

SUMMARY
Cellular proteins rely on reversible redox reactions to establish and maintain biological structure and function. How redox catabolic (NAD+:NADH) and anabolic (NADP+:NADPH) processes integrate during metabolism to maintain cellular redox homeostasis however is unknown. The present work identifies a continuously cycling, mitochondrial membrane potential-dependent redox circuit between the pyruvate dehydrogenase complex (PDHC) and nicotinamide nucleotide transhydrogenase (NNT). PDHC is shown to produce H2O2 in relation to reducing pressure within the complex. The H2O2 produced however is effectively masked by a continuously cycling redox circuit that links, via glutathione/thioredoxin, to NNT, which catalyzes the regeneration of NADPH from NADH at the expense of the mitochondrial membrane potential. The net effect is an automatic fine tuning of NNT-mediated energy expenditure to metabolic balance at the level of PDHC. In mitochondria, genetic or pharmacological disruptions in the PDHC-NNT redox circuit negate counterbalance changes in energy expenditure. At the whole animal level, mice lacking functional NNT (C57BL/6J) are characterized by lower energy expenditure rates, consistent with their well known susceptibility to diet-induced obesity. These findings suggest the integration of redox sensing of metabolic balance with compensatory changes in energy expenditure provides a potential mechanism by which cellular redox homeostasis is maintained and body weight is defended during periods of positive and negative energy balance.
Keywords: mitochondria, redox buffering, energy expenditure, energy balance
INTRODUCTION
Numerous pathological conditions ranging from cancer to diabetes are associated with elevated reactive oxygen species [1]. Specific cause and effect relationships in disease etiology however have proven difficult to establish due to the limited mechanistic understanding of redox-regulated control processes in vivo. At present, the intracellular redox environment is envisaged as the collection of an estimated 80,000 cysteines within the proteome that undergo reversible redox reactions [2]. The majority (~90%) of these redox sensitive cysteines are maintained in a reduced non-equilibrium steady-state by the thiol antioxidant redox couples glutathione (GSSG:GSH) and thioredoxin (TrxSS/TrxSH). Disequilibrium between redox couples (i.e., reduction potentials), and thus electron flow, is made possible by continuous input into the reductive (NADPH) and oxidative (O2, H2O2) arms of the system, thus giving rise to the term “redox circuits” [3]. Mitochondria play a central role in this system by providing both a major source of the reductive power (e.g., NADPH) as well as the counterbalance oxidant (e.g., superoxide anion (O2•−)/hydrogen peroxide (H2O2)) that effectively regulates overall cellular redox charge [4].
Given the potential impact of altered redox homeostasis in the etiology and pathology of disease, considerable effort has been directed at identifying the sites and conditions under which O2•−/H2O2 is produced. The electron transport system (ETS) is widely regarded as the principal source of oxidant generation and, in general, conditions that induce a high reduction state in complex I and/or III (e.g., reducing equivalent supply > respiratory demand, inhibition of electron flow, etc.) accelerate O2•−/H2O2 production [5]. More recently, a number of other mitochondrial enzymes have been identified as sources of oxidant production, including succinate dehydrogenase (complex II; [6]), the electron-transferring flavoprotein/ETF:ubiquinone oxidoreductase [7, 8], glycerol-3-phosphate dehydrogenase [9], dihydroorotate dehydrogenase [9], and the matrix dehydrogenase enzyme complexes α-ketoglutarate dehydrogenase (αKGDH), branched-chain keto-dehydrogenase (BCKDH), and pyruvate dehydrogenase (PDH) [10-15]. The latter three enzyme complexes, all 2-oxoacid dehydrogenases with similar structures and catalytic mechanisms, are particularly intriguing given each occupy a pivotal position in metabolism.
