

Abstract
Fifty years after the discovery of adeno-associated virus (AAV) and more than 30 years after the first gene transfer experiment was conducted, dozens of gene therapy clinical trials are in progress, one vector is approved for use in Europe, and breakthroughs in virus modification and disease modeling are paving the way for a revolution in the treatment of rare diseases, cancer, as well as HIV. This review will provide a historical perspective on the progression of AAV for gene therapy from discovery to the clinic, focusing on contributions from the Samulski lab regarding basic science and cloning of AAV, optimized large-scale production of vectors, preclinical large animal studies and safety data, vector modifications for improved efficacy, and successful clinical applications.
Introduction/Historical Perspective
With the discovery of “virus-like” particles in adenovirus (Ad) preparation by Bob Atchison1 and M. David Hoggan and Wallace Rowe2 in 1965, the journey of a novel parvovirus began.3 Atchison tentatively referred to the virus as adeno-associated virus (AAV), and Rowe continued use of the name (Fig. 1). Early experiments characterized a virus that replicated poorly, if at all, in human cells in the absence of Ad. However, it was clear that AAV was immunologically distinct from any known Ad antigen, and three serotypes were identified, AAV1, 2, and 3.1 The following years saw progress in determining that the AAV capsid contained an ssDNA genome,4,5 that the genome contained inverted terminal repeats (ITRs),6–8 and that these ITRs are structured as self-priming hairpins that act as origins of genome replication.9 Sequencing of the AAV2 serotype ITRs was complete by 1980.10 Concomitantly, the latent state of AAV was observed at NIH when an Ad infection on a panel of cell lines resulted in the production of AAV. This led to the discovery that low numbers of AAV genomes were integrated into the host chromosome,11 thus establishing the foundation for using AAV as a gene therapy vector based on its lytic (co-infection with Ad), latent (establishment of persistence in the absence), bi-phasic life cycle.
FIG. 1.
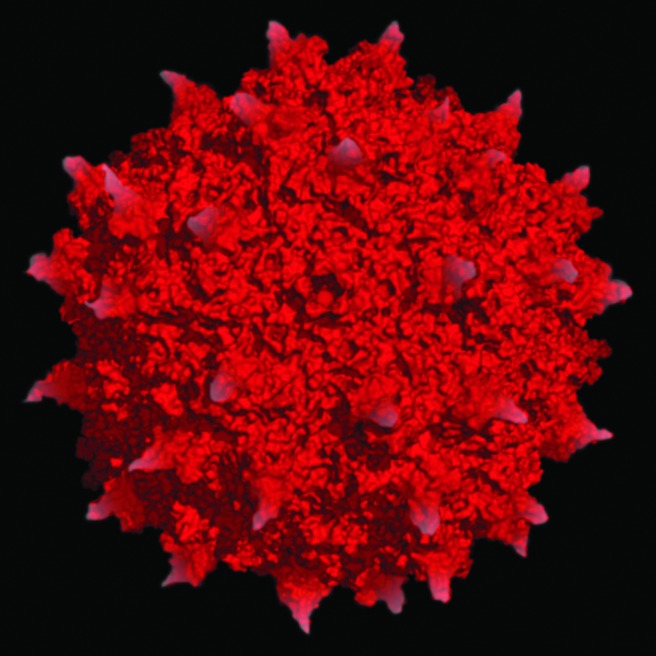
Adeno-associated virus serotype 2 atomic structure. The surface topology and electrostatics of AAV2 are shown (modified from Xie et al.71). The 25 nm virion capsid shown here is composed of 3 viral proteins, VP1, 2, and 3, and encapsidates an approximately 5 kb ssDNA genome that has been used to encode and deliver transgenes of interest for human gene therapy application.
Basic Science
Also in the early 1980s, with all molecular biology experiments being conducted using AAV serotype 2, it was known that the AAV genome was packaged into preformed capsid proteins.12 Further, a transcription map showed three overlapping mRNA families from three promoters, p5, p19, and p40.13,14 The genome appeared divided between left and right, and three capsids, VP1, VP2, and VP3, were produced from the p40 promoter on the right side.15 It was unclear what the left half of the genome encoded, but a replication function was predicted. What was known, however, was that the virus could be produced in excess of 100,000 particles per cell. This ability to produce high yields, the fact that AAV was not a pathogen, and that the virus could persist all encouraged further exploration of its use as a gene therapy delivery platform. Unfortunately, because AAV replicated in the presence of a lytic virus, there was no easy focus-forming assay for selection of isolated mutants. Molecular clones were needed, but the NIH Recombinant DNA Advisory Committee (RAC) guidelines were not clear when it came to cloning AAV DNA into plasmids. According to Barrie Carter, a 1980 ASM meeting in Miami Beach convinced the NIH RAC that recombinant DNA viruses like AAV and Ad could be safely studied in BSL2-approved laboratories.16 Here, cloning of the AAV genome into plasmids became a reality and isolation of wild-type (wt) AAV infectious clones became a major milestone for the field.
In 1978 as a new graduate student in the lab of Nicholas Muzyczka, Jude Samulski was tasked with generating a plasmid containing the AAV genome that could be used to rescue wild-type AAV virions. Based on Nicholas's design, digest of the pBR322 plasmid with PstI generated a linear molecule to which oligo(dG) was attached to the 3′ end of the plasmid, while oligo(dC) was attached to duplex AAV DNA17 and allowed to reanneal after melting out at 70°C. The new plasmid was transfected into HB101 E. coli and 50 colonies were obtained. Three plasmids (pSM501, pSM503, and pSM504) were the correct size for an intact AAV recombinant plasmid and restriction digest with diagnostic PstI enzyme showed that there were no major deletions in the nonrepeated internal sequences of AAV and that the PstI reformed during the cloning process to release the terminal fragments. However, the plasmid preps appeared to contain mixtures of at least two plasmids that differed from each other in the AAV terminal resolution sites (trs). Multiple attempts to subclone from pSM501 mixed parental stock allowed for isolation of a plasmid that had the appropriate cut sites and no trs deletions, pSM620 (named for 620 attempts). Fortunately, this achieved isolation of a plasmid with the complete AAV genome that allowed for the rescue of wt virus by means of plasmid transfection and helper virus infection17 and initiated the current 30-year-long journey to optimize AAV as a viral vector. In fact, supernatant collected from 293-31 cells that were transfected with pSM620 and infected with Ad serotype 5 proved just that, namely, rescue, replication, and packaging of a molecular clone of wt AAV from a plasmid substrate. The Carter lab achieved the same outcome the following year.18 Based on these studies, this work put in place a system to begin site-directed mutagenesis across the AAV genome, which in turn lead to the design of the first-generation of AAV transfer vectors.
