

Abstract
It is challenging to deliver molecules to the brain for diagnosis and treatment of brain diseases. This is primarily due to the presence of the blood-brain barrier (BBB), which restricts the entry of many molecules into the brain. In this study, cyclic ADT peptides (ADTC1, ADTC5, and ADTC6) have been shown to modify the BBB to enhance the delivery of marker molecules (e.g., 14C-mannitol, Gd-DTPA) to the brain via the paracellular pathways of the BBB. The hypothesis is that these peptides modulate cadherin interactions in the adherens junctions of the vascular endothelial cells forming the BBB to increase paracellular drug permeation. In vitro studies indicated that ADTC5 had the best profile to inhibit adherens junction resealing in MDCK cell monolayers in a concentration-dependent manner (IC50 = 0.3 mM) with a maximal response at 0.4 mM. Under the current experimental conditions, ADTC5 improved the delivery of 14C-mannitol to the brain about twofold compared to the negative control in the in situ rat brain perfusion model. Furthermore, ADTC5 peptide increased in vivo delivery of Gd-DTPA to the brain of Balb/c mice when administered intravenously (i.v.). In conclusion, ADTC5 has the potential to improve delivery of diagnostic and therapeutic agents to the brain.
The presence of the blood-brain barrier (BBB) creates a challenge in delivering drugs to the brain for treatment of neurological diseases such as brain tumors and Alzheimer’s and Parkinson’s diseases.1,2 The BBB is formed by endothelial cells lining the microvessels that transport molecules and metabolites in and out of the brain. Unfortunately, the BBB prevents many drug molecules from entering the brain from the systemic circulation. Although most small hydrophobic drugs can partition into cell membranes of the brain microvascular endothelial cells, the presence of efflux pumps can prevent them from crossing the BBB via transcellular pathways.3,4 For those agents entering into the brain via a transcellular route, the existence of highly expressed metabolizing enzymes (e.g., peptidases and cytochrome P450) at the BBB serves as a metabolic barrier to degrade drug molecules and prevent the intact drug molecules from entering the brain.5,6 For many hydrophilic solutes, a paracellular diffusion pathway is the more probable route.7,8 However, the formation of complex tight junctions between the brain endothelial cells acts as a barricade to large molecules attempting to penetrate via paracellular pathway of the BBB.1 Indeed, it has been suggested that molecules with hydrodynamic diameters larger than 11 angstroms or 500 daltons molecular weight are too large to pass through the tight junctions in the BBB.8,9
Many efforts have been made to improve treatment of brain diseases by enhancing the delivery of drugs to the brain.1,10–12 Intra-cerebroventricular injection approach provides high drug bioavailability (close to 100%) in the brain, but the level of drug in the brain can drop drastically due to reduced cerebrospinal fluid (CSF) diffusivity.13 To achieve sufficient drug efficacy, this method requires multiple-site injections.13 Several methods to improve drug permeation through the transcellular pathways of the BBB have been extensively evaluated with limited success. First, cationization of biopharmaceutical drugs was done to improve their adsorptive-mediated transcytosis.14 Second, insulin and transferrin receptors on the endothelial cells of the BBB have been exploited to improve brain delivery of drugs via a receptor-mediated transcytosis process by conjugating the drugs to monoclonal antibodies (mAbs) that bind to respective target receptors.10 Similarly, rabies virus glycoprotein peptide (RVG29) has been used to target the acetylcholine receptor to carry drugs across the BBB.15 The potential drawback of using receptor-mediated transcytosis is that saturation of the receptors could limit their capacity and efficiency to carry drugs through the BBB. Third, a prodrug methodology has also been used to change the physicochemical properties of small molecules and peptides to improve their BBB delivery.16 Finally, inhibition of efflux pump activity (e.g., P-glycoproteins (Pgp)) has also been investigated to improve drug brain delivery.17 Most of these methods have met with limited success in improving drug delivery to the brain in clinical trials.
Alternatively, brain delivery of drugs through the paracellular pathways of the BBB can be enhanced by increasing the porosity of the tight junctions.8,18,19 One method is shrinking the endothelial cells of the BBB with a hypertonic solution of mannitol and disrupting the intercellular junctions of the BBB to allow penetration of the drugs through paracellular pathways.19 A more selective way to increase the porosity of the intercellular junctions is by inhibiting interactions of cell-adhesion proteins at the intercellular junctions (i.e., tight and adherens junctions).8,18 Modulation of protein-protein interactions can be done via extracellular or intracellular mechanisms. The extracellular mechanism involves inhibiting protein-protein interactions using small molecules (e.g., peptides) that bind to the extracellular domain of cell-adhesion proteins (i.e., occludins, claudins, and cadherins) at the intercellular junctions.18 The intracellular mechanism is via regulation or interference of the intracellular signaling pathways in an intracellular space that affects cell-cell adhesion at the intercellular junctions. For example, inhibition of phosphorylation of the cytoplasmic domain of cell adhesion proteins (e.g., occludin, claudin) causes relocation of these proteins to generate opening of the paracellular pathways.20 However, it is difficult to selectively inhibit protein phosphorylation to affect only the endothelial cells of the BBB. Thus, phosphorylation inhibitors as adjuvants to improve drug delivery to the brain may be limited by their non-specificity and the complexity of phosphorylation signaling pathways.
Cadherin peptides have been shown to inhibit E-cadherin-mediated cell-cell adhesion at the biological barriers (i.e., the BBB and intestinal mucosa barrier).8,18 In vivo, an His-Ala-Val (HAV) peptide (HAV6, Table 1) derived from E-cadherin improves the brain delivery of the magnetic resonance imaging (MRI) contrast agent gadolinium-diethylenetriaminepentacetate (Gd-DTPA) in Balb/c mice.21 Similarly, this peptide also increases the BBB penetration of a near infrared fluorescent (NIRF) dye R800 and Rdye 800-cw polyethylene glycols (25 KDa) in Balb/c mice in vivo.21 In the in situ rat brain perfusion model, HAV6 improved the paracellular permeation of 14C-mannitol and 3H-daunomycin through the BBB.22 In vitro, the paracellular permeation of marker molecules (e.g., 14C-mannitol) was enhanced by HAV6 across Madin-Darby Canine Kidney (MDCK) cell monolayers.23,24 In parallel studies, ADT6 peptide (Table 1) from the EC1 domain of E-cadherin was found to modulate the intercellular junctions of MDCK cell monolayers and enhance paracellular permeation of 14C-mannitol through cell monolayers.24
Table 1.
Names and Peptide Sequences
| HAV4 | Ac-Ser-His-Ala-Val-Ala-Ser-NH2 |
| HAV6 | Ac-Ser-His-Ala-Val-Ser-Ser-NH2 |
| ADT6 | Ac-Ala-Asp-Thr-Pro-Pro-Val-NH2 |
| ADTC1 |
|
| ADTC5 |
|
| ADTC6 |
|
| KKLVPR | Ac-Lys-Lys-Leu-Val-Pro-Arg-NH2 |
In this work, cyclic ADT peptides (ADTC1, ADTC5, ADTC6; Table 1) were synthesized and evaluated for their ability to modulate the intercellular junctions in MDCK cells and to enhance paracellular permeation of marker molecules through the BBB. Cyclic ADTC1 peptide was designed from ADT6 by adding two cysteine residues to the N- and C-termini, respectively; then, an intramolecular disulfide bond was formed between the two Cys residues. For cyclic ADTC5, the N-terminus alanine residue was deleted from ADTC1 to evaluate the effects of the Ala residue and cyclic peptide ring size on modulatory activity. Finally, the valine residue was deleted from ADTC5 to produce ADTC6 to test the effect of the C-terminal valine residue on peptide activity. The peptide efficacy in modulating the intercellular junctions was evaluated by measuring the change in transepithelial electrical resistance (TEER) values of MDCK cell monolayers as a function of peptide concentrations. The activities of cyclic peptides in enhancing brain delivery of 14C-mannitol and 14C- or 3H-polyethylene glycols (PEG), and Gd-DTPA were determined in an in situ rat brain perfusion model or an in vivo balb/c mouse model. The brain depositions of 14C/3H-compounds and Gd-DTPA were detected by radioactivity counter and magnetic resonance imaging (MRI), respectively. This study indicates that cadherin peptides can modulate the intercellular junctions of in vitro and in vivo and enhance paracellular transport of marker molecules through the BBB.
Materials and Experimental Methods
Peptide synthesis
Peptides were synthesized using Fmoc chemistry and a Perceptive Peptide synthesizer as previously described.24 The purification was done by reversed-phase HPLC using a semi-preparative column. Mass spectrometry was used to determine the correct molecular weight of each peptide. Cyclic peptides were formed via oxidation of the thiol groups of the N- and C-termini Cys residues using air oxidation in sodium bicarbonate buffer at pH 9.0. All of the peptides had a purity of ≥ 95% as confirmed by reversed-phase analytical HPLC using a C-18 column.
Cell culture
MDCK cell strain-2 obtained from ATCC (Rockville, MD, USA) was grown in 75 cm2 Corning polystyrene flasks (Corning, NY, USA). Dulbecco’s modification of Eagle’s medium (Cellgro, Manassas, VA, USA), which was enriched with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, VA, USA), 100 units/mL penicillin, 100 μg/mL streptomycin (Atlanta Biologicals), and 1.42 g/L HEPES sodium was used to culture the cells. The cells were incubated at 37°C, 95% humidity, and 5% CO2. The medium was changed three times a week, and the cells were trypsinized with 0.25% trypsin in 1.0 mM EDTA (Atlanta Biologicals). One-tenth of the harvest cells were seeded in a new polystyrene flask. The MDCK cells used were between passage number 45 and 65.
Resealing assay
MDCK cells were seeded at 100,000 cells/well in 0.4 μm polyethylene membrane Transwells (Costar, Cambridge, MA, USA) with diameters of 1.12 cm2. The medium was changed every other day until the monolayer had TEER values above 300 Ohm.cm2 between days 5 and 7. Medium A (Hank’s balanced salt solution (HBSS)) containing 10 mM HEPES, 1% glucose, and 2.0 mM CaCl2 at pH 7.4 was used to wash the MDCK monolayers. After three washing of the MDCK monolayers with Medium A, the wells were incubated with Medium A for 1.5 h for equilibration before TEER measurements. TEER values were measured at the end of the equilibration time, which was defined as t = 0 h. Medium B (medium A without CaCl2) was used to open the intercellular junctions. After 1.0 h incubation with medium B, TEER values were measured using EVOM2 (World Precision Instruments, Sarasota, FL, USA) as t = 1 h. Following the junction opening, the apical and basolateral chambers were incubated in 1.0 mM peptide dissolved in buffer A. The change in TEER value during junction resealing was measured every hour up to 6 h. Buffer A and a non-cadherin peptide (i.e., KKVPR) were used as negative controls. The experiment was performed at least in triplicate.
