AIV isolates from the Mississippi Migratory Flyway compared to all available AIV isolates in the Influenza Research Database/GenBank from June 2008 to March 2011
| Factor | No. of high-identity matches |
||
|---|---|---|---|
| Within season | Across two seasonsc | Across three seasons | |
| Distance (km)a | |||
| 0–500 | 1,319 | 417 | 26 |
| 500–1,000 | 467 | 305 | 63 |
| 1,000–1,500 | 317 | 176 | 50 |
| 1,500–2,000 | 103 | 113 | 15 |
| 2,000–2,500 | 154 | 49 | 2 |
| 2,500–3,000 | 123 | 79 | 49 |
| 3,000–3,500 | 3 | 9 | 2 |
| 3,500–4,000 | 0 | 0 | 0 |
| 4,000–4,500 | 8 | 0 | 22 |
| 4,500–5,000 | 52 | 2 | 0 |
| 5,000–5,500 | 10 | 8 | 0 |
| 5,500–6,000 | 0 | 0 | 0 |
| Gene segmentb | |||
| PB2 | 272 | 64 | 0 |
| PB1 | 244 | 77 | 4 |
| PA | 153 | 23 | 0 |
| HA | 189 | 48 | 0 |
| NP | 228 | 67 | 4 |
| NA | 340 | 252 | 84 |
| MP | 347 | 160 | 37 |
| NS | 783 | 466 | 100 |
χ2 = 587.66; df = 18; P < 0.01.
χ2 = 271.27; df = 14; P < 0.01.
Time classes were defined by the number of days between isolations of the two isolate segments included in each pairwise high-identity match (i.e., across two seasons represents a difference of 200 to 400 days between isolation).
FIG 3.

Plots representing all high-identity pairwise comparisons at the 1% cutoff value for each segment over geographic distance (km) and time (days) within the Mississippi Migratory Flyway. Gray points represent pairwise matches between two segments of MMF influenza virus isolates, while orange points represent pairwise matches between an MMF isolate and an isolate from another North American flyway.
TABLE 5.
Counts of all high-identity segment matches at a 1% cutoff for avian-origin AIV isolates from the MMF compared to all available AIV isolates in the Influenza Research Database/GenBank from June 2008 to March 2011
| Factor | No. of high-identity matchesd |
|
|---|---|---|
| Within flyway | Between flyways | |
| No. of shared segmentsa,c | ||
| 1 | 1,813 | 1,012 |
| 2 | 143 | 29 |
| 3 | 34 | 1 |
| 4 | 17 | 1 |
| 5 | 11 | 0 |
| 6 | 9 | 0 |
| 7 | 4 | 0 |
| 8 | 4 | 0 |
| Genetic segmentb | ||
| PB2 | 293 | 43 |
| PB1 | 243 | 82 |
| PA | 163 | 13 |
| HA | 206 | 31 |
| NP | 276 | 23 |
| NA | 532 | 144 |
| MP | 322 | 222 |
| NS | 828 | 521 |
χ2 = 62.83; df = 7; P < 0.01.
χ2 = 302.42; P < 0.01.
Viruses sharing constellations from the same day/same location were removed.
Numbers of matches for shared constellations and segments both within the MMF and for MMF viruses matching isolates outside the flyway.
On analyzing AIV constellations (all eight segments of the isolate), the number of segments sharing high-identity matches with another virus was negatively associated with both distance and time (Fig. 4 [time] and 5 [distance]). As the number of shared segments between two viruses increased, we observed fewer pairwise matches in the data set, ranging from 2,825 comparisons with 1 matching segment to 4 with 8 matching segments (Table 5). In addition, there was a larger number of AIVs within the MMF that shared ≥2 genomic segments in their constellations than between flyways (χ2 = 62.83; P < 0.01) (Table 5). Only four pairwise comparisons that were not collected on the same day at the same time matched at all eight AIV gene segments. Each of these comparisons occurred between isolates collected within 1 week of each other, and only one pair (A/Northern_Shoveler/Mississippi/10OS4526/H6N2/2010 and A/Gadwall/Mississippi/10OS4531/2010) was found at different sampling locations (33 km apart). One comparison sharing all eight segments was removed due to the high likelihood of laboratory contamination (A/Mallard/Ohio/10OS1354/2010/H6N1 and A/American Wigeon/Missouri/10OS4752/2010/H6N1).
FIG 4.