While the success of finding one complete plasmid was encouraging, there were also many other clones identified that contained deletions within the ITR sequences, from the left, right, or both sides of the intact AAV genome. Surprisingly, rescue of plasmids containing only deletion of the left or right ITR resulted in production of capsids containing wt AAV DNA.19 Of the plasmids containing deletions in both ITRs, only one allowed for rescue of the virus, though viability was low. In fact, deletion of the remaining 11 bp from the pSM609 ITR resulted in a complete inability to rescue the virus. This discovery confirmed the necessity of AAV ITRs for replication, confirmed the AAV replication model using “Flip+Flop” terminal repeats, and started the discussion of a model whereby AAV utilizes a mechanism to correct deletions within its ITRs through a process called “gene conversion.” This set the stage for understanding AAV vector concatenation and the recently described fragmented AAV vectors described below.19 Shortly following the generation of AAV clones, the entire sequence of AAV2 was completed19 and allowed for identification of the cis-acting origin of replication function of the ITRs and the trans-acting function of the left and right reading frames of the AAV genome products.3,20
About this time, the first transduction of mammalian cells was demonstrated. Hermonat and Muzyczka3 utilized the ins96(pHM2904) construct, a derivative of pSM620, to generate a plasmid containing a neomycin resistance gene and successfully rescued AAV2 virus. The virus was used to infect human Detroit 6 and KB cells, with antibiotic resistance tested with G41821 and an observed transduction efficacy of 0.4–10%.3 These experiments validated another major milestone in vector development. The authors highlighted areas that required additional understanding, including expression from integrated AAV genomes, improved rescue of AAV, studies of larger DNA inserts, host range mutants, and gene therapy applications, all of which would assume over the next 30 years.
Continuing work on the basic virology of AAV, Samulski working with Patrick Hearing and Thomas Shenk at Princeton reported that deletion of repeated cis-acting sequence allowed for Ad5 genome replication, but prevented packaging of the genome into viral capsids. Further, a series of Ad mutants along with AAV mutants engineered in the infectious molecular clone allowed the lab to determine the sequence required for packaging, successful replication, and integration.22 Importantly, Samulski also continued work on the AAV2 pSM620 vector, generating modifications to facilitate easy swapping of transgenes. The first of these was psub201. In the plasmid, psub201(+), the AAV2 coding domain was flanked with XbaI cleavage sites in a four-fragment ligation allowing for 96% of the viral gene to be removed and replaced with transgene sequence of choice to be easily inserted between the cis-acting ITRs. Additionally, unique PvuII sites allowed for excision of the entire viral sequence from the plasmid.23 These studies define early information concerning the packaging limits of AAV (i.e., 5 kb) and, with respect to further understanding virus biology, confirmed a proposed hypothesis about AAV second-strand DNA synthesis. Specifically, the article demonstrated the ability to isolate plus- and minus-strand AAV2 genomes as molecular clones using single-stranded phage DNA-carrying wt AAV genome and showed that both strands were infectious,23 confirming a hypothesis proposed by Ken Berns and colleagues for AAV self-primed replication using AAV ITRs.
While significant understanding was being acquired for various aspects of AAV biology during this phase of science, the Samulski lab turned their attention to rate-limiting steps involved in AAV vector production. As an early precursor to GMP production, a method was established for generating stocks free of wt helper AAV.24 Using psub201, the recombinant AAV (rAAV) contained only the terminal 191 nucleotides flanking a neomycin or hygromycin resistance marker gene. Trans-acting functions of helper AAV were provided by a construct in which the 191 terminal repeats of AAV were replaced with Ad sequences that did not allow the AAV helper DNA to replicate or facilitate recombination between viral trans and cis sequences.24 As the AAV ITRs are only required in cis for replication, this system allowed for specific generation of rAAV and set forth years of experimental exploration aimed at improving the yields of this robust production system. Infection of human cells with the produced virus resulted in 70% of established colonies expressing the marker gene. This allowed for generation of drug-resistant human cell lines and helped establish for the first time that AAV gene expression was not required for integration into host DNA24 or AAV transduction.
Now in the early 1990s, as integration was thought to be key for prolonged gene expression and therapeutic use, it was important to determine if the vector could be oncogenic. Robert Kotin, working with leading AAV researchers, determined that wt AAV integration occurs specifically on the q arm of chromosome 19, a unique finding among DNA viruses.25,26 Following this work, the Samulski lab and others continued basic virus research demonstrating that AAV Rep protein is required for targeting integration and at the same time began testing AAV for treatment of rare diseases discussed in the next section. Although all labs working on AAV during this time were relying on the latent phase of AAV, namely, integration, no one was prepared for the observation of Xiao and Samulski that AAV persisted as concatenated episomes after vector transduction.27
Preclinical Studies
With 25 years of basic AAV research as a foundation, labs were now attempting gene therapy experiments for treatment of human diseases. With several hemoglobinopathies requiring high-level expression of a transferred globing gene in erythroid cells, β-globin transfer was the first AAV expressed transgene explored by the Samulski lab in collaboration with Chris Walsh at NIH. It was demonstrated that rAAV encoding the Aγ-globin gene could be stably introduced into human K562 eyrthroleukemia cells and that expression was equivalent to that of a native Aγ-globin gene.28 This article also demonstrated how expression could be enhanced with a locus control region (LCR) inserted before the gene and was the first demonstration of tissue-specific promoters in an AAV vector context.28 A follow-up publication made mutations to regions of the hypersensitivity site 2 (HS2) LCR in front of the Aγ-globin gene and demonstrated changes in transcription binding factors that may be useful for regulating gene expression.29 Again, all of these early studies utilized in vitro cell lines and gene expression was tracked with viral integration, which was not very efficient in most of these cell types.