In situ rat brain perfusion model
The in situ rat brain perfusion studies were approved by the IACUC at The University of Kansas. Sprague Dawley rats (55–60 days old) were kept in The Animal Care Unit, The University of Kansas, under free access to food and water. The in situ rat brain perfusion studies were carried out using a method developed by Takasato et al. with some modifications.25 The body temperature of the rats was maintained using a heat lamp. Briefly, the rats were anesthetized with injections of ketamine (100 mg/kg) and xylazine (5 mg/kg). A polyethylene catheter (PE 50) was connected to the left common carotid artery (LCCA) via a cannula, and the arteries at the left pteryopalatine, occipital, and superior thyroid were ligated. Then, a retrograde perfusion with heparin-saline (10 IU/mL) heparin-saline (10 IU/mL) was performed to prevent coagulation.
Cardiac puncture was carried out prior to a perfusion wash of LCAA with heparin-saline solution for 10 s followed by 10 mL perfusion of 1.0 mM peptide dissolved in NaHCO3-buffered physiological saline containing 0.5% v/v Tween 20. The NaHCO3-buffered physiological saline was prepared with 142 μmol/mL NaCl, 28.0 μmol/mL NaHCO3, 4.2 μmol/mL KH2PO4, 1.7 μmol/mL CaSO4, and 1.0 μmol/mL MgSO4. The solution was aerated with 5% CO2 in 95% humidity prior to the experiment at 37°C. After peptide perfusion, 14C-mannitol (10 μCi; Moravek Biochemicals, Brea, CA, USA) was delivered in heparin-saline solution containing 0.5% v/v Tween 20. To remove the excess 14C-mannitol in the brain microvessels, a post-perfusion wash was done using saline for 10 s. The entire perfusion experiment was carried out at 5 mL/min rate using an infusion pump (Model 355 syringe pump, Sage Instruments, Boston, MA, USA). Transport studies with polyethylene glycol (PEG) molecules were performed using the same techniques. 1,2-3H-PEG-1500 and 1,2-14C-PEG-40000 (American Radioactive Chemicals, St. Louis, MO, USA) with strength of 10 μCi/rat were used to evaluate the limits of pore-size openings created by cadherin peptides in in situ rat brain perfusion experiments.
After the rat brain was removed from the skull, it was divided into three parts, followed by weighing and grinding each part of the brain. Into each sample, 4 mL of Ready Solv-MP scintillation cocktail (Beckman Instruments, Fullerton, CA) was added, and the radioactive activity in the scintillation fluid was measured using a dual-double scintillation spectrometer (Beckman LS 6000 IC).
In vivo modulation of the BBB in Mice
Adult female Balb/c mice obtained from the breeding colony at the Central Animal Care Facility, University of Manitoba, were used to evaluate the effect of ADTC5 on modulating the BBB. These mice were kept in a temperature-controlled environment with 12-h dark/light cycles and unlimited access to food and water. All animal experiments were approved by the IACUC at the University of Manitoba (protocol number 11-069). Quantitative determination of BBB disruption was performed using MRI.
Quantitative determination of BBB disruption was performed using MRI. Mice were anesthetized and secured in the MRI probe (Bruker Biospect MR; 7 tesla/21 cm spectrometer; and 2.5 × 2.5 cm2 field of view). A series of T1- and T2-weighted images of the mouse brain were obtained prior to Gd-DTPA administration to acquire background images of the mouse brain and establish structural coordinates as previously described.21 Following the initial T1 and T2 scans, mice were administered Gd-DTPA (0.4 mmol/kg) along with different concentrations of ADTC5 (0.0001–0.032 mmol/kg) or vehicle (PBS). A series of T1-weighted images were obtained at 3 min intervals starting immediately following Gd-DTPA injection. The imaging session lasted 24 min. After the first imaging session, a second