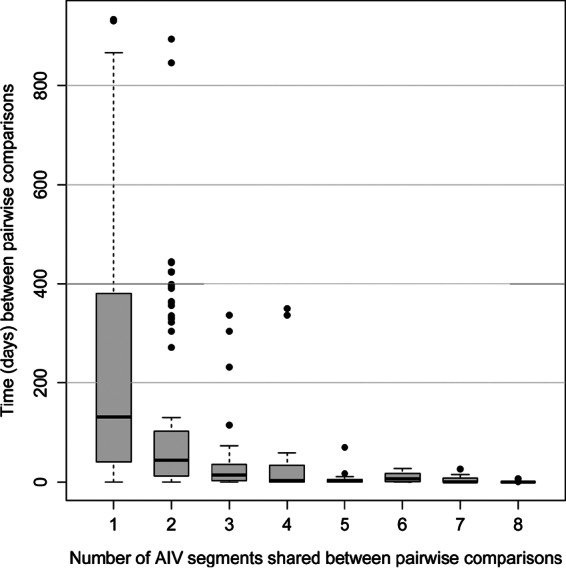
Plot representing the times (days) between samplings for isolates sharing one to eight gene segments with isolates from the Mississippi Migratory Flyway at the 1% cutoff value. The y axis (time) represents the entirety of study sampling, which occurred throughout three migratory seasons (2008 to 2011).
FIG 5.
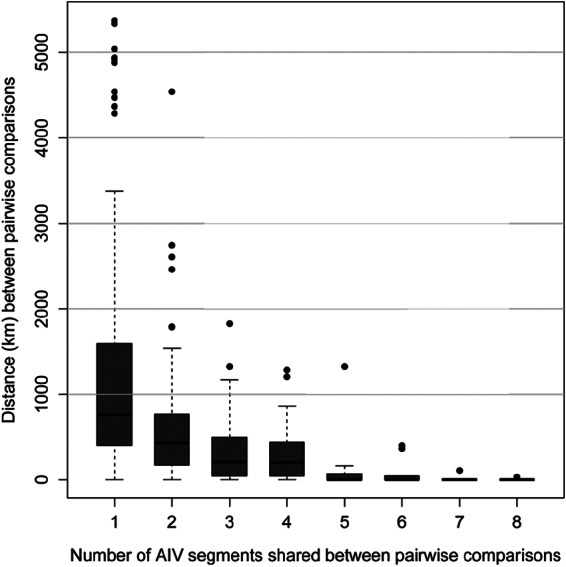
Plot representing the distances (km) between samplings for isolates sharing one to eight gene segments with isolates from the Mississippi Migratory Flyway at the 1% cutoff value. The y axis (distance) represents the entirety of Mississippi Migratory Flyway study sites sampled throughout three migratory seasons (2008 to 2011).
DISCUSSION
Utilizing a novel, systematic strategy of AIV surveillance following the autumn migration of mixed waterfowl populations, we show evidence for increased dispersal of LPAIV gene lineages within the MMF rather than between other migratory flyways. However, while significant gene flow occurs within the MMF over short time frames (i.e., within a migratory season), a pattern of geographic persistence of genetic diversity was apparent in the differential survival of AIV genomic segment lineages over multiple seasons. Furthermore, even though we followed bird populations as they migrated down the river, due to the high levels of reassortment in AIV, we recovered only four isolates containing the same virus constellations. Studies investigating North American AIV movement over much broader geographic and temporal scales suggested that the initial movement of AIVs is likely to occur along migratory routes, but these studies were limited by the availability of surveillance samples from these flyways and may have underestimated the amount of movement occurring within the flyways themselves (19, 20). The present study provides empirical evidence suggesting that waterfowl in the MMF disperse AIV genetic diversity quickly within the migratory corridor and that surveillance efforts to detect AIV threats to human and domestic animal health should pay particular attention to these routes in initially responding to epidemics/epizootics. It should be noted that while our study documents migration patterns of AIV diversity within the MMF, each migratory flyway is unique and warrants further investigations into how these dynamics vary across flyways around the world.
LPAIVs in the MMF.
Studies have shown that LPAIVs can be genetically structured within and between North American migratory flyways (20, 35, 45, 46), even in light of relatively high levels of gene flow measured between flyways (19). In the present study, while we observed a pattern of increased dispersal of AIVs within the MMF over short time frames, we also observed signatures of regional persistence of gene diversity across multiple seasons, which suggests a temporal effect on the genetic structuring of AIV diversity both within and among flyways. In the MMF, the persistence of these regional lineages across migratory seasons (Fig. 3) likely influenced the significant genetic structuring we observed when considering AIVs sampled across all migratory seasons (Table 2). As a result, the sampling of AIVs within the MMF at a few sites in any given year will likely provide a snapshot of the diversity that exists within the flyway and show that movement of these viruses occurs frequently. Traditionally, this is how many very important and noteworthy North American surveillance sites operate (47, 48). However, repeated annual sampling along the MMF as conducted in this study showed that, over time, the movement of these genetic lineages was often unsuccessful and resulted in a pattern of temporal structuring of the AIVs within the MMF. In addition, the MMF segment lineages that showed spillover events to other flyways, in conjunction with measurable (i.e., >0) (Fig. 2) migration rate estimates between the MMF and other flyways, suggest that while less frequent than intraflyway movement, opportunity does exist for AIV lineages to become established in new geographic regions.