Moving from in vitro to in vivo, the first reported use of AAV to express a cystic fibrosis transmembrane conductance regulator (CFTR) protein in an animal model by Terry Flotte showed efficient transduction of rabbit lung tissue, with the AAV-encoded CFTR protein detected in airway epithelium for up to 6 months following vector administration.30 In another in vivo study in rats, for the first time the denervated striatum of the brain was injected with rAAV encoding LacZ (AAVlac) or human tyrosine hydroxylase (AAVth). This treatment resulted in virus-driven gene expression up to 4 months posttreatment and resulted in significant behavioral recovery.31 At this time, many diseases were being addressed by gene therapies and cardiac disease was no exception. Intramuscular (i.m.) injection in rat hearts as well as infusion into porcine circumflex coronary arteries with AAV lac Z with a human cytomegalovirus immediate-early promoter caused limited observable inflammation and gene expression for at least 6 months after infusion.32 These studies and others provided preliminary safety data and highlighted the plausibility of gene therapy success.
What was not directly explored in the early studies was AAV tropism. Studies in the Samulski lab determined that the expression pattern and longevity of the AAV-encoded lacZ reporter depends on the area of the brain being studied.33 Most importantly, for diseases with central nervous system (CNS) pathology, there were no signs of toxicity noted. For treatment of metabolic disorders, such as hemophilia or Duchenne muscular dystrophy (DMD), targeting of liver or muscle tissue will be required, respectively. Previous effort using other vectors (Ad, retrovirus, or naked DNA) had encountered immune clearance of transduced cells, but Xiao's study demonstrated that rAAV could be applied without an adaptive immune clearance of transduced cells and that in vivo gene transfer of at least 1.5 years (life of the mouse) could be achieved.32 This work in collaboration with Xiao and Li was the first study to demonstrate long-term transduction of AAV in an animal model. Similar study showing transduction in muscle for 4 months was published by Barry Byrne and colleagues the same year, and confirmed the unique but hallmark observation that AAV persistence was long lived and consistent with expectations based on viral latent life cycle. Since these initial efforts, significant progress has been observed determining the ability to efficiently deliver transgenes to skeletal muscle in mouse, hamster, canine, and primate models for life of animal, supporting the original studies published by Xiao and Byrne.
Generation of fluorescently labeled rAAV34 allowed for the first-ever in vivo monitoring of rAAV kinetics and provided a basis for targeted application of neuronal populations in human brain therapies following direct infusion with AAV2.35 What was not expected was the perplexing observation described by Xiao that the “in vivo” AAV genomes appeared episomal and not integrated. More confusing was the observation that over time monomeric AAV genomes were transitioning to “head to tail” concatamers. This fascinating observation is believed to occur through AAV ITRs and requires a circular intermediate. Confirmation of nonintegrated high MW episomes was carried out by Reed Clark and Phil Johnson in an elegant but simple analysis of genomic versus nongenomic DNA using DNA nucleases and restriction enzymes. Although the research community was slowly uncoupling the difference between in vitro and in vivo experiments, these findings that rAAVs could be targeted to specific cell types opened a door into viral tropism modification.
The lab continued this focus, first examining regulatory sequences within the ITRs that may determine tissue-specific expression. It was shown that the terminal repeat A/D junction element had promoter activity of its own that may interfere with all regulated expression cassettes.36 This work built on previous studies that explored the use of tissue-specific promoters or enhancer elements to optimize or target rAAV therapies by promoter modifications. Here enters the advent of the self-complementary (sc)AAV vectors. As preclinical studies highlighted a need for increased speed of transgene expression and better dose–response studies, the scAAV eliminated the rate-limiting step of second-strand synthesis.37,38 The scAAV vectors pioneered by Doug McCarty were shown to be up to 140% more effective transducing agents compared with conventional rAAV. These novel “double-stranded” vectors worked in every serotype tested and every tissue analyzed, demonstrating a universal principal of AAV biology with respect to single- versus double-stranded vector DNA genomes. These reagents have now been validated in clinical studies for a number of indications, including hemophilia, demonstrating successful expression of factor IX now out 3 years in hemophilic patients (St. Jude phase I).
One significant drawback was packaging capacity. About half of the ∼5 kb capacity of rAAV vectors is available for transgene substitution. With this caveat, selection of genetic disorders based on transgene size suggested a good place to start when considering AAV vectors. With more than 70% of all cDNAs averaging 5 kb and smaller, this limitation appears suitable for a variety of genes. In an effort to maximize space, the lab carried out a number of promoter studies to find smaller promoters for use in scAAV that would allow for similar gene expression and replaced the traditional 800 bp CMV promoter with a 229 bp fragment of the mouse methyl-CpG-binding protein-2 (MeCP2), leaving extra space for transgene packaging.39
Looking to characterize tissue tropism more, a cross-packaging assay of different serotypes containing the same ITR from AAV2 (hybrid vectors) with the genome packaged in capsids from serotypes 1–5 was evaluated by Joseph Rabinowitz in the lab. Here it was identified that rAAV1 was optimal for transduction of muscle, while serotypes 5 and 4 were most efficient at transduction of the retina with AAV4 only transducing the retinal pigment epithelium.40 Many serotype comparison studies have been done since and provided much needed starting points for optimal capsid designs. With the advent of “hybrid AAV vectors,” an onslaught of studies determining AAV serotype tropism ensued. In the Samulski lab, these studies led to the hypothesis that one could generate chimeric AAV capsids that may process novel tropisms not yet described. This was first described by Joe Rabinowitz, a post doc in the lab, with mixing experiments where different AAV serotype helper plasmids were mixed at varying ratios during vector production (25:75, 50:50, and 75:25 ratios of AAV1 and 2, respectively) and then chimeric AAV capsids were evaluated for transduction tropism.
On the basis of these observations, Dawn Bowles (another post doc) validated that this approach could be achieved using only small fragment contribution of donor parental AAV serotype capsid sequences. One novel variant, a capsid chimera between AAV2 and 3, demonstrated similar binding to the heparin sulfate surface receptor, but demonstrated a useful technique for mapping capsid sequences required for attachment or other functions that could alter tropism.41 Development of more chimeric capsid rAAVs resulted in the ability to change receptor tropism and even virus trafficking.42 Importantly, these works led to the generation of chimeric capsid libraries that could be screened. This “directed evolution” technique carried out by Wupin Li in the lab allowed for generation of rAAV with increased tropism for human airway epithelium eventually extended to in vivo selection by post doc Steve Gray and long-time collaborator Thomas McCown that resulted in new rAAV vectors that cross the seizure-compromised blood–brain barrier.43,44 These studies led to the isolation of the first AAV capsid that has a unique dominant tropism for “oligodendrocytes” and strongly suggest that AAV capsids with selected tropism are limited only by the selection procedure.