Experiments using isolated enzyme complex from bovine heart indicate the PDH complex (PDHC) produces H2O2 directly at relatively low rates in the presence of both the electron donor (i.e., pyruvate) and terminal electron acceptor (i.e., NAD+). However, as the availability of NAD+ decreases (i.e., NAD+/NADH increases), H2O2 generation increases dramatically [14]. This suggests PDHC generates H2O2 as a function of the overall redox state or ‘reducing pressure’ within the complex; i.e., the rate of forward electron flux via E1 and E2 relative to the availability of NAD+. In addition, in intact mitochondria H2O2 production by PDHC increases to extremely high rates as the concentration of the redox buffer glutathione (GSH) declines, suggesting GSH plays a direct role in buffering the H2O2 produced by PDHC [14]. In the present study, the control of PDHC in relation to mitochondrial redox buffering was further explored. PDHC is shown to be part of a continuously cycling redox circuit that links to the NADPH-regenerating, mitochondrial membrane potential-dependent, nicotinamide nucleotide transhydrogenase (NNT). This PDHC-NNT circuit reveals a potential novel mechanism by which energy balance is sensed and integrated in real time to generate corresponding reciprocal changes in energy expenditure.
EXPERIMENTAL
Materials
All animal studies were approved by the East Carolina University Institutional Animal Care and Use Committee. C57BL/6N and C57BL/6J were purchased from Jackson Laboratory. All mice were housed in a temperature (22°C) and light controlled (12 hour light/12 hour dark) room and maintained on standard chow with free access to food and water. For all experiments, mice were fasted 4 hours, anesthetized, and red portions of the gastrocnemius muscle dissected and separated into fiber bundles in Buffer X (50 mM K-MES, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 5.7 mM ATP, 14.3 mM phosphocreatine and 6.56 mM MgCl2 · 6H2O (pH 7.1, 290 mOsm) on ice. Remaining portions of muscle were frozen (liquid N2) for later analysis. All reagents and chemicals were obtained from Sigma-Aldrich with the exception of Amplex Ultra Red reagent (Invitrogen) and the GSH/GSSG assay kit (Oxis International Inc.).
Permeabilized fiber bundles
Fiber bundles were separated with fine forceps in Buffer X under a dissecting microscope to a single layer of interconnected fibers, permeabilized with 50 μg/ml saponin for 30 minutes with continuous rotation at 4oC, and washed in ice-cold Buffer Z (110 mM K-MES, 35 mM KCl, 1 mM EGTA, 5 mM K2HPO4, 3 mM MgCl2-6H2O, 0.5 mg/ml BSA, pH 7.1, 295 mOsm) to remove endogenous substrates. For experiments involving GSH depletion, 1-Chloro-2,4-dinitrobenzene (CDNB, 1 µM) or ethanol was added during saponin permeabilization to deplete matrix GSH by ~75% as previously described [14].
Mitochondrial respiration and H2O2 production/emission
High-resolution O2 consumption measurements were conducted at 37°C in buffer Z, supplemented with creatine monohydrate (25 mM), using the Oroboros Oxygraph-2K (Oroboros Instruments). Mitochondrial H2O2 emission was measured fluorometrically at 37°C via Amplex Ultra Red (10 μM)/horseradish peroxidase (HRP: 3 U/ml) detection system (Ex:Em 565:600). Fluorescence was monitored using a SPEX Fluoromax 3 (HORIBA Jobin Yvon) spectrofluorometer with temperature control and magnetic stirring. For each experiment, resorufin fluorescence was converted to nM H2O2 via an H2O2 standard curve generated under identical substrate conditions as employed for each protocol. Blebbistatin (25 μM) was present during all O2 consumption and H2O2 emission experiments to prevent contraction [16]. At the conclusion of each experiment, fiber bundles were washed in double-distilled H2O to remove salts, freeze-dried in a lyophilizer (Labconco) and weighed using an Orion Cahn C-35 microbalance (Thermo Electron Corporation).
Mitochondrial NADH production
All NADH production assays were carried in a potassium phosphate based buffer (50 mM), containing CaCl2 (10 μM) and MgCl2 (200 μM), pH 7.4. Permeabilized fibers were prepared as indicated above. Following a 15 minute wash in Buffer Z, fibers were incubated with the pore-forming peptide alamethicin (30 μg/ml) to permeabilize the inner mitochondrial membrane [11]. Experiments were carried out in the presence of coenzyme A (100 μM), NAD+ (1 mM), thiamine pyrophosphate (300 μM), rotenone (2 μM) and the indicated substrates. NADH production was tracked via auto-fluorescence (Ex:Em 376/450). Fluorescence values were converted to μM NADH via an NADH standard curve.