It is difficult to determine which mechanisms govern the successful dispersal and subsequent survival of AIV gene lineages within the MMF and between other flyways. One potential explanation is a neutral process by which low-frequency dispersal events that occur over thousands of kilometers are lost to genetic drift, while lineages at higher frequencies at their site of origin may persist for multiple seasons. However, given the enormous population sizes of AIVs and the relative lack of noncoding portions of the AIV RNA genome, it is likely that drift has a negligible effect on AIV (49, 50). Instead, the variability in the survival of different AIV gene segments over time and space observed here (Fig. 3) suggests that selective pressures play a larger role than stochastic processes. Potentially, the same selection dynamics that create the phylogenetic splits of viral lineages at both the continental (51) and between-flyway (20) levels operate within the MMF and other flyways as well. Studies in the North American Pacific Flyway have shown that populations of viruses in northern breeding locations in Alaska are genetically distinct from populations of viruses circulating in the wintering populations in California (45, 46). Some level of competitive exclusion (e.g., host composition or environmental parameters) may operate within the flyway to effectively block migrant lineages from contributing to the dominant lineages circulating in a region (52). While there is evidence of migrant Eurasian lineages of viruses outcompeting and replacing resident North American lineages, these events appear to be rare (53). When these lineage replacement events do occur, they appear to be tied to strong selective sweeps that replace the existing diversity on a much broader, continent-wide level (6, 53).
At the intraflyway level, differing host population immunities and the host species compositions at alternate ends of the flyway are potentially two selective forces influencing any regional genetic structuring and diversity observed within the MMF in this study. The degree of host population immunity is an important driver of the diverse AIV antigenic subtypes that dominate regional sites from year to year (54). For example, antigenic drift likely results in the absence of similar HA gene lineages (i.e., AIV surface protein → host immunity target) across migratory seasons at the regional sites observed in this study (55). Similar patterns observed in the other internal segments (PB2, PB1, PA, and NP) (Fig. 3) may result from the hitchhiking of these regional segment lineages with HA (56). Alternatively, purifying selection resulting in reduced mutation rates and the maintenance of existing diversity in NA, MP, and NS lineages seen in other studies may result in the persistence of these lineages, although sampling of additional migratory seasons is needed (57, 58). Furthermore, the pattern of AIV prevalence for host species can change during migration for species such as northern shovelers (2.0% in northern birds and 26.0% in southern birds within the MMF), which may suggest a role for host ecology in AIV lineage survival among breeding and wintering sites in the MMF, as has been reported in other studies (59, 60). Future studies should consider the roles that differing host complexes at regional localities may play in AIV persistence.
This study showed that virus constellations sharing a large number of segment lineages were transient, existing for short periods and rarely outside sampling localities (Fig. 4 and 5). However, joint estimations of migration rates for five of the internal gene segments provided evidence that when movement of constellations does occur, it is primarily within the flyway (Fig. 2; Table 5). The amount of reassortment suggests that it is unlikely that wild birds move entire AIV constellations from northern breeding grounds to wintering grounds (or vice versa in the next North American spring) during the course of a regular migratory season in the MMF. Instead, AIV gene segment dispersal by wild birds relies on rapid reassortment of constellations along the migratory flyway and, to a greater degree, among flyways (61). The observations of gene dispersal and limited persistence of gene lineages among flyways are likely the result of countless introductions and subsequent removals of single gene lineages introduced by reassortment to AIVs in other flyways (19). The vast majority of reassortment events have negligible effects on AIV fitness and are presumably limited only by the rate at which multiple infections occur in host species (8). However, while it was not observed here, multiple studies have found persistence of entire AIV constellations across migratory seasons, suggesting the possible persistence of a virus in the environment until opportunistic exposure to some naive host species (62–64), although environmental conditions likely influence this persistence (65).
Influenza A virus similarity.
An important aspect of this study was the percent identity values used to define similarity among virus segments, as these values can greatly influence the interpretation of AIV ecology. We defined a conservative criterion for determining virus identity based on the genetic variability observed for each segment. Influenza A virus can mutate at rates of up to 103 mutations/site/year (66). While these rates typically result in many mutations, the population sizes of these viruses allow for efficient selection to remove even marginally deleterious changes (49, 67). In addition, even synonymous changes in influenza A viruses can have significant fitness effects and likely experience some level of selection (68). Therefore, any nucleotide differences observed between two viruses likely represent mutations that have passed through several selective “sieves.” Recently, Reeves et al. (42) showed that a 99% nucleotide identity was a natural cutoff for determining virus similarity among AIVs collected from waterfowl in Alaska, and multiple other studies have built off this work, even exploring nucleotide identity values as low as 97% (63, 64, 69). However, due to the large number of isolates included in this study and the variability among regions, we did not observe a natural cutoff and instead selected the top 1% of all pairwise comparisons within an AIV gene segment to define virus identity (Table 3). While all tests included in this study were significant and showed similar patterns at a 99% nucleotide similarity level, the 1% pairwise cutoff value better defined the amount of genetic variability in this study. A 1% cutoff reduced our variance among matching AIV segments by an order of magnitude (compared to a 99% identity score) and allowed for various levels of nucleotide identity for each independently evolving gene segment (70). Note that while we acknowledge that phylogenetic distances (i.e., patristic distances) (71) may be a more appropriate measure of relationships among viruses, these distances are still subject to arbitrary cutoff values and make measurements of reassortment more difficult (72).