Lastly, an area that is not as explored includes cellular trafficking of rAAV and its effect on tropism and transduction success. Early studies examined the entry pathway, finding rapid internalization by receptor-mediated endocytosis from clathrin-coated pits and a rate-limiting step of nuclear entry.45 Almost 10 years later, confocal microscopy was used to track the subcellular fate of the virus and determined that the microtubule network facilitated rAAV2 movement around the cell.46 It was also reported that a population of rAAV2 enters the nucleus, while empty capsids are excluded. The use of proteasome inhibitors and siRNA against nucleolar protein B23 caused a synergistic increase in transduction more than 50-fold. This work confirmed that rAAV enters the nucleus intact and that entry can be enhanced through inhibition of multiple pathways.47 Additionally, mutation to basic amino acids of rAAV2 capsid VP1 protein resulted in reduced efficacy in transformed cells, but indistinguishable infection to rAAV2 in several nontransformed cell lines and tissue in vivo.48 The outcome of these investigations yielded a new perspective on entry pathways for “in vitro versus in vivo” settings. Additional AAV trafficking studies determined that rAAV2 interacts with importin-β alone or in complex with other karyopherins to facilitate nuclear entry,49 a critical step required for viral and vector infectivity.
Optimizing AAV Production
As study of AAV biology was continued during the 1990s, production of high titers of purified rAAV for therapeutic application required additional research. As mentioned above, preliminary studies determined that second-strand synthesis of the AAV ssDNA genome was the rate-limiting step for efficient transduction by rAAVs.50 Unfortunately, it would be 5 years until the issue was fully addressed with the generation of scAAV vectors in 2001 by Doug McCarty in the lab.37,38 Importantly though, the Ferrari article was the first to demonstrate production of rAAV in a system free from infectious Ad. Transfection with a helper plasmid lacking required Ad replication sequences allowed for expression of Ad genes needed for production of rAAV particles.50 Additional progress toward a production protocol suited for GMP was achieved when Xiao discovery that reduced AAV Rep78/68 expression led to substantially higher yields of rAAV.51 However, most significant for understanding vector production was the advent of the pDD-2 plasmid.52 Focusing on the required components of virus replication, two trans-acting Rep and Cap genes and cis-acting ITRs, a plasmid was generated to contain a novel modified ITR of 165 bp containing flanking D and D′ sequences. The DD ITR is an intermediate structure generated during viral DNA replication. Having this substrate available for the Rep protein nicking activity immediately following transfection facilitated their resolution into linear replication substrates.52 This plasmid intermediate provided direct evidence that resolution of the ITR could be carried out in absence of Rep through a “Holiday” structure resulting in linear fragments with closed “no end” substrates now suitable for replication. Further, Candace Summerford discovered the first receptor used by AAV; namely, that AAV2 host cell receptor was a membrane-associated heparin sulfate proteoglycan. This was also demonstrated to be useful for purification of rAAV particles.53,54
Work by Aravind Asokan, then a post doc, demonstrated that an additional AAV2 receptor was utilized for productive infection; specifically integrin α5β155 adding to growing base knowledge of viral entry. Numerous labs have added to our understanding of AAV receptors, providing a rich resource of information currently being studied with generation of “targeted vectors.” Production development was being carried out in parallel and the first production method completely free of Ad helper virus was demonstrated.56 This method eliminated the need for development of replication-competent Ad assays and provided a more defined AAV reagent for clinical application.56 Additionally, movement away from potentially toxic reagents of CsCl for purification led to the ionic iodixanol gradients followed by ion exchange chromatography developed by colleagues in Florida.57 This method resulted in a 50% increase in recovery from crude lysate and a vector that is 99% pure and is now being used for large-scale production.57 With an optimized vector production method, viral titers of 1012/liter were achieved, whereas previously rescued virus preparation provided titers of 107–8.51 Importantly, vector production had reached yields where quantity and purity were feasible for clinical application and various protocols have now been published supporting these original efforts.58 Production improvements continue with the recent development of the UNC Pro10 suspension cell line by Josh Grieger that is free of all animal and human products.
Translating Preclinical Research to Clinical Trials
Preclinical studies with AAV have led to many clinical trials, reviewed in detail.59–63 The Samulski lab has been a part of a handful of these trials, described below. However, the significant contributions of others toward clinical application of AAV must also be highlighted. On the basis of the initial discovery of AAV2 and the amount of AAV2-specific data, this was the first serotype to be used against numerous diseases, including hemophilia B, cystic fibrosis, Parkinson's, Batten's, Canavan's, rheumatoid arthritis, Leber's congenital amaurosis, and age-related macular degeneration. With the advent of generating “hybrid AAV” vectors, which utilized various AAV serotypes and viral engineering techniques, AAV2 was joined in the clinic by additional serotype vectors. First, to address issues with tropism, serotypes with inherent tissue-specific tropism are used. AAV1, first described by former student Weidong Xiao, with its enhanced muscle tropism compared with AAV2, is being explored for muscular gene delivery to treat α-1 antitrypsin deficiency, lipoprotein lipase deficiency, Pompe's disease, limb girdle muscular dystrophy, and cardiac failure. AAV6 and 9 also demonstrate increased musculoskeletal tropism as well as cardiac tropism. AAV5 and 9 may best facilitate gene transfer to the CNS. Further, AAV4 and 8 are being explored for clinical use in the eye, with AAV8 being utilized for successful factor IX phase I studies. Each serotype with its unique biodistribution provides a potential method for targeted delivery and enhanced efficacy in this first-generation AAV approach and more natural serotypes were being isolated and described by Guangping Gao in Jim Wilson's group.