Mitochondrial membrane potential (ΔΨ)
ΔΨ and respiration rates were measured simultaneously in isolated mitochondria (125 µg protein/ml) using the Oroboros Oxygraph-2k combined with electrodes sensitive to tetraphenylphosponium (TPP+), a membrane-potential dependent probe) and oxygen at 25oC. Mitochondria were isolated and pooled from the entire thigh and calf region of mice using a standard isolation protocol [17]. All experiments were run in Buffer Z in the presence of 1.5 µM carboxyatractyloside, 1.25 µg/ml oligomycin, 0.5 mM GDP, 0.1 µM nigericin, 5 µM rotenone and 6 mM succinate. TPP+ electrode was calibrated by a 5 point titration (1.1-1.7 µM) at the beginning of each experiment. Membrane potential was varied by subsequent titration of the complex II inhibitor malonate (0.25 to 5 mM). ΔΨ was calculated from the Nernst equation based on the distribution of TPP+ using 0.35 mg protein/µl matrix volume to correct for non-specific binding of the probe [18]. In the initial experiments (Figure 2E & 2F), proton conductance was determined during succinate-supported respiration with minimal (2 mM pyruvate) or maximal (2 mM pyruvate + 5 mM carnitine) flux through PDHC. Subsequent experiments (Figure 2G) also included pyruvate and carnitine plus 10 mM glutamate to induce maximal production of NADH.
Figure 2. Effect of inhibiting matrix redox buffering on H2O2 emission by PDHC.

(A) Schematic showing the glutathione and peroxiredoxin/thioredoxin H2O2 buffering circuits. GPx, glutathione peroxidase; Prdx3, mitochondrial peroxiredoxin; GR, glutathioone reductase; Trx2, mitochondrial thioredoxin shown in reduced and oxidized forms; TrxR, thioredoxin reductase. (B) Representative trace of mitochondrial H2O2 emission in response to sequential addition of pyruvate and carnitine in the presence or absence of inhibitors of GR (bis-chloroethylnitrosourea; BCNU) and TrxR (auranofin; AF). (C) Quantified H2O2 emission rates from experiments in panel D. (D) Representative trace of JH2O2 emission in response to sequential addition of pyruvate/carnitine (Pyr/Carn), BCNU and AF. Data are expressed as mean±SEM, N=6-12 per experiment, * p<0.05.
GSH measurements
Frozen red gastrocnemius muscle was homogenized in HEPES (3 mM), sucrose (25 mM), and EGTA (0. 5mM), pH 7.2. Buffer was supplemented on the day of experimentation with 1% Triton and an anti-protease cocktail. Total GSH was measured using the reagents and calibration set provided by the GSH/GSSG assay (Oxis International Inc.).
Whole body calorimetry and body composition
Rates of oxygen consumption (VO2) and carbon dioxide production (VCO2), respiratory exchange ratio, food and water intake were measured using a TSE LabMaster System (TSE Systems, Chesterfield, MO). Energy expenditure was calculated using the equation ((CVO2 * VO2) + (CVCO2 * VCO2)/1000). Constants of the equation include: CVO2 = 3.941 (ml/h) and CVCO2 = 1.106 (ml/h). Infrared sensors were used to record ambulatory activity in three-dimensional axes (X, Y, Z). Counts across all three axes were summed to give total ambulatory activity. After 2 days of acclimation, all rates were averaged over a subsequent 3 day period and expressed per g of body weight or lean body mass. Fat and lean body mass were determined using an EchoMRI-500 (Houston, TX) in accordance with the manufacturer’s instructions.
Glucose tolerance
Whole body glucose tolerance was determined from intraperitoneal injection of glucose (1.5 g glucose/kg body weight) after a 4 h fast (beginning during the last 3h of the dark cycle). Blood samples were obtained from tail nick at 0, 30, 60, and 90 min after injection.