dose of Gd-DTPA was administered and T1-weighted images were collected every 3 min for an additional 27-min imaging session. Quantitative assessment of Gd-DTPA deposition in the cerebral microvasculature was accomplished by determining Gd-DTPA contrast intensity from manually outlined regions of interest (ROI) within the coronal brain slices using Marevisi 3.5 software (Institute for Biodiagnostics, National Research Council, Otawa, Ontario, Canada). Changes in Gd-DTPA intensity in the brain were determined using a percent difference analysis of brain slice images included in the Paravision 3.0 software package according to the following formula:
Effect of ATDC5 on tight junction disruptions of brain microvessels
The effects of ATDC5 on the tight junctions of brain microvessels were also examined using transmission electron microscopy (TEM). For these studies, the mice received an injection of either a vehicle (PBS) or 0.01 mmol/kg of ADTC5. The mice were then sacrificed and perfused with 3% glutaraldehyde in 0.1 M Sorensen’s phosphate buffer (pH 7.3) at 2 h and 4 h. Various organs including the brain were removed and fixed in 3% glutaraldehyde in 0.1 M Sorensen’s phosphate buffer followed by postfixation in 1% osmium tetroxide in 0.1M Sorensen’s phosphate buffer. Tissues were then dehydraded and embedded in Epon 812 using standard techniques. The sections were stained with uranyl acetate and lead citrate, and examined using a Philips CM 10 electron microscope.
Statistical analysis
ANOVA with student-Newman-Keul post hoc comparison of the means were used to analyze the dose-dependent changes in permeability of Gd-DTPA in different brain regions at different time points (Fig. 7B–D) as well as the dose-dependent effects of ADTC5 on Gd-DTPA accumulations over a period of 51 min (Fig. 7E). Statistical significance was set at p < 0.05 unless otherwise stated.
Figure 7.
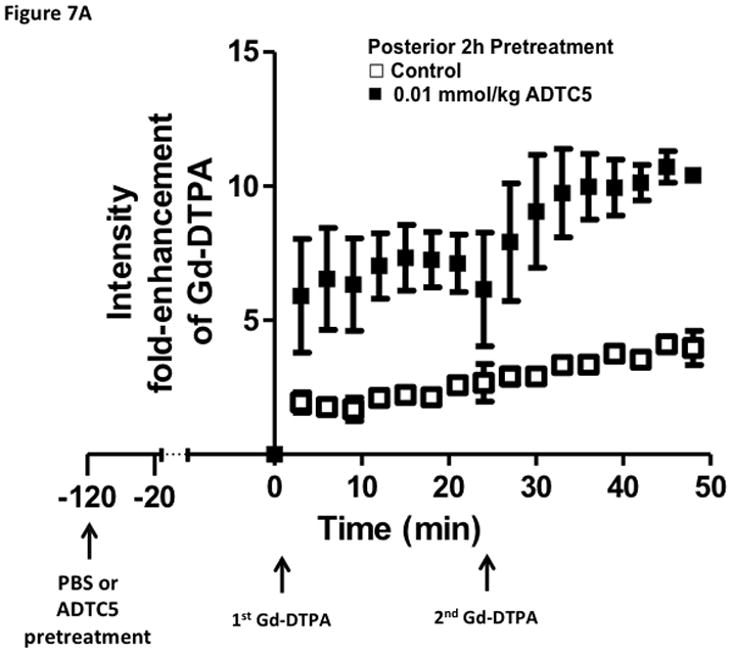
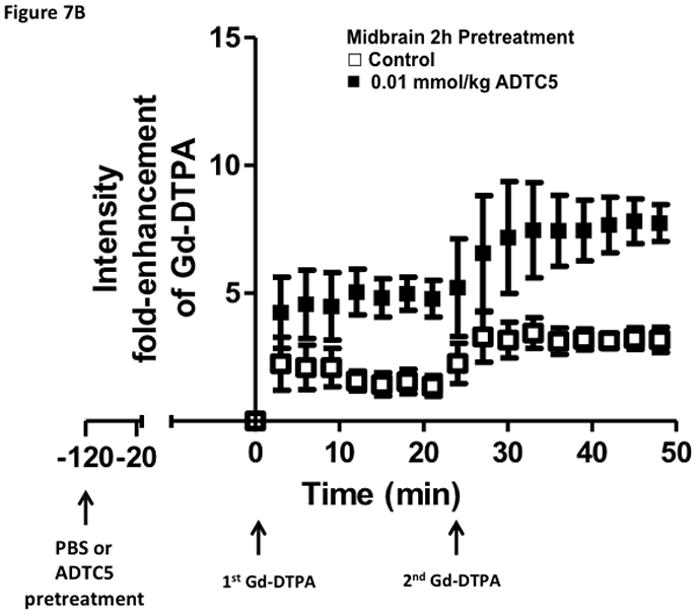
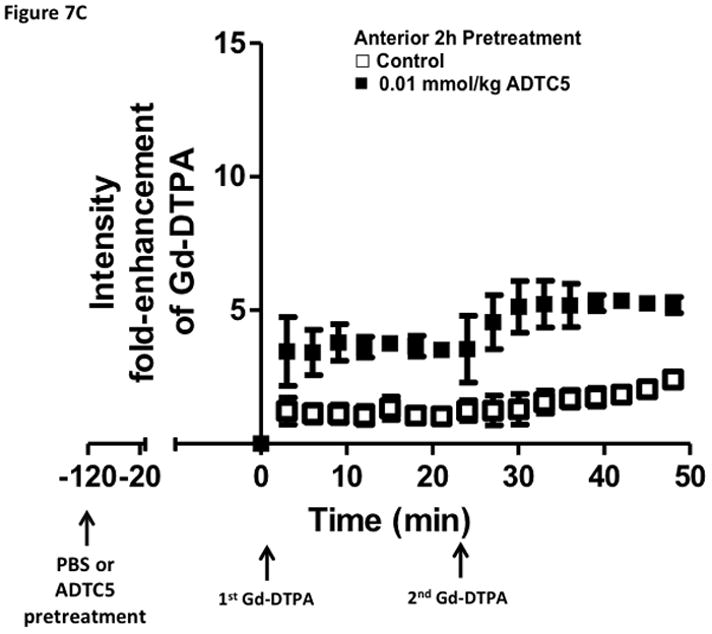
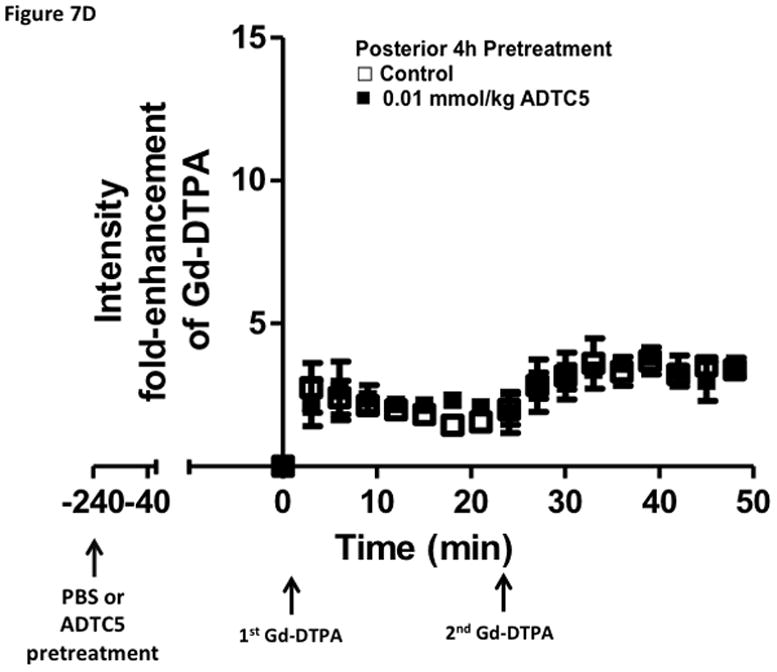
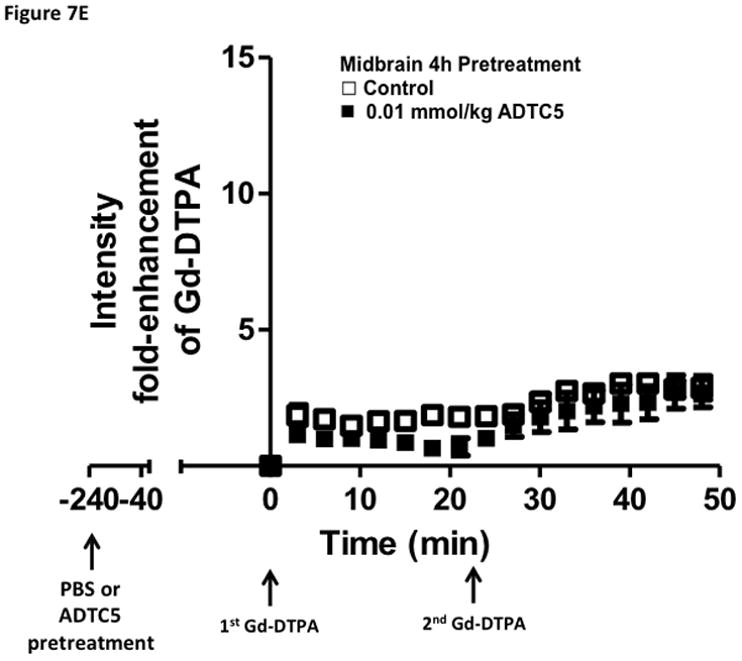
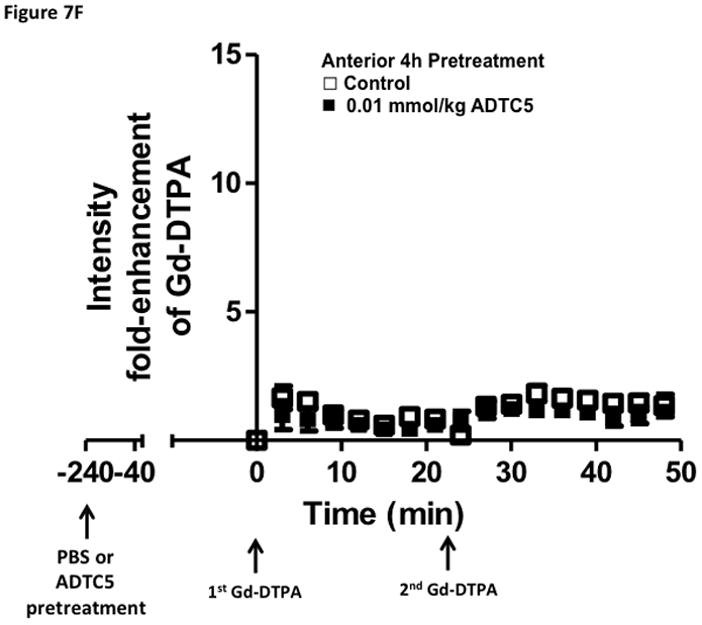
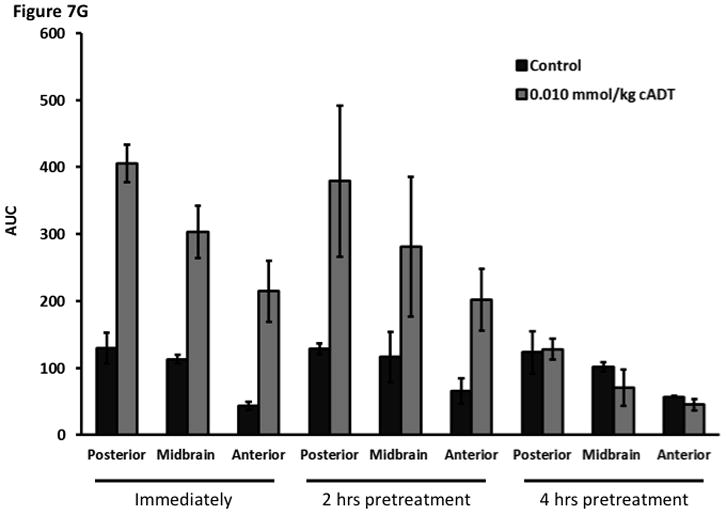
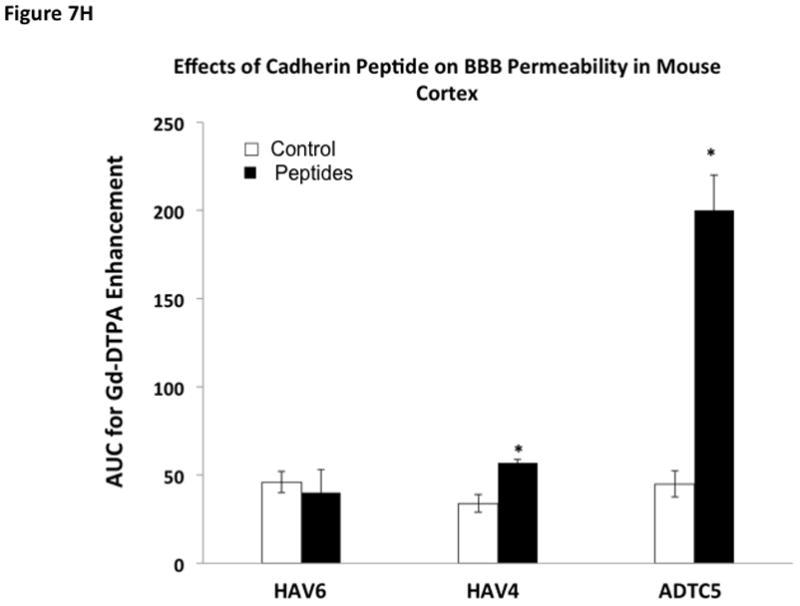
Time-dependent opening and closing of the BBB influenced by ADTC5 as measured by the amount of Gd-DTPA in the brain using MRI. The ADTC5 peptide (0.01 mml/kg) or PBS was delivered 2 h (A–C) and 4 h (D–F) before the delivery of Gd-DTPA at time 0 and 21 min. The brain was scanned every 3 min to determine the amount of Gd-DTPA deposition at the posterior (A & D), midbrain (B & E), and anterior (C & F). (G) Comparison of area under the curve (AUC) of the amounts of brain deposition of Gd-DTPA at the posterior, midbrain, and anterior when Gd-DTPA delivered immediately after the peptide, 2 h after the peptide, and 4 h after the peptide. (H) Comparison of the activity of HAV6, HAV4, and ADTC5 at 0.001 mmol/kg in enhancing Gd-DTPA brain deposition. The AUCs were collected from the anterior regions (cortex). (*, p < 0.05)
RESULTS
In vitro junction modulatory activity of cyclic ADT peptides
The junction modulatory activities of linear ADT6 and cyclic ADT peptides were evaluated in MDCK cell monolayers. In these studies, the peptides were compared to a negative control (HBSS medium) in blocking the resealing of the intercellular junctions of MDCK cell monolayers (Fig. 1). At 1.0 mM concentration, linear ADT6 and cyclic ADTC1 and ADTC5 peptides significantly inhibited the junction resealing from the 4- to 6-h time points (p<0.05) compared to HBSS control (Fig. 1). In contrast, no significant difference was seen between cyclic ADTC6 and HBSS (Fig. 1). The effects of 1.0 mM ADTC6 was similar to that of the scrambled KKLVPR peptide used as a negative control (data not shown).
Figure 1.
