

Abstract
Pontocerebellar hypoplasia consists of a rare heterogeneous group of congenital neurodevelopmental disorders characterized by hypoplasia and atrophy of the cerebellar cortex, dentate and pontine nuclei, and inferior olives. The very low density lipoprotein receptor protein is an integral part of the reelin signaling pathway, which guides neuroblast migration in the cerebral cortex and cerebellum. Mutations in this receptor cause nonprogressive cerebellar ataxia, mental retardation, and cerebellar hypoplasia. In this report, we present 3 patients from 2 different families displaying very low density lipoprotein receptor–associated pontocerebellar hypoplasia, cortical dysplasia, mental retardation, and bipedal gait. One of the siblings has also displayed dysmorphic features, as we previously reported before the identification of the genetic defect in this family.
Keywords: pontocerebellar hypoplasia, VLDLR mutation, dysmorphic features, reelin
Very low density lipoprotein receptor (VLDLR)–associated pontocerebellar hypoplasia (a subgroup of dysequilibrium syndrome) is a rare, autosomal recessive, nonprogressive cerebellar disorder characterized by nonprogressive cerebellar ataxia, delayed ambulation, and moderate to profound mental retardation.1–6 The cause of the disease is mutations in the VLDLR gene that encodes very low density lipoprotein receptor on chromosome 9p24, and it was first described in the Hutterite population by Boycott et al in 2005.1 Until now, several mutations were described in Iranian,3 Turkish,2,6,7 and white people.5
The gene consists of 19 exons and encodes an 873–amino acid receptor protein that belongs to the low-density lipoprotein receptor superfamily.8 In humans, 2 isoforms of the receptor have been described.9 Type I is mainly expressed in heart and skeletal muscles and takes place in active fatty acid metabolism and type II is predominant in nonmuscular tissue such as the kidney, spleen, adrenal gland, lung, uterus, testis, ovary, and brain.9,10
We present 3 patients from 2 different families displaying very low density lipoprotein receptor–associated pontocerebellar hypoplasia, cortical dysplasia, mental retardation, and bipedal gait. One of the siblings displayed dysmorphic features, as we previously reported11 before the identification of the genetic defect in this family.
Case Summary
Case 1
A 26-year-old woman, who was born to first-cousin parents, was first admitted with complaints of microcephaly and mental and motor retardation at 8 months of age. Head control was achieved at 7 months. At age 30 months, she was unable to sit and walk. At the age of 17 years, physical examination revealed a high arched palate, triangular-shaped face, thoracolumbar scoliosis, pectus carinatum, kyphosis, cubitus valgus, arachnodactyly, and long extremities (Figure 1). Neurodevelopmental evaluation revealed microcephaly (49.8 cm, −4 SD), bilateral strabismus and nystagmus, truncal ataxia, spasticity and hyperreflexia of the lower limbs, and poor cognitive development. She could walk without support and had a monotone voice with unintelligible speech. We reported this case, along with her sibling, as “pontocerebellar hypoplasia in 2 siblings with dysmorphic features.”11 She is now 26 years old, has similar neurologic findings, bipedal ataxic gait, and can only speak in single words.
Figure 1.

General appearance of patient 1.
Case 2
The sibling of the first patient was born as the fifth child of the same family following an uneventful gestation and delivery. He was first seen at 3 months of age with complaints similar to those of the first child. He had microcephaly (36 cm, −4 SD) and lack of head control and social smile. At the age of 30 months, he could sit but was unable to walk without support.
He also had bilateral strabismus, nystagmus, truncal ataxia, and hyperreflexia of the lower limbs. He had a monotone voice without speech. He is now 11 years old and can walk without support for short distances and speak in single words. Metabolic investigation of the patient, including blood and cerebrospinal fluid lactic and pyruvic acid, blood ammonia, serum and urinary amino acids, urine organic acids, lysosomal and peroxisomal screening tests, and transferrin pattern and karyotypes of the patients were normal. Electroencephalogram, brainstem auditory evoked responses, electromyogram, and nerve conductance studies showed no abnormalities.