Implications for wild bird surveillance.
This study offers support for the AIV spread by migratory wild birds evident in the dispersal of AIV segment genetic diversity within the migratory flyway over short periods. However, the apparently stochastic nature by which AIV lineages disperse, in addition to the selective pressures limiting most dispersal events, makes it difficult to determine the ecological factors influencing AIV gene flow in wild birds within the MMF (73). Ultimately, surveillance efforts in wild birds should be conducted with the goal of capturing AIV diversity, both antigenic and genetic, to prevent and prepare for pandemic threats and potential spillover to domestic animals (7). In order to capture this diversity, surveillance should be conducted annually given the rate at which these viruses appear to move with wild birds and among strategically placed (both temporally and geographically) sites along migratory corridors. This result is especially relevant for surveillance projects conducting surveillance for highly pathogenic strains in wild birds (74). Given the degree of reassortment in AIVs observed here, surveillance sites for strains of interest (e.g., HPH5N1, HPH7N3, and LPH7N9) are ideally situated within migratory routes, although gene segments from any one of these viruses may be found in other flyways (61, 75–77). While targeted surveillance efforts are necessary for rapid responses to disease threats, continued longitudinal studies examining the persistence and evolutionary patterns at regional sites are essential for elucidating the natural history of these viruses in their wild bird hosts (78, 79).
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the Minnesota Center of Excellence for Influenza Research and Surveillance (MCEIRS) with federal funds from the Centers of Excellence for Influenza Research and Surveillance (CEIRS), National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract HHSN266200700007C. This project was also funded by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract HHSN272200900007C.
We thank Chad Courtney, Lindsey Long, John Siekerski, Dave Sherman, Elizabeth St. James, Joe Lancaster, Jasmine Batten, Rose Foster, Joe Baker, Guy Zenner, Omari Nkullo, and Justin Dickey for assistance with sample collection. We greatly appreciate the cooperation of federal, state, and private employees at all of our surveillance locations, as well as the waterfowl hunters who kindly offered up their birds. Martha Nelson and Andrew Rambaut provided helpful suggestions on analyses. We particularly thank all those researchers who deposited the tremendous wealth of AIV sequences in shared public databases, which added immeasurably to this study.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03249-14.
REFERENCES
- 1.Real LA, Henderson JC, Biek R, Snaman J, Jack TL, Childs JE, Stahl E, Waller L, Tinline R, Nadin-Davis S. 2005. Unifying the spatial population dynamics and molecular evolution of epidemic rabies virus. Proc Natl Acad Sci U S A 102:12107–12111. doi: 10.1073/pnas.0500057102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altizer S, Bartel R, Han BA. 2011. Animal migration and infectious disease risk. Science 331:296–302. doi: 10.1126/science.1194694. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Smith GJD, Zhang SY, Qin K, Wang J, Li KS, Webster RG, Peiris JSM, Guan Y. 2005. H5N1 virus outbreak in migratory waterfowl. Nature 436:191–192. doi: 10.1038/nature03974. [DOI] [PubMed] [Google Scholar]
- 4.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slemons RD, Johnson DC, Osborn JS, Hayes F. 1974. Type-A influenza-viruses isolated from wild free-flying ducks in California. Avian Dis 18:119–124. doi: 10.2307/1589250. [DOI] [PubMed] [Google Scholar]