Statistics
Data are presented as mean ± SEM. Statistical analysis was performed using t-tests or one-way ANOVA with Student-Newman-Keuls methods for analysis of significance among groups. The level of significance was set at P < 0.05.
RESULTS
Removal of Acetyl-CoA Accelerates Flux but Decreases H2O2 Production by PDHC in Intact Mitochondria
Flux through PDHC is inhibited allosterically by acetyl-CoA. Acetyl-CoA accumulates in the mitochondria when production by PDHC (or other enzymes generating acetyl-CoA) exceeds the availability of oxaloacetate and thus limits entry into the TCA cycle. In striated muscle, the accumulation of acetyl-CoA is counterbalanced by the activity of carnitine acetyltransferase (CrAT), a mitochondrial enzyme that converts excess acetyl-CoA to membrane permeable acetylcarnitine esters which then efflux out of the organelle and cell (Figure 1A) [19]. To test whether H2O2 production by PDHC is directly related to reducing pressure within the complex, a series of experiments were conducted to manipulate flux through the complex using permeabilized skeletal muscle fiber bundles. In agreement with previous findings [19], addition of carnitine during basal respiration supported by pyruvate increased mitochondrial O2 consumption (JO2) and NADH production (JNADH) rates in fiber bundles from wild-type but not muscle-specific CrAT−/− mice (Figure 1B and 1C), confirming at least partial removal of acetyl-CoA via CrAT accelerates catalytic flux through PDHC [19]. Surprisingly however, in contrast to the sharp increase in H2O2 production rate (JH2O2) that occurs when catalytic flux is accelerated through isolated PDHC [14], increased flux induced by addition of carnitine reduced PDHC-mediated JH2O2 emission in permeabilized fibers, an effect that was especially evident when mitochondrial GSH was partially depleted by pretreatment with CDNB (Figure 1D). These findings reveal a clear discrepancy between the control of JH2O2 generation in the isolated enzyme [14] versus intact mitochondria, and suggest that the net rate of H2O2 production by PDHC in intact mitochondria does not simply reflect a balance between catalytic flux and buffering by GSH.
Figure 1. Effect of carnitine on PDHC flux and H2O2 emission.

Permeabilized fiber bundles were prepared from red portions of the gastrocnemius muscle. (A) Schematic showing product inhibition of PDHC by acetyl-CoA and removal of acetyl-CoA via CrAT in the presence of carnitine [28] (B) Mitochondrial oxygen consumption rate in permeabilized skeletal muscle fiber bundles from C57BL/6N wild-type (WT) and muscle-specific CrAT knockout (CrATm−/−) mice during respiration supported by pyruvate or pyruvate plus carnitine. (C) Rate of NADH production under the same experimental conditions as in B. Fibers were pre-treated with alamethicin (30 μg/ml) to permeabilize mitochondria prior to assay. (D) Mitochondrial H2O2 emission rate during respiration supported by pyruvate or pyruvate plus carnitine in fibers from WT mice plus or minus pre-treatment with 1-Chloro-2,4-dinitrobenzene (CDNB, 1 µM during permeabilization) to partially deplete (~75%) mitochondrial GSH [14].
H2O2 Generation by PDHC Is Mitigated by Direct Coupling to Redox Buffering System
The mitochondrial pools of reduced GSH and Trx2, which regulate and protect protein thiols, are established and maintained by the combined actions of glutathione reductase and thioredoxin reductase (Figure 2A). To determine whether PDHC may be integrated with the mitochondrial redox buffering system, PDHC-mediated H2O2 production was studied in the absence or presence of inhibitors to both glutathione reductase and thioredoxin reductase without depleting matrix GSH. With both arms of the matrix redox buffering system inhibited, the increase in PDHC catalytic flux induced by carnitine failed to suppress PDHC-mediated JH2O2 emission (i.e., pyruvate sole substrate); in fact, addition of carnitine generated a marked increase in pyruvate-supported JH2O2 emission (Figure 2B & 2C compared with Figure 1D), with loss of thioredoxin reductase accounting for most of the effect (Figure 2D). The TrxSS:TrxSH redox couple has previously been linked to redox buffering of αKGDH [20], an enzyme complex structurally similar to PDHC. Interestingly, αKGDH is not sensitive to matrix GSH levels [14] but, similar to PDHC, inhibition of thioredoxin reductase increases αKGDH-supported JH2O2 emission, particularly when catalytic flux is accelerated by the addition of ADP (data not shown). Collectively, these findings suggest PDHC, and likely the other 2-oxoacid dehydrogenase complexes, are capable of generating high rates of H2O2, but the net rate of H2O2 release is kept in check by direct coupling with the matrix redox buffering system.