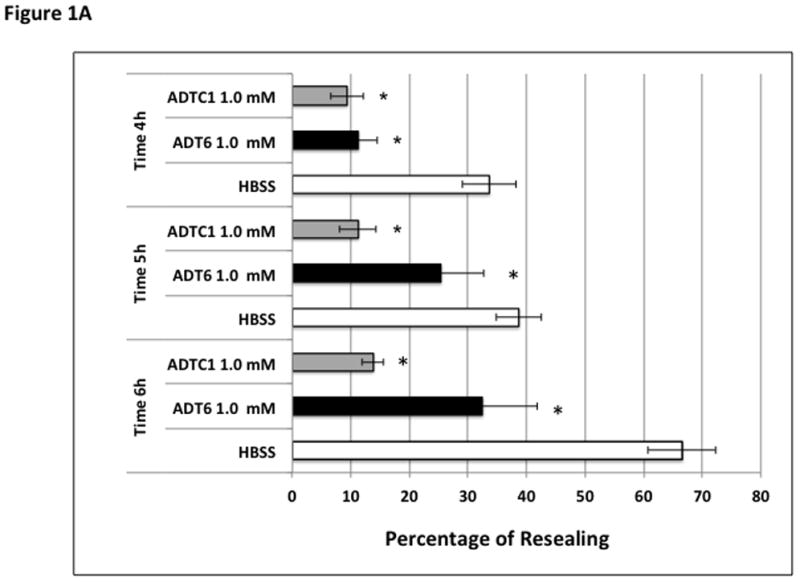
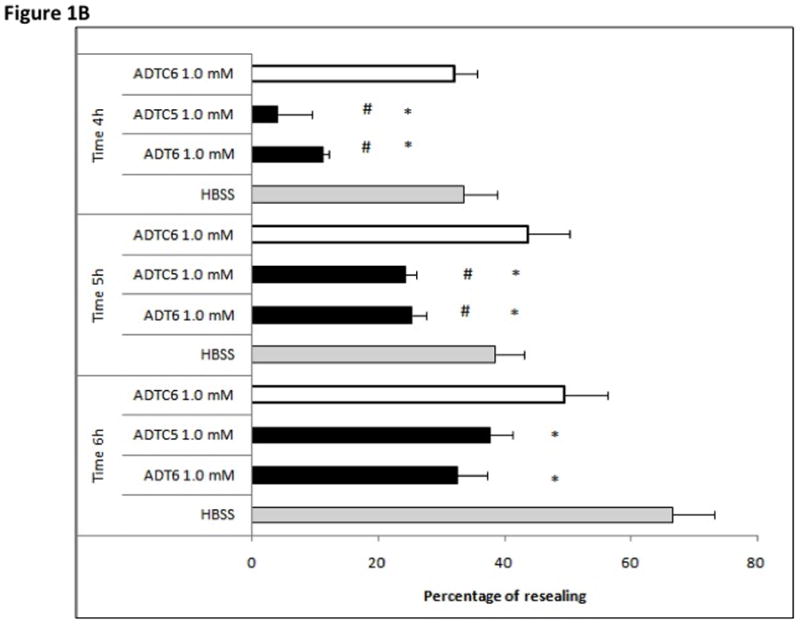
Effect of incubation time on percent change of TEER values during resealing inhibited by 1.0 mM of peptides at different time points (4–6 h). (A) Inhibition activity comparison of ADT6, ADTC1, and control HBSS at 4-, 5-, and 6-h time points (*, p <0.05 compared to HBSS control). (B) Inhibition activity comparison for ADT6, ADTC5, ADTC6, and control HBSS at time points 4-, 5-, and 6-h. ADT6 and ADTC5 have significant inhibitory activity compared to HBSS (*, p <0.05). ADTC6 did not show any difference in percent of resealing compared to HBSS at any time point. Significant differences were observed between ADT6 and ADTC6 and between ADTC5 and ADTC6 at a concentration of 1.0 mM at 4- and 5-h time points (#, p <0.02). Generally, there was no significant difference between linear ADT6 and ADTC5 at 1.0 mM concentration. The experiments were done in triplicate, and the values are the mean ± SE. Star (*) means significantly different from HBSS; number sign (#) means significantly different from 1.0 mM ADTC6.
Concentration-dependent activity of ADTC5
The activity of ADTC5 peptide at two different concentrations (0.5 and 1.0 mM) was compared to that of a control peptide (KKLVPR) (Fig. 2). A trend showing that KKLVPR causes a lower percentage of resealing compared to HBSS was observed; however, there was no statistically significant difference between HBSS and KKLVPR treatments (p>0.05) at the 6-h time point. In contrast, 1.0 mM ADTC5 significantly reduced the resealing of intercellular junctions compared to either 1.0 mM KKLVPR (p <0.05) or HBSS (p <0.01). As similar inhibitory activity was observed with the lower concentration of ADTC5 (0.5 mM) examined, these studies suggest the maximum activity of ADTC5 occurs at concentrations as low as 0.5 mM. This was confirmed with more extensive concentration-dependent studies to determine the efficacy and potency of the inhibitory effects of ADTC5 on the resealing of MDCK intercellular junctions (Fig. 3). In these studies, different concentrations of ADTC5 and KKLVPR were exposed to the cell monolayers, and the TEER values were measured up to the 6-h time point. The TEER value changes caused by the peptide were compared to that of HBSS to determine the percent of resealing, which was assumed to be 100%. The results showed that inhibition of intercellular junction resealing by ADTC5 had an IC50 of approximately 0.3 mM with was maximal effects observed at concentrations above 0.4 mM (Fig. 3). In contrast, KKLVPR had no significant activity in inhibiting the resealing compared to the HBSS medium at any concentration.
Figure 2.
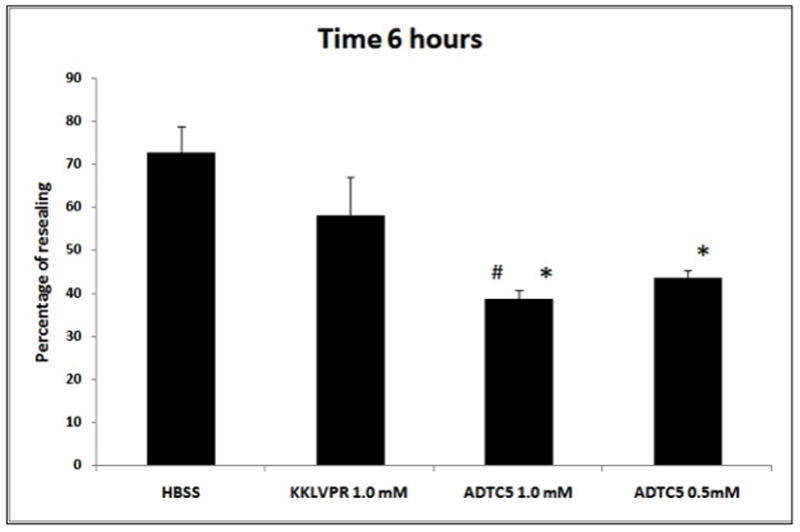
Inhibitory activity for junction resealing of ADTC5 (0.5 and 1.0 mM) compared to 1.0 mM control peptide (KKLVPR) and HBSS in MDCK monolayers. At the 6-h time point, the ADTC5 peptide (at 0.5 and 1.0 mM) blocked the resealing of MDCK cell monolayers significantly better than 1.0 mM KKLVPR peptide and HBSS. There was no significant difference between KKLVPR and HBSS (p >0.05). No significant difference in activity was observed between 1.0 mM and 0.5 mM ADTC5 at any time points (p >0.05). Star (*) means significantly different from HBSS; number sign (#) means significantly different from 1.0 mM KKLVPR.
Figure 3.
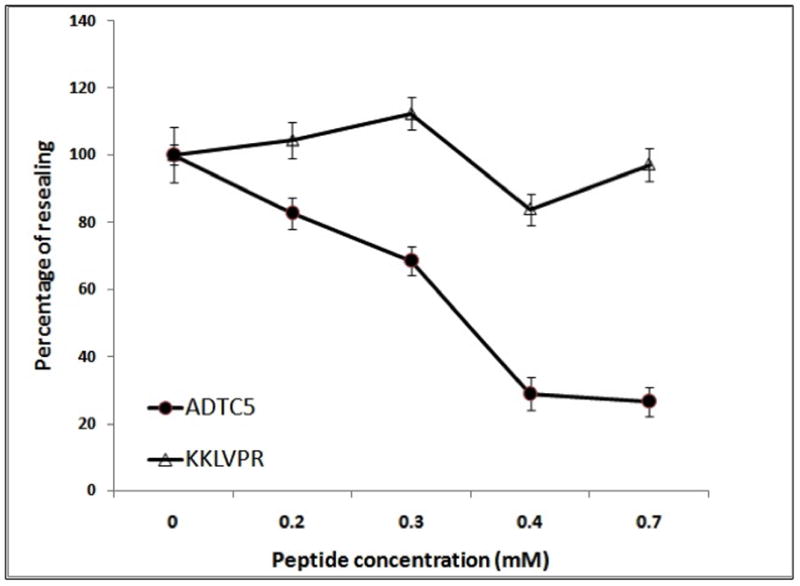
The concentration-dependent activity of ADTC5 (●) and KKLVPR (△) to inhibit the junction resealing of MDCK cell monolayers. Each data point represents the relative TEER values between the peptide-treated and medium-treated cell monolayers. The value is the mean ± SE of six replicates. The inhibition of MDCK resealing by ADTC5 was saturated at a concentration of 0.4 mM, and KKLVPR did not affect the resealing up to a concentration of 0.7 mM.
Improving BBB permeation of 14C-mannitol, 3H-PEG-1,500 and 14C-PEG-40,000 in the in situ rat brain perfusion model
In the in situ rat brain perfusion model, ADTC5 (1.93-fold) and HAV4 peptides significantly enhance the transport of 14C-mannitol compared to vehicle control (p<0.01) (Fig. 4). To evaluate the limit of pore size opening created by ADTC5, the BBB permeation of large paracellular marker molecules (i.e., 3H-PEG-1500 and 14C-PEG-40000) in in situ rat brain perfusion was determined in the presence and absence of ADTC5 peptide. There was no significant difference between the transport of PEG-1500 in the presence and absence of ADTC5 peptide (Fig. 5). The transport of PEG-1500 with or without peptide was 0.34 × 10−3 and 0.42 × 10−3 mL.min−1.gram−1, respectively. Similarly, there was no statistically significant difference in the transport of PEG-40000 in the presence and absence of ADTC5 peptide (Fig. 5); the Kin values of PEG-40000 with or without peptide were 0.07 × 10−3 and 0.08 × 10−3 mL.min−1.gram−1, respectively. As expected due to their respective size, the Kin of PEG-1500 is higher than that of PEG-40000.
Figure 4.