However, with the ability to clone and produce AAV2 came the ability to engineer the next generation of clinical-grade rAAVs that were rationally designed to address issues with clinical application such as toxicity, evasion of immune response, de-targeting, and enhanced tissue tropism. Shown by Zhong, Srivastava, and others, AAV capsid proteins with tyrosine-to-phenylalanine substitutions have shown enhanced transduction with reduced toxicity.64 The volume of AAV structural data generated almost exclusively by Mavis Agbandje-McKenna's lab has set the backdrop to carry out rational capsid mutagenesis. With exciting articles from David Schaffer and Dirk Grimm in Mark Kay's lab, as well as others, the portfolio of AAV capsid options exploded. Engineering of hybrid capsids also facilitates reduced toxicity and the ability to home tissue tropism. In general, vectors that allow for enhanced gene delivery and expression can be used in reduced concentrations. One such example engineered in the Samulski lab resulted in AAV2.5, where the muscle tropism determinants of AAV1 are combined with parental AAV2. This hybrid vector also allows for immune evasion of neutralizing antibodies to AAV1 and 2.65 This led to the first and only clinical trial using a chimeric AAV capsid for a DMD phase I study. Currently, chimeric AAV capsids are lining up to enter clinical testing now that extensive preclinical data have been generated. Additional hybrids have emerged: (1) AAV6.2 that may prove beneficial for intravenous delivery,66 (2) AAV2i8 designed by Aravind Asokan in the lab that demonstrates attenuate liver sequestration,67 and (3) AAVrh32.33 that induces an impaired T-cell response in the clinic.68,69
Beyond these initial hybrid vectors, chimeric capsid proteins are a product of sequence or peptide libraries first generated by Muller et al.70 Use of these libraries allows for engineering and selection of rAAVs that are resistant to neutralizing antibodies, have enhanced/targeted tissue tropism, or can be used at reduced concentrations to produce similar outcomes to parental vectors. Rational design also led to scAAV. As mentioned previously, compared with ssAAV, these vectors have the ability to produce more rapid gene expression, but are limited to smaller genes or those that have been optimized for the smaller vector genome size of 2.5 kb. What these collaborative works mean for clinical success is that we are now armed with limitless options to discover and provide disease-specific rAAV.
Regarding the clinical trials involving the Samulski lab and the UNC Gene Therapy Center, preclinical studies in rodent and primate systems had demonstrated safety of AAV therapy in the CNS, and the first clinical trial using rAAV2 (Fig. 1)71 for gene therapy in the human CNS was produced at UNC vector core and conducted by Dr. Leone and colleagues.72
Canavan's disease, a rare leukodystrophy with no treatment options, is caused by mutation to the aspartoacylase (ASPA) gene and prevents normal myelination as a result of increased levels of N-acetylaspartate (NAA). The trial enlisted pediatric patients naïve to previous nonviral gene transfer and administered AAV2-ASPA via neurosurgical administration. Neutralizing antibodies were not found in 7/10 patients and in all patients minimal signs of inflammation or immune stimulation were noted. Long-term monitoring of patients was conducted to determine efficacy.73 Significantly, no adverse effects from vector administration were observed. Moreover, in patients treated with rAAV2-ASPA, there were decreased levels of NAA and slowed progression of brain atrophy, improved seizure frequency, and stabilized clinical status.73 This clinical study benefited from the concomitant development of vector design and production, both of which initiated a hand-in-hand approach that would be adopted for future clinical efforts. More importantly, from these early clinical studies, it has now been determined that targeting gene transfer to oligos is the ideal target, and phase II studies utilizing novel oligo tropic AAV capsid for targeted delivery are being contemplated and should demonstrate significant improvement based on preclinical data. A single theme emerging is that identifying the correct AAV capsid for a genetic disorder (e.g., muscle versus liver tropic) will provide the best opportunity to obtain meaningful clinical results.
Focusing next on hemophilia, in a preclinical canine model carried out by Paul Monahan in the lab, i.m. delivery of rAAV encoding hFIX under control of a CMV promoter was well tolerated and resulted in transient reduction in whole blood clotting time.74 Antibodies were generated against the hFIX, and expression continued at least 10 weeks after vector administration. This first large animal study suggested safety following i.m. delivery and encouraged development of gene therapy for human diseases. Additional large animal research with rAAV and nonhuman primates demonstrated successful transduction without toxicity.75
Follow-up work to the canine hFIX study began examining the effect that viral capsid modification could have on murine muscle cell transduction and human or canine FIX secretion.76 This work highlighted the use of alternative serotypes for tissue-specific treatments, demonstrating that rAAV2, while best for in vitro transduction, was not optimal in vivo compared with rAAVs 1, 3, or 5.76 In fact, this led to work where rAAV1 encoding FIX treatment of hemophilia B mice causes a sustained and complete phenotype correction.77 Work by other labs in a phase I/II dose escalation clinical trial from 2001 to 2004 determined that the therapy was safe, but that transient FIX expression caused by immune clearance of transduced cells prevented a permanent solution.78 Follow-up work examined the prevalence of NAbs in 62 children with hemophilia A. This study demonstrated that prior exposure to rAAV2 produced NAbs with partial cross reactivity for rAAV5 and 8 and may guide rational design of future trials.79 The information gathered from the previous trial and patient samples, coupled with the advent of scAAV, led to an ongoing phase I/II clinical using scAAV8FIX led by Amit Nathwani and scAAV8.FIXR338L trial lead by Paul Monahan.80 The R338L variant of FIX allows for increased specific activity and can achieve therapeutic success at lower concentrations than wt FIX. The study by Nathwani supports long-term correction (now more than 3 years). This novel UNC vector is in phase 1 studies and preliminary results are supportive of the mouse studies. The UNC FIX experience strongly supports the value of the “bench to bedside, back to bench, etc.” approach, and the hope is that this novel vector should yield a commercial product within the next 5–7 years.
As briefly described above, building on the work of the capsid modification for enhanced efficacy, the first clinical trial using a chimeric rAAV2.5 (a chimera of AAV2 with five mutations from AAV1) that combines improved muscle transduction with reduced antigenic cross reactivity against both serotypes was carried out.65 DMD is caused by a mutation to the dystrophin gene and was predicted to be correctable with gene therapy. rAAV2.5-mediated delivery of a novel mini-dystrophin gene developed by Xiao was shown to improve the lifespan of dystrophin-deficient mice without significant toxicity.81,82 In the trial, vector injection was done via the bicep muscle in one arm and a saline control or empty vector in the contralateral arm. Injection of empty vector to patients was meant to allow for distinguished immune response between the rAAV or the minidystrophin transgene. Significantly, no immune response to the rAAV2.5 capsid was detected and the safety of the vector was established.65,83 Long-term monitoring is ongoing with new phase II studies looking at limb infusion with long-term objective of rescuing upper limb mobility followed by systemic whole-body gene transfer in DMD patients. Additional chimeric capsids are being developed to assist these clinical efforts and optimization is high.