Brain magnetic resonance imaging (MRI) of all patients was performed on a 1.5-T unit with the use of a head coil. The conventional MRI protocol included T2-weighted turbo spin-echo sequence (TR/TE, 8820/126; voxel size, 0.9 × 0.7 × 5.0 mm) in the axial and sagittal plane and fluid-attenuated inversion recovery sequence (TR/TE/TI, 8650/126/2500; voxel size, 1.0 × 0.9 × 5.0 mm) in the axial plane. Brain MR images of the children showed bilateral diffuse cerebral cortical thickening and decreased gyration in accordance with pachygyria and cerebellar and pontine hypoplasia (Figure 2A and B and Figure 3A and B).
Figure 2.
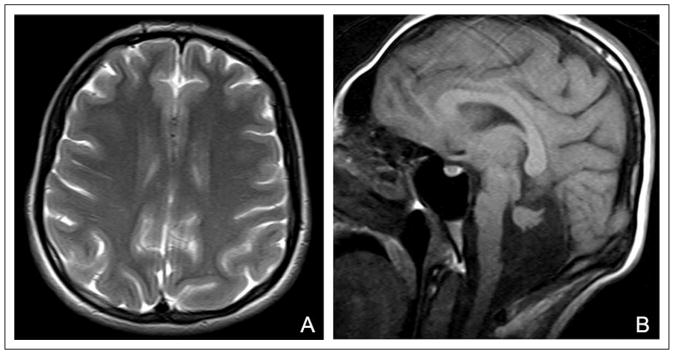
Cranial magnetic resonance imaging (MRI) appearance of patient 1. (A) Axial T2-weighted image shows diffuse pachygyria of the cerebrum and (B) sagittal T1-weighted image shows hypoplasia of the pons and cerebellum.
Figure 3.
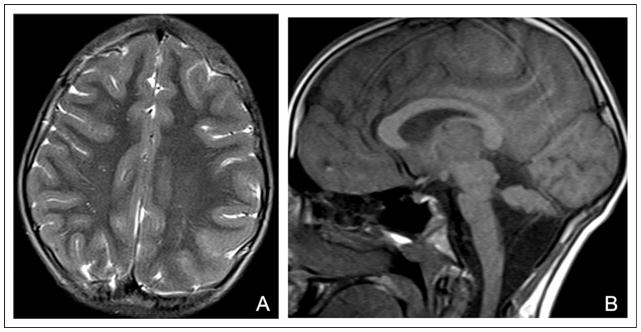
Axial T2-weighted magnetic resonance imaging (MRI) of patient 2 shows diffuse pachygyria of the cerebrum (A) and sagittal T1-weighted image shows hypoplasia of the pons and cerebellum (B).
Molecular genetic investigations, including linkage analysis using 5 K genome-wide SNP genotyping followed by linkage analysis, pointed to several potential linkage peaks. Whole exome sequencing analysis showed coverage of 94.7% of the targeted bases at >10X, from which 21,693 variants were identified. Bioinformatics analysis under the assumption of a rare homozygous deleterious allele occurring within one of the homozygous intervals identified 1 deletion and 6 nonsynonymous variants.
Further prioritization of the variants pointed to a homozygous 5-bp deletion within the open reading frame of the VLDLR gene on human chromosome 9. This c.1247_53delGTTACAA results in a p.G1246fsX1305 frame shift, followed by a premature stop codon in the protein.
Case 3
A 4-year-old male patient was born to first-degree parents. He was first seen at 10 months of age for mental and motor retardation. He had microcephaly and head control. He was able to sit without support at 17 months of age. At the present time, he is 4 years old and still has microcephaly (48 cm, −4 SD), strabismus, nystagmus, spasticity on the right arm, and intentional tremor. He can say some words unintentionally and walk with support. All of the laboratory investigations including lactic and pyruvic acid levels in blood, urine and blood amino acids, tandem mass spectrometry, urine organic acids, lysosomal and peroxisomal screenings, and transferrin focusing were normal. Cranial MRI showed bilateral diffuse cerebral cortical thickening and decreased gyration in accordance with pachygyria and cerebellar and pontine hypoplasia (Figure 4A and B).