PDHC and Nicotinamide Nucleotide Transhydrogenase are Integrated Via a Continuously Cycling Redox Circuit
The matrix redox buffering system is regenerated by, and thus derives its reducing power from, NADPH. The principal source of NADPH in the matrix is nicotinamide nucleotide transhydrogenase (NNT), an inner mitochondrial trans-membrane protein that utilizes the mitochondrial membrane potential (ΔΨm) to drive the reduction of NADP+ from NADH [21]. Thus, NADH is both a substrate for NNT and a source of fuel for the ETS to generate the ΔΨm needed to support NNT activity.
In the experiments described above, PDHC was the only source of NADH when respiration was supported by pyruvate plus carnitine. It was therefore reasoned that PDHC-derived NADH may contribute to NADPH provision via an NNT-dependent mechanism. This would explain why accelerating flux through PDHC by addition of carnitine reduces JH2O2 production when the redox buffering system is intact (due to increased JNADH production and therefore substrate for NNT; Figure 1D) but increases JH2O2 production when the redox buffering circuit is inhibited (Figure 2C). To test this hypothesis, pyruvate-supported JH2O2 emission was measured in permeabilized fibers from C57BL/6J mice, which do not express NNT due to a spontaneous in-frame 5-exon deletion [22-24], and compared with permeabilized fibers from C57BL/6N mice, which express functional NNT. Similar to the increase in JH2O2 production observed when redox buffering was compromised by inhibition of both glutathione and thioredoxin reductase (Figure 2B & 2C), the absence of NNT in C57BL/6J mice resulted in ~2-fold higher JH2O2 emission during respiration supported by pyruvate, which increased to ~5-fold higher when flux through PDHC was increased by addition of carnitine (Figure 3A and 3B). By contrast, increasing flux through PDHC had no effect on JH2O2 emission in fibers from C57BL/6N mice, indicating that continuous regeneration of NADPH via NNT is critical to buffering the H2O2 produced by PDHC. In fibers lacking NNT, providing an alternative means of regenerating NADPH via NADP-linked isocitrate dehydrogenase (i.e., increased flux of acetyl-CoA into TCA cycle via addition of malate) during maximal flux through PDHC reduced H2O2 emission back to baseline (Figure 3C), indicating restoration of the buffering network. Subsequent inhibition of glutathione and thioredoxin reductases reversed the effect of malate, confirming the effect of malate was mediated via restoration of NADPH synthesis. These findings emphasize the dependence of PDHC on an NNT-linked redox buffering network to regulate the net H2O2 emission by PDHC.
Figure 3. Comparison of H2O2 production by PDHC in mitochondria from C57BL/6N vs C57BL/6J mice.

(A-D) Permeabilized fiber bundles were prepared from red portions of the gastrocnemius muscle from C57BL/6N (6N) or C57BL/6J (6J) mice. (A) Representative trace of H2O2 emission during respiration supported by sequential addition of pyruvate and carnitine in fibers from 6N (+NNT) vs 6J (-NNT) mice. (B) Quantified H2O2 emission rates from experiments in panel A. (C) Rates of H2O2 emission in fibers from 6J (-NNT) mice in response to sequential addition of pyruvate, carnitine, malate, and the glutathione/thioredoxin reductase inhibitors (BCNU/AF). (D) Pyruvate (1 mM) and carnitine (5 mM)-supported H2O2 emission in response to increasing concentrations of ADP (0, 5, 50, 500, 2000 μM). Assay buffer was supplemented with hexokinase (1 U/ml) and 2-deoxyglucose (5 mM) to clamp ADP at each desired concentration.