More recently, work with giant axonal neuropathy (GAN) is moving forward toward early clinical testing. Caused by loss of function of the gigaxonin protein, GAN is characterized by aggregation of intermediate filaments (IF) that results in fatal neuropathy. This was the first study to demonstrate that re-introduction of the GAN gene to deficient cells would restore cytoskeleton IF homeostasis. Preliminary testing was conducted on GAN patient primary skin fibroblast cultures. Treatment with rAAV2 encoding the human GAN transgene caused significant reduction in the number of cells displaying IF aggregation.84 Moving in vivo, GAN mice that received intracisternal injection of rAAV9 encoding a CMV promoter and the GAN gene resulted in global deliver to the CNS. By 3 weeks postinjection, treated animals showed a nearly complete clearance of IF accumulation.84 To address concerns of GAN overexpression-related toxicity, a weak synthetic JeT promoter will be tested against CMV and to increase transduction efficiency an scAAV has been generated. This phase 1 study was recently approved and is recruiting patients for initiation of clinical studies February this year. While numerous AAV capsids and clinical indications are marching into the clinic, the UNC vector core generated GMP vector for the first-ever gene transfer study in Australia for wet age-related macular degeneration. This clinical focus was related to ocular gene transfer for macular degeneration using the original AAV2 vector generated in 1982 in the lab of Nick Muzyczka at University of Florida and still appears to be a suitable workhorse for some clinical indications.
Ongoing Challenges and Future Application
With the many successes of AAV gene therapy in the last 50 years, key discoveries about virus biology, improved methods of vector production, and positive effects seen in long-term clinical trials, it remains clear that there is always room for improvement. Ongoing challenges to evade neutralizing antibody responses to treatment are still being met with novel capsid generation as well as improved vector delivery. A lot of continued work is examining the immune clearance of transduced cells and has noted that rAAV capsid antigen presentation is dependent on endosomal escape.85 Further, single amino acid changes in the capsid proteins result in vectors with less immunogenicity and improved transduction.86 Limitations to vector packaging size also continue to be explored and may soon result in generation of vectors with room for larger transgene inserts.87 Finally, in preclinical exploration and clinical testing, rAAV-mediated therapies have only been applied to the tip of the iceberg regarding the more than 7,000 rare diseases that remain a scourge. Significant advances have been made in retinal diseases and approximately 40% of ongoing clinical trials are focused on ocular-related disease. One certainty with all of this ongoing research is that the future of AAV, on the cusp of clinical approval in the United States, is bright and will have a lasting impact on the many individuals amendable to AAV-mediated gene therapy.
Acknowledgments
These studies would not have been possible without the work of numerous talented lab members, the majority listed below: Lynn Hunter, Xiao Xiao, Xiaodong Zhu, Fabienne Rolling, Weidong Xiao, Doug McCarty, Jeffrey Barlett, David Ansardi, Richard Snyder, Patrick Hearing, Forrest Ferrari, Candace Summerford, Charles Yang, Gabriele, Kroner-Lux, Chris Walsh, Joseph Rabinowitz, Sam Young, Jackie Stilwell, Denise Gavin, Anna Skulimowski, Rebecca Haberman, Pamela Carter, Terry Amiss, Chengwen Li, Dawn Bowles, Vivian Choi, Nathan Allen, Zhijian Wu, Steve Snowdy, Josh Grieger, Hongzhe Dong, Xiaohuai Zhou, Matthew Hirsch, Steve Gray, Long-Sheng Chang, Stacey Foti, Curtis Hewitt, Wuping Li, David Dismuke, Aravind Asokan, Jarrod Johnson, Nina DiPrimio, Pingjie Xiao, Angela Mitchell, Sara Nicolson, Thomas Lentz, Max Salganik, Marc Weinberg, Jayme Warischalk, Katherine Liang, Kenton Woodard, Scott McPhee, Liliane Tenenbaum, Eugene Redmond, Thomas McCown, Paul Monahan, Jude Samulski, Thaddeus V. Samulski, and especially Ken Berns, Arun Srivastava, and Nicholas Muzyczka for more than 30 years of rewarding AAV collaboration. We would like to acknowledge funding support for R.J.S from University ODC (MPSI-13-010-01) and NIH (R01 AI080726, R01 DK084033, P01 HL112761, R01 AI072176, R01 AR064369).
Author Disclosure Statement
R.J.S. holds patents related to AAV vector technology and is a founder of companies that are developing commercial applications for AAV gene therapy.