Figure 4.
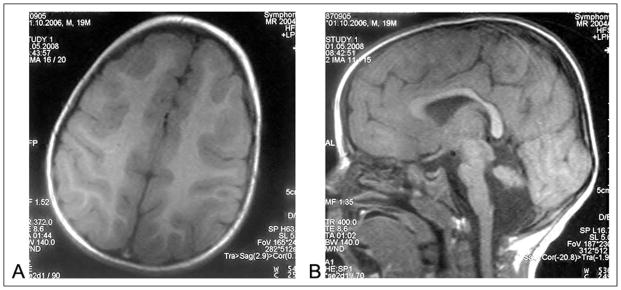
Bilateral diffuse cerebral cortical thickening and decreased gyration in accordance with pachygyria (A) and cerebellar and pontine hypoplasia (B) was described in axial and sagittal section magnetic resonance imaging (MRI) of patient 3.
Based on the imaging findings typical for the disease, genomic DNA was isolated and tested directly for a mutation in this gene. Molecular genetic investigation revealed a homozygous mutation in VLDLR exon 6 resulting in a c.C835T p.R279X homozygous stop codon.
Discussion
The VLDLR protein plays an important role in fatty acid metabolism; it is also an integral part of the reelin signaling pathway.12 Human reelin is a glycoprotein and is expressed in the fetal and postnatal brain as well as in the liver. Reelin regulates neuronal positioning in the cortical brain structures and the migration of neurons along the radial glial fiber network for proper positioning of neurons within the laminated nervous system parenchyma by binding to the lipoprotein receptors VLDLR and ApoER2 and the adapter protein disabled-1.13,14 These receptors are transmembrane coreceptors for reelin.15
The first human VLDLR mutation was observed in the Hutterite population.1 These patients had nonprogressive cerebellar ataxia, mental retardation, and cerebellar hypoplasia and a 199-kb homozygous deletion encompassing the entire gene.1 Since then, several different mutations affecting exons 5, 17,2 10,3 11, 12,5 and 1 to 57 were reported. We similarly encountered likely null mutations, suggesting a loss of function of the VLDLR as the disease mechanism.
Dysequilibrium syndrome secondary to mutations in VLDLR is the first human lipoprotein receptor malformation syndrome. The major diagnostic features of the disease are a nonprogressive congenital ataxia that is predominantly truncal and which results in delayed ambulation, moderate to profound mental retardation, and dysarthria. The characteristic MRI features include hypoplasia of the inferior portion of the cerebellar vermis and hemispheres, simplified gyration of the cerebral hemispheres with minimally thickened but uniform cortex, and a lack of clear anteroposterior gradients and small brain stem, particularly the pons. Strabismus, seizures, pes planus, and short stature are supportive features.4
We described different dysmorphic features from the literature in 1 of our patients including a high arched palate, triangular-shaped face, thoracolumbar scoliosis, pectus carinatus, kyphosis, cubitus valgus, arachnodactyly, and long extremities that were not previously reported in this disease.
All of our patients have a bipedal gait similar to the patient reported by Kolb et al7 and different from the other Turkish patients reported by Tan,16 Ozcelik et al,2 and Turkmen et al.6 When we asked the parents about their children’s quadripedal gait, they said if they did not provide any effort to support the children’s walking with physical therapy and orthopedics, their children would display a quadripedal gait, which they observed on occasion when the children were left unsupervised. Therefore, we concluded that the quadripedal gait is likely to be a physical adaptation: if these children did not receive adequate medical attention and support, they would probably display a quadripedal gait to compensate for their ataxia.
We suggest that the clinical and MRI findings of VLDLR mutations may show different features and in the future, the studies including large series can explain the cause of these differences.
Acknowledgments
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Author Contributions
The cases were diagnosed and followed by FMS; the DNA isolation of the patients was performed by FC; the molecular genetic analyses of the samples were done by JGG in Gleeson Lab, San Diego; and the radiologic evaluation was done by SK. The article was written by FMS.