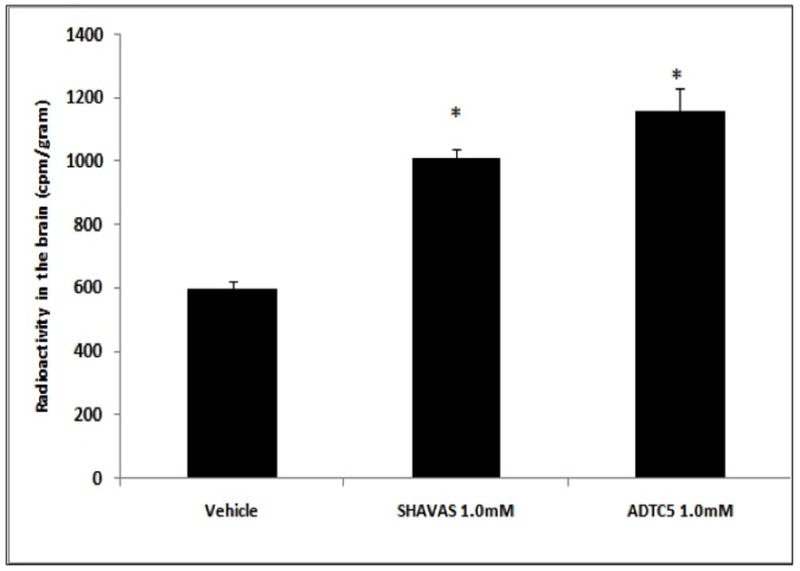
Level of radioactive 14C-mannitol in the brain after in situ rat brain perfusion following perfusion with vehicle, HAV4, and ADTC5. The brain was perfused with 1.0 mM of peptide (HAV4 and ADTC5) prior to delivery of 10 mL of 14C-mannitol 1 μCi/mL at a rate of 5 mL/min. The value was the mean ± SE of three replications. Star (*) indicates a significant difference from the vehicle.
Figure 5.
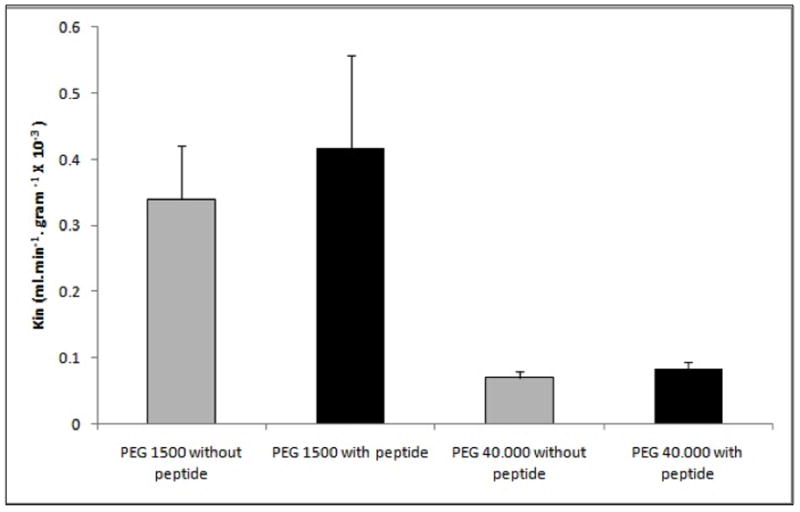
Kin of 3H-PEG-1500 and 14C-PEG-40000 calculated after perfusion in the presence and absence of ADTC5 peptide. ADTC5 (1.0 mM) was perfused at a rate of 5 mL/min for 2 min followed by a 2 -min perfusion of 1.0 μCi/mL radioactive PEG at the same rate. The experiment was done in triplicate. Kin value was the mean ± SE. There was no significant improvement in PEG-1500 and PEG-40000 transport to the brain with or without peptide.
In vivo improvement of BBB penetration of Gd-DTPA by ADTC5
The effects of ADTC5 peptide in disruption of the BBB in vivo were evaluated in Balb/c mice by tail vein injections of Gd-DTPA contrast imaging agent either alone or in combination with various concentrations (0.0001–0.032 mmol/kg) of ADTC5 peptide. The deposition of Gd-DTPA in the brain was quantified by MRI. Representative T1-weighted MR images of Gd-DTPA contrast enhancement of coronal brain slices taken from posterior region of the brain in both control and ADTC5-treated mice are shown in Figure 6A. In the control mice, there was limited deposition of Gd-DTPA in the brain, and no change in contrast enhancement was observed over time (Fig. 6A). A similar Gd-DTPA contrast enhancement was observed in mid-brain and anterior regions of the brain in mice receiving vehicle (data not shown). Indeed, the only brain region showing appreciable accumulation of Gd-DTPA were the ventricles and circumventricular organs residing outside of the BBB. In contrast, in mice receiving ADTC5 peptide, Gd-DTPA contrast enhancement was increased within the brain as early as 15-min post treatment and remained elevated for the 51-min duration of the imaging session.
Figure 6.
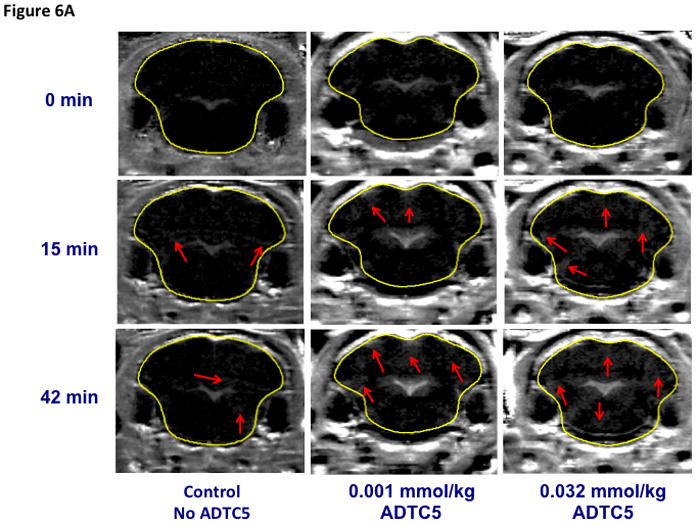
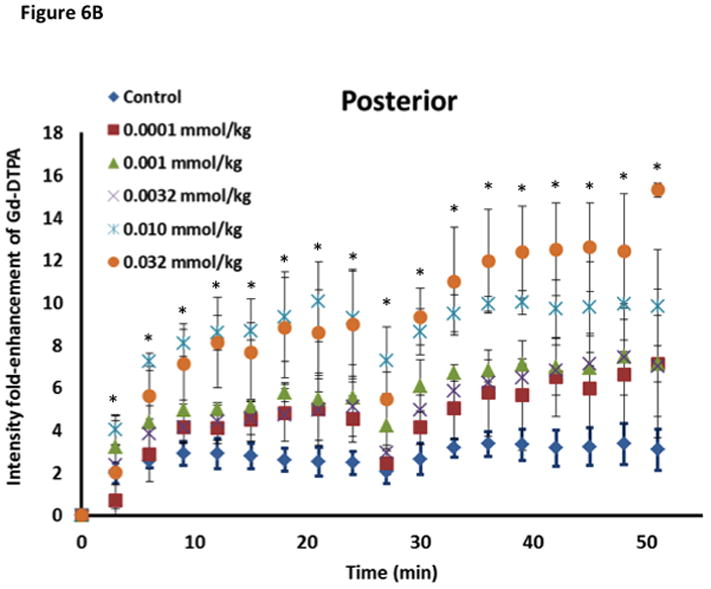
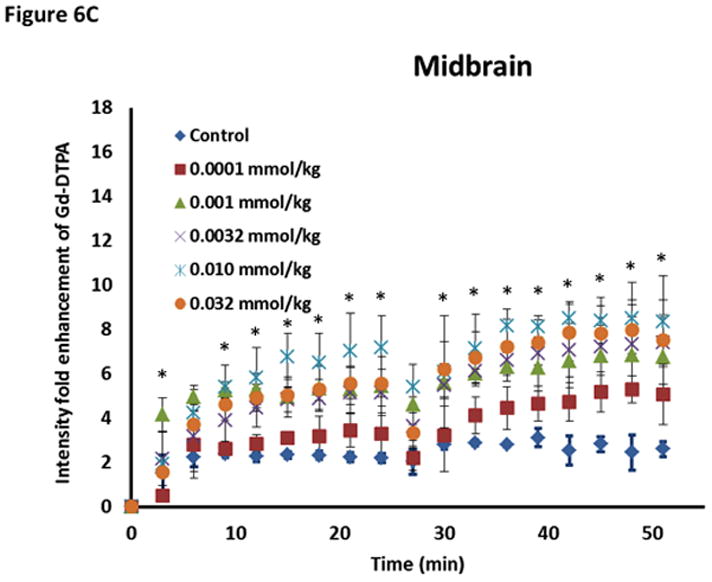
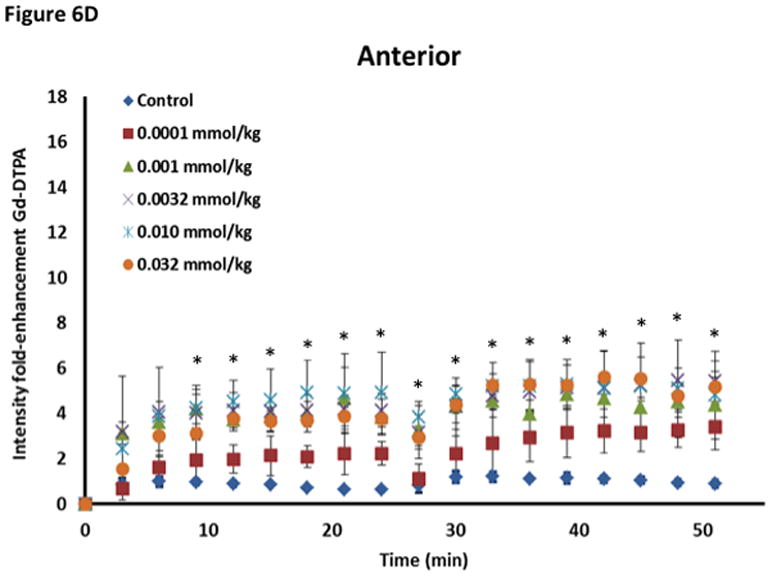
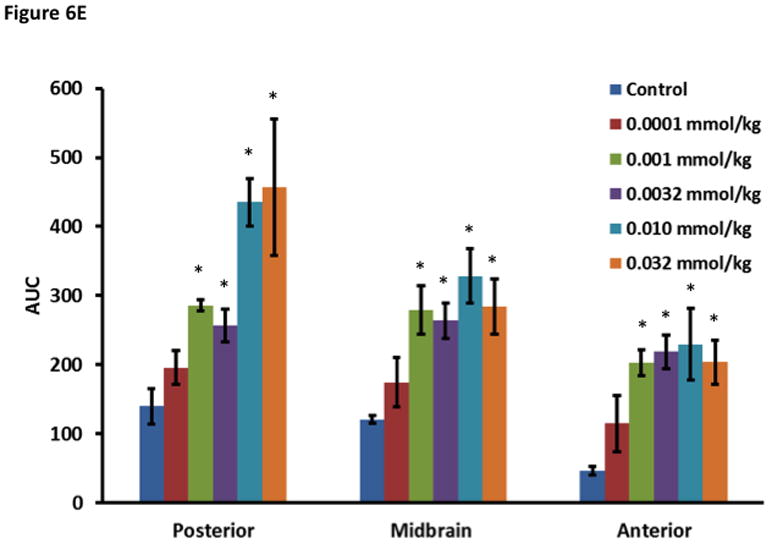
The effect of ADTC5 on the enhanced brain delivery of Gd-DTPA in Balb/c mice after i.v. administration. (A) Representative of T1-weighted MR images of Gd-DTPA deposition in the posterior region of the brain after i.v. administration of Gd-DTPA with vehicle and ADTC5 peptide at doses 0.01 and 0.032 mmol/kg at time points 0, 15, and 42 min. The arrows show representatives of contrast regions where deposition of Gd-DTPA resides in the brain. (B–D) The effect of doses of ADTC5 (0, 0.0001, 0.001, 0.032, 0.01, 0.032 mmol/kg) on the quantitative fold enhancements of brain delivery of Gd-DTPA in the (B) posterior, (C) mid-brain, and (D) anterior regions of the brain. (E) The effect of different doses of ADTC5 (0, 0.0001, 0.001, 0.032, 0.01, 0.032 mmol/kg) in enhancing Gd-DTPA brain depositions represented as area under the curve (AUC) over the combined imaging session of 51 min. * p < 0.05 compared to control mice in the same region. Values represent the mean ± SEM for 4 mice per treatment group.