- 6.Worobey M, Han G-Z, Rambaut A. 2014. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature 508:254–257. doi: 10.1038/nature13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Runstadler J, Hill N, Hussein ITM, Puryear W, Keogh M. 2013. Connecting the study of wild influenza with the potential for pandemic disease. Infect Genet Evol 17:162–187. doi: 10.1016/j.meegid.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marshall N, Priyamvada L, Ende Z, Steel J, Lowen AC. 2013. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog 9:e1003421. doi: 10.1371/journal.ppat.1003421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latorre-Margalef N, Gunnarsson G, Munster VJ, Fouchier RAM, Osterhaus A, Elmberg J, Olsen B, Wallensten A, Haemig PD, Fransson T, Brudin L, Waldenstrom J. 2009. Effects of influenza A virus infection on migrating mallard ducks. Proc Biol Sci 276:1029–1036. doi: 10.1098/rspb.2008.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jourdain E, Gunnarsson G, Wahlgren J, Latorre-Margalef N, Bröjer C, Sahlin S, Svensson L, Waldenström J, Lundkvist Å, Olsen B. 2010. Influenza virus in a natural host, the mallard: experimental infection data. PLoS One 5:e8935. doi: 10.1371/journal.pone.0008935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krauss S, Walker D, Pryor SP, Niles L, Li CH, Hinshaw VS, Webster RG. 2004. Influenza A viruses of migrating wild aquatic birds in North America. Vector Borne Zoonotic Dis 4:177–189. doi: 10.1089/vbz.2004.4.177. [DOI] [PubMed] [Google Scholar]
- 12.Prosser DJ, Cui P, Takekawa JY, Tang M, Hou Y, Collins BM, Yan B, Hill NJ, Li T, Li Y, Lei F, Guo S, Xing Z, He Y, Zhou Y, Douglas DC, Perry WM, Newman SH. 2011. Wild bird migration across the Qinghai-Tibetan Plateau: a transmission route for highly pathogenic H5N1. PLoS One 6:e17622. doi: 10.1371/journal.pone.0017622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou J-Y, Shen H-G, Chen H-X, Tong G-Z, Liao M, Yang H-C, Liu J-X. 2006. Characterization of a highly pathogenic H5N1 influenza virus derived from bar-headed geese in China. J Gen Virol 87:1823–1833. doi: 10.1099/vir.0.81800-0. [DOI] [PubMed] [Google Scholar]
- 14.Fries AC, Nolting JM, Danner A, Webster RG, Bowman AS, Krauss S, Slemons RD. 2013. Evidence for the circulation and inter-hemispheric movement of the H14 subtype influenza A virus. PLoS One 8:e59216. doi: 10.1371/journal.pone.0059216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kilpatrick AM, Chmura AA, Gibbons DW, Fleischer RC, Marra PP, Daszak P. 2006. Predicting the global spread of H5N1 avian influenza. Proc Natl Acad Sci U S A 103:19368–19373. doi: 10.1073/pnas.0609227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaidet N, Cattoli G, Hammoumi S, Newman SH, Hagemeijer W, Takekawa JY, Cappelle J, Dodman T, Joannis T, Gil P, Monne I, Fusaro A, Capua I, Manu S, Micheloni P, Ottosson U, Mshelbwala JH, Lubroth J, Domenech J, Monicat F. 2008. Evidence of infection by H5N2 highly pathogenic avian influenza viruses in healthy wild waterfowl. PLoS Pathog 4:e1000127. doi: 10.1371/journal.ppat.1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munster VJ, Baas C, Lexmond P, Waldenstrom J, Wallensten A, Fransson T, Rimmelzwaan GF, Beyer WEP, Schutten M, Olsen B, Osterhaus A, Fouchier RAM. 2007. Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog 3:e61. doi: 10.1371/journal.ppat.0030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaidet N, Cappelle J, Takekawa JY, Prosser DJ, Iverson SA, Douglas DC, Perry WM, Mundkur T, Newman SH. 2010. Potential spread of highly pathogenic avian influenza H5N1 by wildfowl: dispersal ranges and rates determined from large-scale satellite telemetry. J Appl Ecol 47:1147–1157. doi: 10.1111/j.1365-2664.2010.01845.x. [DOI] [Google Scholar]