PDHC Produces H2O2 Continuously
The experiments described above were carried out under non-ADP-stimulated conditions (i.e., low demand). To determine whether H2O2 is produced by PDHC in a more physiological context, JH2O2 was followed in response to titration of ADP in fibers from C57BL/6J mice supported by pyruvate plus carnitine. Titration of ADP led to a dose-dependent decrease in JH2O2 emission (Figure 3D), presumably reflecting a progressive increase the oxidation rate of NADH. Interestingly however, even during maximal respiration, the rate of H2O2 production was considerably higher in fibers from C57BL/6J (~22 pmol/min/mg dry wt) versus C57BL/6N (<5 pmol/min/mg dry wt.) mice. However, when redox buffering was blocked in fibers from C57BL/6N mice (as in Figure 2), JH2O2 production during maximum ADP-stimulated respiration was similar (~18 pmol/min/mg dry wt; data not shown) to that seen in fibers from C57BL/6J mice. Taken together, these findings suggest PDHC produces H2O2 continuously, even under ADP-stimulated conditions, but that production is effectively masked by a membrane potential-dependent redox circuit that couples PDHC-mediated NADH production with NNT-mediated NADPH regeneration, thereby regulating the net rate of H2O2 release from PDHC.
PDHC - NNT Constitute an Energy Consuming Redox Circuit
The fact that NNT-mediated NADPH generation consumes ΔΨm suggests that continuous flux through NNT-linked redox circuits contributes to energetic demand [25]. To directly test this hypothesis, JO2 was measured as a function of ΔΨm under conditions designed to stimulate maximal H2O2 production and thus demand on NNT activity. In these experiments, a higher JO2 for a given ΔΨm reflects a greater rate of proton conductance across the inner mitochondrial membrane and generates a leftward shift in the curve [5].
Figure 4A shows data from mitochondria isolated from C57BL/6N mice (i.e. NNT intact). With the substrate combination of succinate+pyruvate+carnitine, the highest membrane potential achieved (145 mV) is associated with a JO2 of 239 pmol/sec/mg. By contrast, with succinate or succinate+pyruvate as substrates, the same membrane potential (145 mV) is associated with a significantly lower JO2 (150-160 pmol/sec/mg), indicating inclusion of carnitine accelerates proton conductance, consistent with an increased rate of flux through PDHC (due to carnitine catalyzed removal of acetyl-CoA), increased JH2O2 production, and increased flux through the NNT redox circuit.
Figure 4. Effect of accelerating flux through PDHC-NNT circuit on mitochondrial proton conductance.
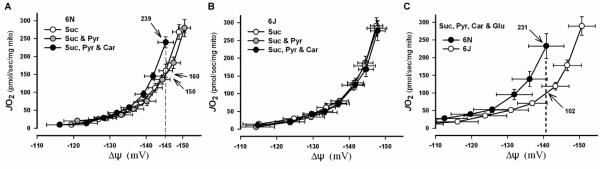
(A-C) Mitochondria were isolated from gastrocnemius and quadriceps muscle from C57BL/6N or C57BL/6J mice. (A & B) Graphs show rates of O2 consumption as a function of membrane potential (ΔΨm) in mitochondria isolated from C57BL/6N (+NNT; E) and C57BL/6J (-NNT; F) mice during respiration with minimal (succinate and succinate + pyruvate) and maximal (succinate + pyruvate + carnitine) flux through PDHC. Note the differences in JO2 (numbers with arrows) at a given ΔΨm (dotted line) indicating differences in proton conductance. (C) Direct comparison of proton conductance in isolated mitochondria from C57BL/6N and C57BL/6J mice under substrate conditions inducing maximal H2O2 production from PDHC and complex I (succinate + pyruvate + carnitine + glutamate). Note greater proton conductance in C57BL/6N mice at highest common ΔΨm.