The concentration of ADTC5 peptide has an effect on the brain delivery of Gd-DTPA (Fig. 6B–E), and the amounts of Gd-DTPA in the presence of peptides were significantly different than control. The Gd-DTPA brain depositions were increased as the doses of ADTC5 peptides increased from 0.0001 to 0.032 mmol/kg (Fig. 6B–D). The modulation of the BBB by ADTC5 peptide occurred within minutes, and the first maximum brain deposition of Gd-DTPA was observed around 15 min. The second delivery of Gd-DTPA without the peptide at 24-min showed an increase in Gd-DTPA deposition in different parts of the brain up to 51-min observation (Figs. 6B–D), suggesting that the effect of the peptide to open the intercellular junctions lasts longer than 51 min. Integrations of the areas under the curve for deposition of Gd-DTPA in posterior, mid-brain, and anterior regions of the brain were consistent, indicating enhancements of Gd-DTPA in the ADTC5-treated mice compared to vehicle-treated mice (Fig. 6E). While Gd-DTPA contrast enhancement demonstrated regional differences in basal BBB permeability, increases in BBB permeability in response to the peptide were similar in all brain regions (Fig. 6E). Maximal responses to ADTC5 peptide resulted in an approximately 4-to-5-fold increase in Gd-DTPA brain deposition in posterior, mid-brain, and anterior regions of the brain.
The time-dependent effect of ADTC5 peptide in opening intercellular junctions of the BBB was evaluated by administering the peptide or PBS control at 2 h (Fig. 7A–C) and 4 h (Fig. 7D–F) before administering Gd-DTPA at 0 and 21 min time points. For the 2 h time lapse experiment, there were significant differences in the amounts of Gd-DTPA in the posterior, midbrain, and anterior sections in the animals treated with ADTC5 compared to PBS. In contrast, there were no differences in the Gd-DTPA depositions in all three parts of the brain of animals treated with ADTC5 and PBS when ADTC5 peptide was delivered 4 h prior to Gd-DTPA injection. While the areas under the curve of Gd-DTPA contrast enhancement were significantly greater in mice receiving ADTC5 at time of imaging or as a 2-h pretreatment prior to imaging, there were no significant differences compared to vehicle control following a 4-hr pretreatment with ADTC5 (Fig. 7G).
The effects of ADTC5, HAV6, and HAV4 peptides in enhancing BBB penetration Gd-DTPA were compared at peptide dose of 0.001 mmol/kg (Fig. 7H). The amount of Gd-DTPA in the anterior (cortex) region of the brain of ADTC5-treated mice was about 400% higher than the PBS-treated control while the Gd-DTPA deposition in HAV4-treated mice was approximately 67% higher than PBS-treated mice. At this peptide dose, there was no enhancement of the brain deposition of Gd-DTPA caused by HAV6 peptide.
The effect of ADTC5 peptide on brain microvessel endothelial cell morphology was evaluated using transmission electron microscopy (TEM). Representative micrographs from vehicle-treated mice show brain microvessels with intact tight junctions and limited vesicular activity (Fig. 8). The electron micrographs of the brain microvessels from ADTC5-treated mice are similar in morphology to those of vehicle-treated mice (Fig. 8A) at both 2 h (Fig. 8B) and 4 h (Fig 8C) post-treatment.
Figure 8.
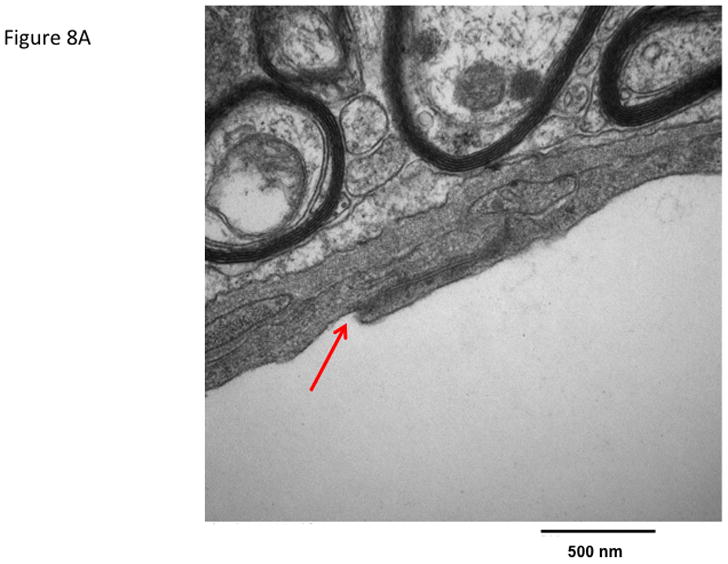
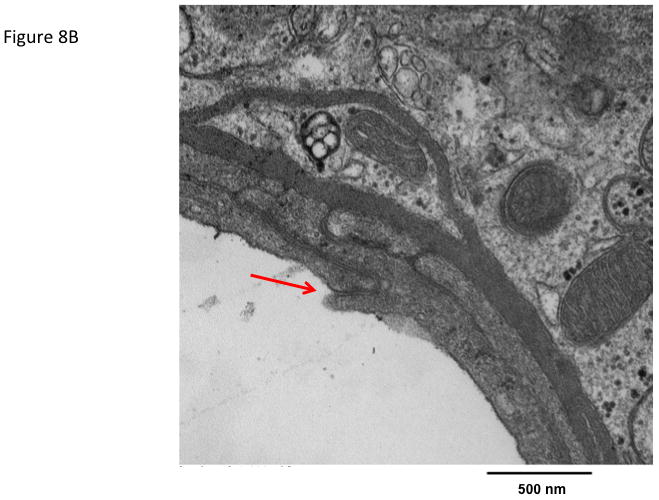
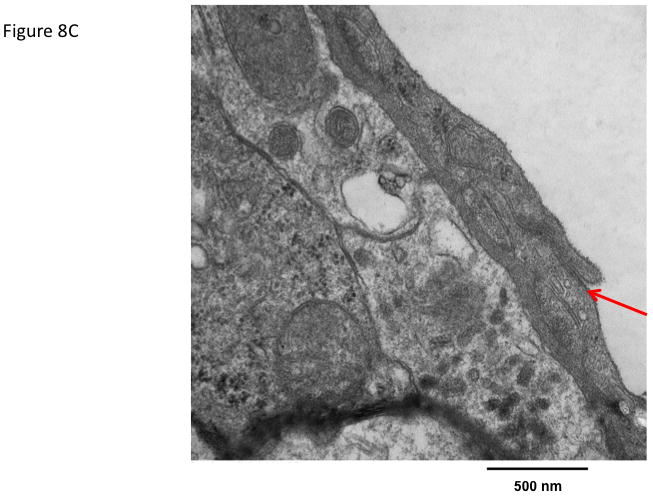
The effect of ADTC5 peptide on cell morphology of the brain microvessel endothelial cells of the BBB observed using transmission electron microscopy (TEM). The micrograph images were representatives from brain microvessels of mice (A) treated with control vehicle, (B) 2 h after treatment with 0.01 mmol/kg of ADTC5, and (C) 4 h after treatment with 0.01 mmol/kg of ADTC5. The arrows indicate the intact tight junctions of the brain microvessels.
DISCUSSION
One of the problems in diagnosing and treating brain diseases is the difficulty in delivering diagnostic and therapeutic molecules to the brain. The BBB represents a significant obstacle, and there is much interest in approaches for increasing brain penetration of diagnostic and therapeutic agents. One tactic that has been used for enhancing brain penetration of drugs is transient disruption of the BBB using osmotic agents such as mannitol26 or pharmacological agents such as the bradykinin receptor agonist Cereport™ (RMP-7).27 In our previous studies, we have demonstrated that a linear peptide (i.e., HAV6) that binds to the external domain of cadherin can alter paracellular diffusion of various marker molecules in both in vitro and in vivo BBB models.21–23 In the present study, a cyclic peptide, ADTC5, targeting the external domain of cadherin was examined for its effect on BBB permeability. Although there is no significant difference in the activity of ADTC5 compared to ADTC1 in the MDCK cell monolayer systems, the binding affinity of ADTC5 is stronger than ADTC1 to the EC1 domain of E-cadherin as determined by NMR and CD spectroscopy. In addition, the size of ADTC5 is a smaller than ADTC1; therefore, the conformation of ADTC5 is more restricted than ADTC1. A restricted backbone conformation of ADTC5 contributes to peptide selectivity to bind the EC1 domain of E-cadherin. In general, cyclic peptides have been shown to have better plasma stability than the parent linear peptides.