- 19.Bahl J, Krauss S, Kühnert D, Fourment M, Raven G, Pryor SP, Niles LJ, Danner A, Walker D, Mendenhall IH, Su YCF, Dugan VG, Halpin RA, Stockwell TB, Webby RJ, Wentworth DE, Drummond AJ, Smith GJD, Webster RG. 2013. Influenza A virus migration and persistence in North American wild birds. PLoS Pathog 9:e1003570. doi: 10.1371/journal.ppat.1003570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam TT-Y, Ip HS, Ghedin E, Wentworth DE, Halpin RA, Stockwell TB, Spiro DJ, Dusek RJ, Bortner JB, Hoskins J, Bales BD, Yparraguirre DR, Holmes EC. 2012. Migratory flyway and geographical distance are barriers to the gene flow of influenza virus among North American birds. Ecol Lett 15:24–33. doi: 10.1111/j.1461-0248.2011.01703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam TT-Y, Wang J, Shen Y, Zhou B, Duan L, Cheung C-L, Ma C, Lycett SJ, Leung CY-H, Chen X, Li L, Hong W, Chai Y, Zhou L, Liang H, Ou Z, Liu Y, Farooqui A, Kelvin DJ, Poon LLM, Smith DK, Pybus OG, Leung GM, Shu Y, Webster RG, Webby RJ, Peiris JSM, Rambaut A, Zhu H, Guan Y. 2013. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 502:241–244. doi: 10.1038/nature12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jhung MA, Nelson DI. 2015. Outbreaks of avian influenza A (H5N2), (H5N8), and (H5N1) among birds—United States, December 2014–January 2015. MMWR Morb Mortal Wkly Rep 64:111. [PMC free article] [PubMed] [Google Scholar]
- 23.Bellrose FC, Kortright FH. 1976. Ducks, geese & swans of North America, 2nd ed Stackpole Books, Harrisburg, PA. [Google Scholar]
- 24.Kawaoka Y, Chambers TM, Sladen WL, Webster RG. 1988. Is the gene pool of influenza viruses in shorebirds and gulls different from that in wild ducks? Virology 163:247–250. doi: 10.1016/0042-6822(88)90260-7. [DOI] [PubMed] [Google Scholar]
- 25.Hinshaw VS, Webster RG, Turner B. 1980. The perpetuation of orthomyxoviruses and paramyxoviruses in Canadian waterfowl. Can J Microbiol 26:622–629. doi: 10.1139/m80-108. [DOI] [PubMed] [Google Scholar]
- 26.Stallknecht DE, Brown JD. 2007. Wild birds and the epidemiology of avian influenza. J Wildl Dis 43:S15–S20. [DOI] [PubMed] [Google Scholar]
- 27.Hinshaw VS, Wood JM, Webster RG, Deibel R, Turner B. 1985. Circulation of influenza-viruses and paramyxoviruses in waterfowl originating from 2 different areas of North-America. Bull World Health Organ 63:711–719. [PMC free article] [PubMed] [Google Scholar]
- 28.Slemons RD, Easterday BC. 1978. Virus replication in the digestive tract of ducks exposed by aerosol to type-A influenza. Avian Dis 22:367–377. doi: 10.2307/1589291. [DOI] [PubMed] [Google Scholar]
- 29.WHO. 2002. WHO manual on animal influenza diagnosis and surveillance. WHO, Geneva, Switzerland. [Google Scholar]
- 30.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou B, Wentworth D. 2012. Influenza A virus molecular virology techniques. Methods Mol Biol 865:175–192. doi: 10.1007/978-1-61779-621-0_11. [DOI] [PubMed] [Google Scholar]
- 32.Djikeng A, Halpin R, Kuzmickas R, DePasse J, Feldblyum J, Sengamalay N, Afonso C, Zhang X, Anderson N, Ghedin E, Spiro D. 2008. Viral genome sequencing by random priming methods. BMC Genomics 9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98. [Google Scholar]
- 34.Squires RB, Noronha J, Hunt V, García-Sastre A, Macken C, Baumgarth N, Suarez D, Pickett BE, Zhang Y, Larsen CN, Ramsey A, Zhou L, Zaremba S, Kumar S, Deitrich J, Klem E, Scheuermann RH. 2012. Influenza Research Database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respir Viruses 6:404–416. doi: 10.1111/j.1750-2659.2011.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen R, Holmes EC. 2009. Frequent inter-species transmission and geographic subdivision in avian influenza viruses from wild birds. Virology 383:156–161. doi: 10.1016/j.virol.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 37.Posada D, Crandall KA. 1998. Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 38.Slatkin M, Maddison WP. 1989. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 123:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maddison WP, Maddison DR. 2011. Mesquite: a modular system for evolutionary analysis, version 2.75. http://mesquiteproject.org.