References
- 1.Atchison RW, Casto BC, Hammon WM. Adenovirus-associated defective virus particles. Science 1965;149:754–756 [DOI] [PubMed] [Google Scholar]
- 2.Hoggan MD, Blacklow NR, Rowe WP. Studies of small DNA viruses found in various adenovirus preparations: Physical, biological, and immunological characteristics. Proc Natl Acad Sci USA 1966;55:1467–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hermonat PL, Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: Transduction of neomycin resistance into mammalian tissue culture cells. Proc Natl Acad Sci USA 1984;81:6466–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rose JA, Berns KI, Hoggan MD, et al. Evidence for a single-stranded adenovirus-associated virus genome: Formation of a DNA density hybrid on release of viral DNA. Proc Natl Acad Sci USA 1969;64:863–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crawford LV, Follett EA, Burdon MG, et al. The DNA of a minute virus of mice. J Gen Virol 1969;4:37–46 [DOI] [PubMed] [Google Scholar]
- 6.Carter BJ, Khoury G, Denhardt DT. Physical map and strand polarity of specific fragments of adenovirus-associated virus DNA produced by endonuclease r-ecori. J Virol 1975;16:559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter BJ, Khoury G, Rose JA. Adenovirus-associated virus multiplication. Ix. Extent of transcription of the viral genome in vivo. J Virol 1972;10:1118–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koczot FJ, Carter BJ, Garon CF, et al. Self-complementarity of terminal sequences within plus or minus strands of adenovirus-associated virus DNA. Proc Natl Acad Sci USA 1973;70:215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Straus SE, Sebring ED, Rose JA. Concatemers of alternating plus and minus strands are intermediates in adenovirus-associated virus DNA synthesis. Proc Natl Acad Sci USA 1976;73:742–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lusby E, Fife KH, Berns KI. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. J Virol 1980;34:402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung AK, Hoggan MD, Hauswirth WW, et al. Integration of the adeno-associated virus genome into cellular DNA in latently infected human detroit 6 cells. J Virol 1980;33:739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myers MW, Carter BJ. Assembly of adeno-associated virus. Virology 1980;102:71–82 [DOI] [PubMed] [Google Scholar]
- 13.Laughlin CA, Westphal H, Carter BJ. Spliced adenovirus-associated virus rna. Proc Natl Acad Sci USA 1979;76:5567–5571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcus CJ, Laughlin CA, Carter BJ. Adeno-associated virus rna transcription in vivo. Eur J Biochem 1981;121:147–154 [DOI] [PubMed] [Google Scholar]
- 15.Jay FT, Laughlin CA, Carter BJ. Eukaryotic translational control: Adeno-associated virus protein synthesis is affected by a mutation in the adenovirus DNA-binding protein. Proc Natl Acad Sci USA 1981;78:2927–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter BJ. Adeno-associated virus and the development of adeno-associated virus vectors: A historical perspective. Mol Ther 2004;10:981–989 [DOI] [PubMed] [Google Scholar]
- 17.Samulski RJ, Berns KI, Tan M, et al. Cloning of adeno-associated virus into pbr322: Rescue of intact virus from the recombinant plasmid in human cells. Proc Natl Acad Sci USA 1982;79:2077–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laughlin CA, Tratschin JD, Coon H, et al. Cloning of infectious adeno-associated virus genomes in bacterial plasmids. Gene 1983;23:65–73 [DOI] [PubMed] [Google Scholar]
- 19.Samulski RJ, Srivastava A, Berns KI, et al. Rescue of adeno-associated virus from recombinant plasmids: Gene correction within the terminal repeats of AAV. Cell 1983;33:135–143 [DOI] [PubMed] [Google Scholar]
- 20.Tratschin JD, Miller IL, Carter BJ. Genetic analysis of adeno-associated virus: Properties of deletion mutants constructed in vitro and evidence for an adeno-associated virus replication function. J Virol 1984;51:611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermonat PL. The first adeno-associated virus gene transfer experiment, 1983. Hum Gene Ther 2014;25:486–487 [DOI] [PubMed] [Google Scholar]
- 22.Hearing P, Samulski RJ, Wishart WL, et al. Identification of a repeated sequence element required for efficient encapsidation of the adenovirus type 5 chromosome. J Virol 1987;61:2555–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samulski RJ, Chang LS, Shenk T. A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. J Virol 1987;61:3096–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samulski RJ, Chang LS, Shenk T. Helper-free stocks of recombinant adeno-associated viruses: Normal integration does not require viral gene expression. J Virol 1989;63:3822–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotin RM, Siniscalco M, Samulski RJ, et al. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci USA 1990;87:2211–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samulski RJ, Zhu X, Xiao X, et al. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J 1991;10:3941–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol 1996;70:8098–8108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walsh CE, Liu JM, Xiao X, et al. Regulated high level expression of a human gamma-globin gene introduced into erythroid cells by an adeno-associated virus vector. Proc Natl Acad Sci USA 1992;89:7257–7261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller JL, Walsh CE, Ney PA, et al. Single-copy transduction and expression of human gamma-globin in k562 erythroleukemia cells using recombinant adeno-associated virus vectors: The effect of mutations in nf-e2 and gata-1 binding motifs within the hypersensitivity site 2 enhancer. Blood 1993;82:1900–1906 [PubMed] [Google Scholar]
- 30.Flotte TR, Afione SA, Conrad C, et al. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc Natl Acad Sci USA 1993;90:10613–10617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaplitt MG, Leone P, Samulski RJ, et al. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet 1994;8:148–154 [DOI] [PubMed] [Google Scholar]
- 32.Kaplitt MG, Xiao X, Samulski RJ, et al. Long-term gene transfer in porcine myocardium after coronary infusion of an adeno-associated virus vector. Ann Thorac Surg 1996;62:1669–1676 [DOI] [PubMed] [Google Scholar]
- 33.Mccown TJ, Xiao X, Li J, et al. Differential and persistent expression patterns of cns gene transfer by an adeno-associated virus (AAV) vector. Brain Res 1996;713:99–107 [DOI] [PubMed] [Google Scholar]
- 34.Bartlett JS, Samulski RJ. Fluorescent viral vectors: A new technique for the pharmacological analysis of gene therapy. Nat Med 1998;4:635–637 [DOI] [PubMed] [Google Scholar]
- 35.Bartlett JS, Samulski RJ, Mccown TJ. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther 1998;9:1181–1186 [DOI] [PubMed] [Google Scholar]
- 36.Haberman RP, Mccown TJ, Samulski RJ. Novel transcriptional regulatory signals in the adeno-associated virus terminal repeat a/d junction element. J Virol 2000;74:8732–8739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mccarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther 2001;8:1248–1254 [DOI] [PubMed] [Google Scholar]
- 38.Mccarty DM, Fu H, Monahan PE, et al. Adeno-associated virus terminal repeat (tr) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther 2003;10:2112–2118 [DOI] [PubMed] [Google Scholar]