This study shows that ADTC5 cyclic peptide can enhance the delivery of marker molecules such as 14C-mannitol (Fig. 4) and an MRI contrast agent Gd-DTPA (Fig. 6) to the brain various animal models. Using the in situ rat brain perfusion model (Fig. 4), the brain deposition of 14C-mannitol was increased 1.93-fold by ADTC5 compared to control, suggesting that the peptide increased the porosity of the intercellular junctions of the BBB. The linear cadherin binding peptide, HAV4, also significantly enhanced 14C-mannitol transport (1.72-fold) compared to control (Fig. 4). The effect of ADTC5 in enhancing the brain delivery of Gd-DTPA is concentration-dependent, and the low dose of peptide (0.0001 mmol/kg) can improve the brain delivery of the MRI contrast agent. While the magnitude of BBB opening observed with ADTC5 in the present study is similar to that reported previously with the linear HAV6 peptide, the duration of BBB opening was approximately 4 h (Fig. 7C–G) compared to less than one hour duration with HAV6.21 It is interesting to find that ADTC5 peptide has better BBB modulatory activity than linear HAV6 and HAV4 peptides as indicated by higher brain deposition of Gd-DTPA generated by ADTC5 compare to those of HAV6 and HAV4 (Fig. 7H). This may be due in part to the improved metabolic stability and binding selectivity/affinity to the EC domain(s) of E-cadherin of the cyclic peptide ADTC5 compared to the linear peptides (i.e., HAV6 and HAV4).
It is proposed that the activity of cadherin peptide is due to binding to the EC domain of E-cadherin to block cadherin-cadherin interactions. Using the NMR residue assignments of the EC1 domain of human E-cadherin (hE-cadherin),28 the titration of ADTC5 caused chemical shift changes in the NH atoms in some residues, suggesting that the ADTC5 binds to a certain binding site at the EC1 domain. In addition, the chemical shift changes depended on the concentrations of ADTC5 peptide and the changes were plateaued at high peptide concentrations, indicating peptide binding saturation. Similarly, NMR and CD studies showed that the HAV6 peptide (Ac-SHAVSS-NH2) binds to the EC1 domain of E-cadherin. In the future, we will test a hypothesis that the activity of cadherin peptides (ADT and HAV) is due to binding to different EC domains (EC1, EC2, EC3, EC4, and EC5) within E-cadherin.
To evaluate the pore-size limit of the intercellular junction opening mediated by ADTC5 peptide, the transport of larger permeability markers including PEG-1500 and PEG-40000 was evaluated in the absence and presence of peptide in the in situ rat brain perfusion model. A very large opening of the intercellular junctions of the BBB by cadherin peptides may create a problem of diffusion of unwanted large proteins, particles, and immune cells into the central nervous system (CNS) to generate toxicity in the brain. Although there is a trend of increase of PEG-1500 and PEG-40000 in the presence of ADTC5 compared to the vehicle negative control, these increases were not statistically significant. Due to the hydrogen-bonding potential of PEG molecules, the hydrodynamic radius of either PEG-1500 or PEG-40000 is larger than the size of the molecule itself; for example, PEG-40000 has a hydrodynamic radius 14 times larger than the actual radius of the molecule alone.29 This indicates that, under the current experimental conditions used in the in situ rat brain perfusion model, the pore opening caused by ADTC5 is not large enough to allow molecules the size of PEG-1500 or larger to pass through the BBB. In contrast, previous in vivo experiments using Balb/c mice indicated that linear HAV6 peptide enhanced brain delivery of large molecular weight 800CW PEG (25 KDa).21 The lack of significant enhancement of brain delivery of PEG-1500 and PEG-40000 by ADTC5 in the in situ brain perfusion model could be due to experimental conditions. The in situ rat brain perfusion has a short duration (2 min) of exposure to the peptide and marker molecules, while the in vivo experiments captured changes in BBB permeability over longer periods of time (i.e., 15 min or longer). This suggests that peptide exposure time of two minutes in the in situ rat brain perfusion experiment may be too short to create large size pore openings that would allow enhancement of permeation of PEG-1500 through the BBB. Therefore, lengthening the peptide incubation time in the in situ rat brain perfusion experiment may allow a more accurate assessment of the magnitude of paracellular pore size opening of the BBB produced by the cadherin peptides. Alternatively, the differences in large molecular permeability in the two animal models could be dose related. The ability of the cadherin peptides to modulate BBB opening to fit the desired molecule would be an improvement over existing transient disruption approaches. Characterization of the types of molecules (e.g., size, shape, and charges) that can undergo improved BBB permeation through modulation of the intercellular junctions with cadherin peptides is currently under investigation in both the in situ and in vivo BBB models.
The formation of the cyclic peptide restricts the backbone conformation to stabilize the secondary structure for selective binding to receptors on the cell surface, and it can also improve chemical and enzymatic stabilities of cyclic peptides.30 However, formation of cyclic peptides could also result in a loss of activity due to the creation of an unfavorable conformation for binding to the receptor.31 Here, the formation of a cyclic peptide ADTC1 from ADT6 maintained its bioactivity in inhibiting the junction resealing. The deletion of the N-terminus alanine (Ala) residue from ADTC1 to make ADTC5 still maintained the activity of ADTC5 in in inhibiting the junction resealing of MDCK monolayers. These findings suggest that the N-terminus alanine residue is not important for peptide activity and binding to E-cadherin. The alternative explanation is that the side chain of the N-terminus cysteine residue in ADTC5 could act as a substitute for the side chain of the alanine. In contrast, further deletion of the C-terminal valine residue in ADTC5 to make ADTC6 resulted in a complete loss of biological activity. This indicates that the C-terminal valine residue in ADTC5 peptide is essential for binding to E-cadherin to exert its biological activity.
While the brain microvessel endothelial cells of the BBB express N-, E- and VE-cadherin, it is still a question whether cadherin peptides from E-cadherin could also bind and modulate VE-cadherin interactions. While E- and VE-cadherin are responsible for homotypic interactions in the adherens junctions of the BBB, N-cadherin’s role has been suggested to be anchoring vascular endothelial cells to its surrounding cells.32 Our previous studies indicated that fluorescence-labeled HAV peptide and anti-E-cadherin antibody bind to and decorate the intercellular junctions of bovine-brain microvessel cell (BBMEC) monolayers, suggesting the presence of E-cadherin in the adherens junctions.33 Western blot experiments indicated that the primary cell culture of BBMEC contains both E-cadherin and VE-cadherin (Cadherin-5).33 It needs to be determined whether peptides from E-cadherin can modulate VE-cadherin interactions in the BBB. Although ADT sequences were not available in N- and VE-cadherin, the HAV sequence does exist in E- and N-cadherin peptides (i.e., AHAVSS and AHAVDI, respectively) while VE-cadherin has a homology sequence AVIVDK.34,35 We are currently expressing the EC1 domain of VE-cadherin to evaluate whether HAV and ADT peptides can bind to the EC1 domain of VE-cadherin. These binding studies will be carried out using NMR and other spectroscopic methods (i.e., CD, fluorescence and FTIR). Furthermore, the binding affinities of HAV and ADT peptides to the EC1 domain of E- and VE-cadherin will be compared.
Because of the close cellular association of tight junctions and adherens junctions and the growing evidence for crosstalk between the two, the present study examined the effects of ADCT5 on brain endothelial cell morphology. Compared to vehicle-treated mice, there were no detectable differences in the morphology of brain microvessel endothelial cells following ADTC5 exposure. Tight junction complexes were present and intact in both vehicle- and peptide-treated mice. Furthermore, vesicular activity appeared similar in both vehicle and peptide treated mice. Together, the electron micrograph analysis of brain capillaries showed no overt changes. This is in stark contrast to the resulting endothelial morphology changes observed in pathological conditions where there is BBB disruption such as brain tumors or multiple sclerosis.36,37 Even compared to other transient BBB disruption methods, such as osmotic disruption where there is conflicting evidence of structural breakdown of tight junctions, there is an altered morphology present as increased vesicular activity is observed.38–40 Such ultrastructural changes to the brain microvasculature were not observed in the present study with ADTC5. The absence of anatomical changes in the brain microvasculature following cadherin peptide exposure suggests enhanced drug delivery across the BBB may be possible while minimizing potential CNS toxicity.
In conclusion, the results show that ADTC5 can improve the delivery of molecules in vitro through cell monolayers and in vivo through the BBB. The in vivo studies showed that cyclic ADTC5 was better enhancer than linear HAV6 and HAV4. The activity of ADTC5 is sequence-specific, and removal of the Val residue in ADTC6 abolished the modulatory activity of the peptide. In the future, ADTC5 peptide will be used to enhance brain delivery of diagnostic and therapeutic agents in animal models for brain tumors, multiple sclerosis, and Parkinson’s and Alzheimer’s diseases. Finally, the binding site(s) of ADT peptides on the EC1 domain of E-cadherin are being investigated using 3D NMR and molecular modeling studies. The results from these binding studies will be used to design an effective modulator of the BBB for improving in vivo drug delivery to the brain.
Acknowledgments
We thank the National Institutes of Health (R01-NS075374) for supporting this work. ML was also supported by The Fulbright US Student Program. We are grateful for the help of Nancy Harmony in proofreading the manuscript.
Footnotes
Disclosure: The authors do not have personal financial conflict of interests related to this manuscript.