- 40.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemey P, Rambaut A, Drummond AJ, Suchard MA. 2009. Bayesian phylogeography finds its roots. PLoS Comput Biol 5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reeves AB, Pearce JM, Ramey AM, Meixell BW, Runstadler JA. 2011. Interspecies transmission and limited persistence of low pathogenic avian influenza genomes among Alaska dabbling ducks. Infect Genet Evol 11:2004–2010. doi: 10.1016/j.meegid.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Legendre P, Legendre L. 1998. Numerical ecology, 2nd ed Elsevier Science, Amsterdam, Netherlands. [Google Scholar]
- 45.Girard YA, Runstadler JA, Aldehoff F, Boyce W. 2012. Genetic structure of Pacific Flyway avian influenza viruses is shaped by geographic location, host species, and sampling period. Virus Genes 44:415–428. doi: 10.1007/s11262-011-0706-5. [DOI] [PubMed] [Google Scholar]
- 46.Pearce JM, Ramey AM, Flint PL, Koehler AV, Fleskes JP, Franson JC, Hall JS, Derksen DV, Ip HS. 2009. Avian influenza at both ends of a migratory flyway: characterizing viral genomic diversity to optimize surveillance plans for North America. Evol Appl 2:457–468. doi: 10.1111/j.1752-4571.2009.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stallknecht DE, Luttrell MP, Poulson R, Goekjian V, Niles L, Dey A, Krauss S, Webster RG. 2012. Detection of avian influenza viruses from shorebirds: evaluation of surveillance and testing approaches. J Wildl Dis 48:382–393. doi: 10.7589/0090-3558-48.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharp GB, Kawaoka Y, Wright SM, Turner B, Hinshaw V, Webster RG. 1993. Wild ducks are the reservoir for only a limited number of influenza A subtypes. Epidemiol Infect 110:161–176. doi: 10.1017/S0950268800050780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holmes EC. 2009. The evolutionary genetics of emerging viruses. Annu Rev Ecol Evol Syst 40:353–372. doi: 10.1146/annurev.ecolsys.110308.120248. [DOI] [Google Scholar]
- 50.Kimura M, Ohta T. 1969. The average number of generations until fixation of a mutant gene in a finite population. Genetics 61:763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obenauer JC, Denson J, Mehta PK, Su XP, Mukatira S, Finkelstein DB, Xu XQ, Wang JH, Ma J, Fan YP, Rakestraw KM, Webster RG, Hoffmann E, Krauss S, Zheng J, Zhang ZW, Naeve CW. 2006. Large-scale sequence analysis of avian influenza isolates. Science 311:1576–1580. doi: 10.1126/science.1121586. [DOI] [PubMed] [Google Scholar]
- 52.Hill NJ, Takekawa JY, Ackerman JT, Hobson KA, Herring G, Cardona CJ, Runstadler JA, Boyce WM. 2012. Migration strategy affects avian influenza dynamics in mallards (Anas platyrhynchos). Mol Ecol 21:5986–5999. doi: 10.1111/j.1365-294X.2012.05735.x. [DOI] [PubMed] [Google Scholar]
- 53.Bahl J, Vijaykrishna D, Holmes EC, Smith GJD, Guan Y. 2009. Gene flow and competitive exclusion of avian influenza A virus in natural reservoir hosts. Virology 390:289–297. doi: 10.1016/j.virol.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Latorre-Margalef N, Tolf C, Grosbois V, Avril A, Bengtsson D, Wille M, Osterhaus ADME, Fouchier RAM, Olsen B, Waldenström J. 2014. Long-term variation in influenza A virus prevalence and subtype diversity in migratory mallards in northern Europe. Proc Biol Sci 281:20140098. doi: 10.1098/rspb.2014.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ferguson NM, Galvani AP, Bush RM. 2003. Ecological and immunological determinants of influenza evolution. Nature 422:428–433. doi: 10.1038/nature01509. [DOI] [PubMed] [Google Scholar]
- 56.Chen RB, Holmes EC. 2010. Hitchhiking and the population genetic structure of avian influenza virus. J Mol Evol 70:98–105. doi: 10.1007/s00239-009-9312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito T, Gorman OT, Kawaoka Y, Bean WJ, Webster RG. 1991. Evolutionary analysis of the influenza A virus M gene with comparison of the M1 and M2 proteins. J Virol 65:5491–5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wertheim JO, Kosakovsky Pond SL. 2011. Purifying selection can obscure the ancient age of viral lineages. Mol Biol Evol 28:3355–3365. doi: 10.1093/molbev/msr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hill NJ, Takekawa JY, Cardona CJ, Ackerman JT, Schultz AK, Spragens KA, Boyce WM. 2010. Waterfowl ecology and avian influenza in California: do host traits inform us about viral occurrence? Avian Dis 54:426–432. doi: 10.1637/8912-043009-Reg.1. [DOI] [PubMed] [Google Scholar]
- 60.Stallknecht DE, Shane SM, Zwank PJ, Senne DA, Kearney MT. 1990. Avian influenza-viruses from migratory and resident ducks of coastal Louisiana. Avian Dis 34:398–405. doi: 10.2307/1591427. [DOI] [PubMed] [Google Scholar]