- 39.Gray SJ, Foti SB, Schwartz JW, et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther 2011;22:1143–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rabinowitz JE, Rolling F, Li C, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol 2002;76:791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bowles DE, Rabinowitz JE, Samulski RJ. Marker rescue of adeno-associated virus (AAV) capsid mutants: A novel approach for chimeric AAV production. J Virol 2003;77:423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rabinowitz JE, Bowles DE, Faust SM, et al. Cross-dressing the virion: The transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J Virol 2004;78:4421–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Zhang L, Johnson JS, et al. Generation of novel AAV variants by directed evolution for improved cftr delivery to human ciliated airway epithelium. Mol Ther 2009;17:2067–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray SJ, Blake BL, Criswell HE, et al. Directed evolution of a novel adeno-associated virus (AAV) vector that crosses the seizure-compromised blood-brain barrier (bbb). Mol Ther 2010;18:570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartlett JS, Wilcher R, Samulski RJ. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J Virol 2000;74:2777–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao PJ, Samulski RJ. Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. J Virol 2012;86:10462–10473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson JS, Samulski RJ. Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. J Virol 2009;83:2632–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson JS, Li C, Diprimio N, et al. Mutagenesis of adeno-associated virus type 2 capsid protein vp1 uncovers new roles for basic amino acids in trafficking and cell-specific transduction. J Virol 2010;84:8888–8902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicolson SC, Samulski RJ. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J Virol 2014;88:4132–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferrari FK, Samulski T, Shenk T, et al. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol 1996;70:3227–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J, Samulski RJ, Xiao X. Role for highly regulated rep gene expression in adeno-associated virus vector production. J Virol 1997;71:5236–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao X, Xiao W, Li J, et al. A novel 165-base-pair terminal repeat sequence is the sole cis requirement for the adeno-associated virus life cycle. J Virol 1997;71:941–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol 1998;72:1438–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Summerford C, Samulski RJ. Viral receptors and vector purification: New approaches for generating clinical-grade reagents. Nat Med 1999;5:587–588 [DOI] [PubMed] [Google Scholar]
- 55.Asokan A, Hamra JB, Govindasamy L, et al. Adeno-associated virus type 2 contains an integrin alpha5beta1 binding domain essential for viral cell entry. J Virol 2006;80:8961–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol 1998;72:2224–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zolotukhin S, Byrne BJ, Mason E, et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 1999;6:973–985 [DOI] [PubMed] [Google Scholar]
- 58.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc 2006;1:1412–1428 [DOI] [PubMed] [Google Scholar]
- 59.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat Rev Genet 2011;12:341–355 [DOI] [PubMed] [Google Scholar]
- 60.Agbandje-Mckenna M, Kleinschmidt J. AAV capsid structure and cell interactions. Methods Mol Biol 2011;807:47–92 [DOI] [PubMed] [Google Scholar]
- 61.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: Poised at the clinical crossroads. Mol Ther 2012;20:699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Asokan A, Samulski RJ. An emerging adeno-associated viral vector pipeline for cardiac gene therapy. Hum Gene Ther 2013;24:906–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mueller C, Flotte TR. Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Ther 2008;15:858–863 [DOI] [PubMed] [Google Scholar]
- 64.Zhong L, Li B, Mah CS, et al. Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA 2008;105:7827–7832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bowles DE, Mcphee SW, Li C, et al. Phase 1 gene therapy for duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther 2012;20:443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vandenberghe LH, Breous E, Nam HJ, et al. Naturally occurring singleton residues in AAV capsid impact vector performance and illustrate structural constraints. Gene Ther 2009;16:1416–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asokan A, Conway JC, Phillips JL, et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol 2010;28:79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mays LE, Vandenberghe LH, Xiao R, et al. Adeno-associated virus capsid structure drives cd4-dependent cd8+ t cell response to vector encoded proteins. J Immunol 2009;182:6051–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin SW, Hensley SE, Tatsis N, et al. Recombinant adeno-associated virus vectors induce functionally impaired transgene product-specific cd8+ t cells in mice. J Clin Invest 2007;117:3958–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muller OJ, Kaul F, Weitzman MD, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol 2003;21:1040–1046 [DOI] [PubMed] [Google Scholar]
- 71.Xie Q, Bu W, Bhatia S, et al. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci USA 2002;99:10405–10410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mcphee SW, Janson CG, Li C, et al. Immune responses to AAV in a phase I study for Canavan disease. J Gene Med 2006;8:577–588 [DOI] [PubMed] [Google Scholar]
- 73.Leone P, Shera D, Mcphee SW, et al. Long-term follow-up after gene therapy for Canavan disease. Sci Transl Med 2012;4:165ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Monahan PE, Samulski RJ, Tazelaar J, et al. Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophilia. Gene Ther 1998;5:40–49 [DOI] [PubMed] [Google Scholar]
- 75.During MJ, Samulski RJ, Elsworth JD, et al. In vivo expression of therapeutic human genes for dopamine production in the caudates of mptp-treated monkeys using an AAV vector. Gene Ther 1998;5:820–827 [DOI] [PubMed] [Google Scholar]
- 76.Chao H, Liu Y, Rabinowitz J, et al. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol Ther 2000;2:619–623 [DOI] [PubMed] [Google Scholar]
- 77.Chao H, Monahan PE, Liu Y, et al. Sustained and complete phenotype correction of hemophilia b mice following intramuscular injection of AAV1 serotype vectors. Mol Ther 2001;4:217–222 [DOI] [PubMed] [Google Scholar]
- 78.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-factor ix and limitations imposed by the host immune response. Nat Med 2006;12:342–347 [DOI] [PubMed] [Google Scholar]
- 79.Li C, Narkbunnam N, Samulski RJ, et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther 2012;19:288–294 [DOI] [PubMed] [Google Scholar]
- 80.Monahan PE, Sun J, Gui T, et al. Employing a gain-of-function factor IX variant r338l to advance the efficacy and safety of hemophilia B human gene therapy: Preclinical evaluation supporting an ongoing AAV clinical trial. Hum Gene Ther 2015;26:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA 2000;97:13714–13719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang B, Li J, Fu FH, et al. Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J Orthop Res 2009;27:421–426 [DOI] [PubMed] [Google Scholar]
- 83.Mendell JR, Campbell K, Rodino-Klapac L, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med 2010;363:1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mussche S, Devreese B, Nagabhushan Kalburgi S, et al. Restoration of cytoskeleton homeostasis after gigaxonin gene transfer for giant axonal neuropathy. Hum Gene Ther 2013;24:209–219 [DOI] [PubMed] [Google Scholar]
- 85.Li C, He Y, Nicolson S, et al. Adeno-associated virus capsid antigen presentation is dependent on endosomal escape. J Clin Invest 2013;123:1390–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li C, Diprimio N, Bowles DE, et al. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol 2012;86:7752–7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hirsch ML, Storici F, Li C, et al. AAV recombineering with single strand oligonucleotides. PLoS One 2009;4:e7705. [DOI] [PMC free article] [PubMed] [Google Scholar]