References
- 1.Gabathuler R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol Dis. 2010;37(1):48–57. doi: 10.1016/j.nbd.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 2012;64(7):640–665. doi: 10.1016/j.addr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 3.McCaffrey G, Davis TP. Physiology and pathophysiology of the blood-brain barrier: P-glycoprotein and occludin trafficking as therapeutic targets to optimize central nervous system drug delivery. J Investig Med. 2012;60(8):1131–1140. doi: 10.231/JIM.0b013e318276de79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandery K, Glaeser H, Fromm MF. Interaction of innovative small molecule drugs used for cancer therapy with drug transporters. Br J Pharmacol. 2012;165(2):345–362. doi: 10.1111/j.1476-5381.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownlees J, Williams CH. Peptidases, peptides, and the mammalian blood-brain barrier. J Neurochem. 1993;60(3):793–803. doi: 10.1111/j.1471-4159.1993.tb03223.x. [DOI] [PubMed] [Google Scholar]
- 6.Meyer RP, Gehlhaus M, Knoth R, Volk B. Expression and function of cytochrome p450 in brain drug metabolism. Curr Drug Metab. 2007;8(4):297–306. doi: 10.2174/138920007780655478. [DOI] [PubMed] [Google Scholar]
- 7.Deli MA. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim Biophys Acta. 2009;1788(4):892–910. doi: 10.1016/j.bbamem.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Zheng K, Trivedi M, Siahaan TJ. Structure and function of the intercellular junctions: barrier of paracellular drug delivery. Curr Pharm Des. 2006;12(22):2813–2824. doi: 10.2174/138161206777947722. [DOI] [PubMed] [Google Scholar]
- 9.Van Itallie CM, Anderson JM. Measuring size-dependent permeability of the tight junction using PEG profiling. Methods Mol Biol. 2011;762:1–11. doi: 10.1007/978-1-61779-185-7_1. [DOI] [PubMed] [Google Scholar]
- 10.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007;12(1–2):54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, Engelhardt B, Grammas P, Nedergaard M, Nutt J, Pardridge W, Rosenberg GA, Smith Q, Drewes LR. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7(1):84–96. doi: 10.1016/S1474-4422(07)70326-5. [DOI] [PubMed] [Google Scholar]
- 12.Frank RT, Aboody KS, Najbauer J. Strategies for enhancing antibody delivery to the brain. Biochim Biophys Acta. 2011;1816(2):191–198. doi: 10.1016/j.bbcan.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Haller MF, Saltzman WM. Localized delivery of proteins in the brain: can transport be customized? Pharm Res. 1998;15(3):377–385. doi: 10.1023/a:1011911912174. [DOI] [PubMed] [Google Scholar]
- 14.Elmquist A, Lindgren M, Bartfai T, Langel U. VE-cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp Cell Res. 2001;269(2):237–244. doi: 10.1006/excr.2001.5316. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Huang R, Han L, Ke W, Shao K, Ye L, Lou J, Jiang C. Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles. Biomaterials. 2009;30(25):4195–4202. doi: 10.1016/j.biomaterials.2009.02.051. [DOI] [PubMed] [Google Scholar]
- 16.Peura L, Malmioja K, Huttunen K, Leppanen J, Hamalainen M, Forsberg MM, Rautio J, Laine K. Design, synthesis and brain uptake of LAT1-targeted amino acid prodrugs of dopamine. Pharm Res. 2013;30(10):2523–2537. doi: 10.1007/s11095-012-0966-3. [DOI] [PubMed] [Google Scholar]
- 17.Namanja HA, Emmert D, Davis DA, Campos C, Miller DS, Hrycyna CA, Chmielewski J. Toward eradicating HIV reservoirs in the brain: inhibiting P-glycoprotein at the blood-brain barrier with prodrug abacavir dimers. J Am Chem Soc. 2012;134(6):2976–2980. doi: 10.1021/ja206867t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiptoo P, Laksitorini MD, Siahaan TJ. Peptide Delivery. In: Kastin AJ, editor. Handbook of Biologically Active Peptides. San Diego: Academic Press; 2013. pp. 1702–1710. [Google Scholar]
- 19.Neuwelt EA, Maravilla KR, Frenkel EP, Rapaport SI, Hill SA, Barnett PA. Osmotic blood-brain barrier disruption. Computerized tomographic monitoring of chemotherapeutic agent delivery. J Clin Invest. 1979;64(2):684–688. doi: 10.1172/JCI109509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao R. Occludin phosphorylation in regulation of epithelial tight junctions. Ann N Y Acad Sci. 2009;1165:62–68. doi: 10.1111/j.1749-6632.2009.04054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.On NH, Kiptoo P, Siahaan TJ, Miller DW. Modulation of blood-brain barrier permeability in mice using synthetic E-cadherin peptide. Mol Pharm. 2014;1(3):974–981. doi: 10.1021/mp400624v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiptoo P, Sinaga E, Calcagno AM, Zhao H, Kobayashi N, Tambunan US, Siahaan TJ. Enhancement of drug absorption through the blood-brain barrier and inhibition of intercellular tight junction resealing by E-cadherin peptides. Mol Pharm. 2011;8(1):239–249. doi: 10.1021/mp100293m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makagiansar IT, Avery M, Hu Y, Audus KL, Siahaan TJ. Improving the selectivity of HAV-peptides in modulating E-cadherin-E-cadherin interactions in the intercellular junction of MDCK cell monolayers. Pharm Res. 2001;18(4):446–453. doi: 10.1023/a:1011094025008. [DOI] [PubMed] [Google Scholar]
- 24.Sinaga E, Jois SD, Avery M, Makagiansar IT, Tambunan US, Audus KL, Siahaan TJ. Increasing paracellular porosity by E-cadherin peptides: Discovery of bulge and groove regions in the EC1-domain of E-cadherin. Pharm Res. 2002;19(8):1170–1179. doi: 10.1023/a:1019850226631. [DOI] [PubMed] [Google Scholar]
- 25.Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol. 1984;247(3 Pt 2):H484–493. doi: 10.1152/ajpheart.1984.247.3.H484. [DOI] [PubMed] [Google Scholar]
- 26.Neuwelt EA, Hill SA, Frenkel EP. Osmotic blood-brain barrier modification and combination chemotherapy: concurrent tumor regression in areas of barrier opening and progression in brain regions distant to barrier opening. Neurosurgery. 1984;15(3):362–366. doi: 10.1227/00006123-198409000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Dean RL, Emerich DF, Hasler BP, Bartus RT. Cereport (RMP-7) increases carboplatin levels in brain tumors after pretreatment with dexamethasone. Neuro Oncol. 1999;1(4):268–274. doi: 10.1093/neuonc/1.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prasasty VD, Krause ME, Tambunan US, Anbanandam A, Laurence JS, Siahaan TJ. 1-H, 13-C and 15-N backbone assignment of the EC-1 domain of human E-cadherin. Biomol NMR Assign. 2014 Feb 8; doi: 10.1007/s12104-013-9539-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu A, Yang K, Wang M, Liu S, Chintala R, Palm T, Zhao H, Peng P, Wu D, Zhang Z, Hua J, Hsieh MC, Zhou J, Petti G, Li X, Janjua A, Mendez M, Liu J, Longley C, Zhang Z, Mehlig M, Borowski V, Viswanathan M, Filpula D. Structure-function engineering of interferon-beta-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug Chem. 2006;17(3):618–630. doi: 10.1021/bc050322y. [DOI] [PubMed] [Google Scholar]
- 30.Bogdanowich-Knipp SJ, Chakrabarti S, Williams TD, Dillman RK, Siahaan TJ. Solution stability of linear vs. cyclic RGD peptides. J Pept Res. 1999;53(5):530–541. doi: 10.1034/j.1399-3011.1999.00052.x. [DOI] [PubMed] [Google Scholar]
- 31.Pakkala M, Hekim C, Soininen P, Leinonen J, Koistinen H, Weisell J, Stenman UH, Vepsalainen J, Narvanen A. Activity and stability of human kallikrein-2-specific linear and cyclic peptide inhibitors. J Pept Sci. 2007;13(5):348–353. doi: 10.1002/psc.849. [DOI] [PubMed] [Google Scholar]
- 32.Navarro P, Ruco L, Dejana E. Differential localization of VE- and N-cadherins in human endothelial cells: VE-cadherin competes with N-cadherin for junctional localization. The Journal of cell biology. 1998;140(6):1475–1484. doi: 10.1083/jcb.140.6.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pal D, Audus KL, Siahaan TJ. Modulation of cellular adhesion in bovine brain microvessel endothelial cells by a decapeptide. Brain Res. 1997;747(1):103–113. doi: 10.1016/s0006-8993(96)01223-1. [DOI] [PubMed] [Google Scholar]
- 34.Williams E, Williams G, Gour BJ, Blaschuk OW, Doherty P. A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J Biol Chem. 2000;275(6):4007–4012. doi: 10.1074/jbc.275.6.4007. [DOI] [PubMed] [Google Scholar]
- 35.Brasch J, Harrison OJ, Ahlsen G, Carnally SM, Henderson RM, Honig B, Shapiro L. Structure and binding mechanism of vascular endothelial cadherin: a divergent classical cadherin. J Mol Biol. 2011;408(1):57–73. doi: 10.1016/j.jmb.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greig NH. Brain tumors and the blood-tumor barrier. In: Neuwelt EA, editor. Implications of the Blood-Brain Barrier and Its Manipulation: Clinical Aspects. New York: Plenum Publishers; 1989. pp. 77–106. [Google Scholar]
- 37.Engelhardt B, Liebner S. Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res. 2014;355(3):687–699. doi: 10.1007/s00441-014-1811-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cosolo WC, Martinello P, Louis WJ, Christophidis N. Blood-brain barrier disruption using mannitol: time course and electron microscopy studies. Am J Physiol. 1989;256(2 Pt 2):R443–447. doi: 10.1152/ajpregu.1989.256.2.R443. [DOI] [PubMed] [Google Scholar]
- 39.Farrell CL, Shivers RR. Capillary junctions of the rat are not affected by osmotic opening of the blood-brain barrier. Acta Neuropathol. 1984;63(3):179–189. doi: 10.1007/BF00685243. [DOI] [PubMed] [Google Scholar]
- 40.Samak G, Narayanan D, Jaggar JH, Rao R. CaV1.3 channels and intracellular calcium mediate osmotic stress-induced N-terminal c-Jun kinase activation and disruption of tight junctions in Caco-2 cell monolayers. J Biol Chem. 2011;286(34):30232–30243. doi: 10.1074/jbc.M111.240358. [DOI] [PMC free article] [PubMed] [Google Scholar]