- 61.Dusek RJ, Hallgrimsson GT, Ip HS, Jónsson JE, Sreevatsan S, Nashold SW, Teslaa JL, Enomoto S, Halpin RA, Lin X, Fedorova N, Stockwell TB, Dugan VG, Wentworth DE, Hall JS. 2014. North Atlantic migratory bird flyways provide routes for intercontinental movement of avian influenza viruses. PLoS One 9:e92075. doi: 10.1371/journal.pone.0092075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lebarbenchon C, Yang M, Keeler SP, Ramakrishnan MA, Brown JD, Stallknecht DE, Sreevatsan S. 2011. Viral replication, persistence in water and genetic characterization of two influenza A viruses isolated from surface lake water. PLoS One 6:e26566. doi: 10.1371/journal.pone.0026566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wille M, Tolf C, Avril A, Latorre-Margalef N, Wallerström S, Olsen B, Waldenström J. 2013. Frequency and patterns of reassortment in natural influenza A virus infection in a reservoir host. Virology 443:150–160. doi: 10.1016/j.virol.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 64.Reeves AB, Pearce JM, Ramey AM, Ely CR, Schmutz JA, Flint PL, Derksen DV, Ip HS, Trust KA. 2013. Genomic analysis of avian influenza viruses from waterfowl in western Alaska, USA. J Wildl Dis 49:600–610. doi: 10.7589/2012-04-108. [DOI] [PubMed] [Google Scholar]
- 65.Stallknecht DE, Kearney MT, Shane SM, Zwank PJ. 1990. Effects of pH, temperature, and salinity on persistence of avian influenza-viruses in water. Avian Dis 34:412–418. doi: 10.2307/1591429. [DOI] [PubMed] [Google Scholar]
- 66.Duffy S, Shackelton LA, Holmes EC. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet 9:267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 67.Nobusawa E, Sato K. 2006. Comparison of the mutation rates of human influenza A and B viruses. J Virol 80:3675–3678. doi: 10.1128/JVI.80.7.3675-3678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marsh GA, Rabadán R, Levine AJ, Palese P. 2008. Highly conserved regions of influenza A virus polymerase gene segments are critical for efficient viral RNA packaging. J Virol 82:2295–2304. doi: 10.1128/JVI.02267-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang Y, Wille M, Dobbin A, Walzthöni NM, Robertson GJ, Ojkic D, Whitney H, Lang AS. 2014. Genetic structure of avian influenza viruses from ducks of the Atlantic Flyway of North America. PLoS One 9:e86999. doi: 10.1371/journal.pone.0086999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suarez DL. 2000. Evolution of avian influenza viruses. Vet Microbiol 74:15–27. doi: 10.1016/S0378-1135(00)00161-9. [DOI] [PubMed] [Google Scholar]
- 71.Carrel MA, Emch M, Jobe RT, Moody A, Wan XF. 2010. Spatiotemporal structure of molecular evolution of H5N1 highly pathogenic avian influenza viruses in Vietnam. PLoS One 5:e8631. doi: 10.1371/journal.pone.0008631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dugan VG, Chen R, Sprio DJ, Sengamalay N, Zaborsky J, Ghedin E, Nolting J, Swayne DE, Runstadler JA, Happ GM, Senne DA, Wang R, Slemons RD, Holmes EC, Taubenberger JK. 2008. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLoS Pathog 4:e1000076. doi: 10.1371/journal.ppat.1000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Farnsworth ML, Miller RS, Pedersen K, Lutman MW, Swafford SR, Riggs PD, Webb CT. 2012. Environmental and demographic determinants of avian influenza viruses in waterfowl across the contiguous United States. PLoS One 7:e32729. doi: 10.1371/journal.pone.0032729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jones JC, Sonnberg S, Kocer ZA, Shanmuganatham K, Seiler P, Shu Y, Zhu HC, Guan Y, Peiris M, Webby RJ, Webster RG. 2014. Possible role of songbirds and parakeets in transmission of influenza A (H7N9) virus to humans. Emerg Infect Dis 20:380–385. doi: 10.3201/eid2003.131271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pearce JM, Reeves AB, Ramey AM, Hupp JW, Ip HS, Bertram M, Petrula MJ, Scotton BD, Trust KA, Meixell BW, Runstadler JA. 2011. Interspecific exchange of avian influenza virus genes in Alaska: the influence of trans-hemispheric migratory tendency and breeding ground sympatry. Mol Ecol 20:1015–1025. doi: 10.1111/j.1365-294X.2010.04908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fries AC, Nolting JM, Bowman AS, Killian ML, Wentworth DE, Slemons RD. 2014. Genomic analyses detect Eurasian-lineage H10 and additional H14 influenza A viruses recovered from waterfowl in the central United States. Influenza Other Respir Viruses 8:493–498. doi: 10.1111/irv.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu L, Lycett SJ, Leigh Brown AJ. 2014. Determining the phylogenetic and phylogeographic origin of highly pathogenic avian influenza (H7N3) in Mexico. PLoS One 9:e107330. doi: 10.1371/journal.pone.0107330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krauss S, Obert CA, Franks J, Walker D, Jones K, Seiler P, Niles L, Pryor SP, Obenauer JC, Naeve CW, Widjaja L, Webby RJ, Webster RG. 2007. Influenza in migratory birds and evidence of limited intercontinental virus exchange. PLoS Pathog 3:e167. doi: 10.1371/journal.ppat.0030167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wallensten A, Munster VJ, Latorre-Margalef N, Brytting M, Elmberg J, Fouchier RAM, Fransson T, Haemig PD, Karlsson M, Lundkvist A, Osterhaus A, Stervander M, Waldenstrom J, Olsen B. 2007. Surveillance of influenza A virus in migratory waterfowl in northern Europe. Emerg Infect Dis 13:404–411. doi: 10.3201/eid1303.061130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.