These cases will be presented as, oral presentation in the 17th Mediterranean Child Neurology Congress, September 14–17 2011, Slovenia, Piran.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This work was deemed exempt from formal review by the institutional review board of Fatih University, Faculty of Medicine.
References
- 1.Boycott KM, Flavelle S, Bureau A, et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77:477–483. doi: 10.1086/444400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ozcelik T, Akarsu N, Uz E, et al. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadripedal locomoton in humans. Proc Natl Acad Sci U S A. 2008;105:4232–4236. doi: 10.1073/pnas.0710010105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moheb LA, Tzschach A, Garshasbi M, et al. Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an Iranian family with dysequilibrium syndrome. Eur J Hum Genet. 2008;16:270–273. doi: 10.1038/sj.ejhg.5201967. [DOI] [PubMed] [Google Scholar]
- 4.Boycott KM, Parboosingh JS. VLDLR-associated cerebellar hypoplasia. In: Pagon RA, Bird TD, Dalan CR, Stephens K, editors. Gene Reviews (Internet) Seattle, WA: University of Washington; 1993–2008. [Google Scholar]
- 5.Boycott KM, Bonnemann C, Herz J, et al. Mutations in VLDLR as a cause for autosomalrecessive cerebellar ataxia with metal retardation (dysequilibrium syndrome) J Child Neurol. 2009;24:1310–1315. doi: 10.1177/0883073809332696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turkmen S, Hoffmann K, Demirhan O, Aruoba D, Humphrey N, Mundlos S. Cerebellar hypoplasia, with quadripedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene. Eur J Hum Genet. 2008;16:1070–1074. doi: 10.1038/ejhg.2008.73. [DOI] [PubMed] [Google Scholar]
- 7.Kolb LE, Arlier Z, Yalcinkaya C, et al. Novel VLDLR micro-deletion identified in two Turkish siblings with pachygyria and pontocerebellar atrophy. Neurogenetics. 2010;11:319–325. doi: 10.1007/s10048-009-0232-y. [DOI] [PubMed] [Google Scholar]
- 8.Strickland DK, Kounnas MZ, Argraves WS. LDL receptor-related protein: a multiligand receptor for lipoprotein and proteinase catabolism. FASEB J. 1995;9:890–898. doi: 10.1096/fasebj.9.10.7615159. [DOI] [PubMed] [Google Scholar]
- 9.Sakai J, Hoshino A, Takahashi S, et al. Structure, chromosome location and expression of the human very low-density lipoprotein receptor gene. J Biol Chem. 1994;269:2173–2182. [PubMed] [Google Scholar]
- 10.Takahashi S, Suzuki J, Kohno M, et al. Enhancement of the binding of triglyceride-rich lipoproteins to the very low-density lipoprotein receptor by apolipoprotein E and lipoprotein lipase. J Biol Chem. 1995;270:15747–15754. doi: 10.1074/jbc.270.26.15747. [DOI] [PubMed] [Google Scholar]
- 11.Dilber E, Aynaci FM, Ahmetoglu A. Pontocerebellar hypoplasia in two siblings with dysmorphic features. J Child Neurol. 2002;17:64–66. doi: 10.1177/088307380201700119. [DOI] [PubMed] [Google Scholar]
- 12.Tissir F, Gofinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- 13.Hiesberger T, Trommsdorff M, Howell BW, et al. Direct binding of reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 14.Zaki M, Shehab M, El-Aleem A, et al. Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am J Med Genet. 2007;143A:939–944. doi: 10.1002/ajmg.a.31667. [DOI] [PubMed] [Google Scholar]
- 15.DeSilva U, D’Arcangelo G, Braden VV, et al. The human reelin gene: isolation, sequencing, and mapping on chromosome 7. Genome Res. 1997;7:157–164. doi: 10.1101/gr.7.2.157. [DOI] [PubMed] [Google Scholar]
- 16.Tan U. A new syndrome with quadripedal gait, primitive speech, and severe mental retardation as a live model for human evolution. Int J Neurosci. 2006;116:361–369. doi: 10.1080/00207450500455330. [DOI] [PubMed] [Google Scholar]