Figure 4B shows identical experiments conducted on mitochondria isolated from C57BL/6J mice. The three substrate conditions generate identical curves, indicating no difference in proton conductance. This finding is consistent with the fact that J mice lack NNT and therefore lack the capacity to increase proton conductance when flux through PDHC is elevated by the presence of carnitine. Thus, the measured net JH2O2 is markedly higher (Figure 3A and 3B) because the mechanism that normally buffers the H2O2 is absent. In C57BL/6N mice, the same JH2O2 rate is generated but is masked by cycling of the redox buffering circuit (Figure 3B, 3C, 4A)
Figure 4C shows data in which proton conductance experiments were repeated to provide a direct comparison between mitochondria isolated from C57BL/6N vs C57BL/6J mice. Again, at the highest common ΔΨ (142 mV), JO2 was much higher in mitochondria from C57BL/6N than C57BL/6J mice (231 vs 102 pmol/sec/mg). Glutamate was included as a substrate in these experiments in attempt to also maximize JH2O2 production from complex I and/or αKGDH; however, the impact on proton conductance over succinate+pyruvate+carnitine was minimal (not shown). Collectively, these findings suggest that PDHC, as well as other sources of H2O2, link through redox buffering systems to NNT to form continuously cycling energy consuming redox circuits.
Mice Lacking NNT Have Lower Energy Expenditure
The establishment and maintenance of a relatively reduced redox environment throughout the mitochondrial and cellular proteome depends on the continuous generation of NADPH, implying that NNT activity may represent a significant source of energy expenditure. NNT, similar to other proteins/processes requiring an input of energy (e.g., ATP synthase, adenine nucleotide translocase, calcium uptake, etc.), consumes ΔΨm to drive its activity. From a whole organism perspective, a greater rate of O2 consumption corresponds to a greater rate of ΔΨm dissipation (i.e., higher demand), whereas reduced O2 consumption reflects a slower rate of ΔΨm dissipation (i.e., lower demand). In mice lacking functional NNT (C57BL/6J), rates of whole body O2 consumption, CO2 production (not shown), and energy expenditure were lower than in mice with NNT intact (C57BL/6N), whether normalized to total body mass (Figure 5A & 5B) or fat free mass (Figure 5C & 5D). Total activity, food intake on a standard diet, and respiratory exchange ratio were identical between the two strains (data not shown) and thus could not account for the difference in energy expenditure. Mice lacking NNT were also characterized by a higher percent body fat (Figure 5E) and, consistent with previous reports [22, 23], lower glucose tolerance (Figure 5F). Although the C57BL/6J strain also displays impaired insulin secretion and harbors a number of other genetic mutations [22, 23], the current findings provide evidence that NNT activity is a significant contributor to whole body energy expenditure, the absence of which likely contributes to the increased susceptibility of the widely used C57BL/6J strain to diet-induced obesity and insulin resistance.
Figure 5. Mice lacking NNT have lower energy expenditure and glucose tolerance.

(A-D) Indirect metabolic calorimetry in C57BL/6N and C57BL/6J mice. (A) Oxygen consumption and (B) calculated energy expenditure per gram of body mass. (C) Oxygen consumption and (D) calculated energy expenditure per gram of fat free mass. *Different from 6N mice within either the light or dark cycle (p < 0.05), n=10-13/group. (E) Fat mass, lean body mass, percent fat mass, and (F) whole body glucose tolerance determined in 6N and 6J mice at ~10-12 weeks of age. Data are mean ± SEM, n=10/group. *Different from 6N (p < 0.05), n=10/group.
DISCUSSION
The present study reveals a continuously cycling redox circuit between PDHC and NNT. PDHC produces H2O2 at substantial rates as a natural byproduct of catalysis, even under conditions when the ‘reducing pressure’ within the complex is minimal (i.e., high NAD+ availability, ADP-stimulated respiration). Under normal circumstances, the H2O2 is reduced to H2O and thus masked by the cycling of the redox circuit. The reducing power for the circuit is derived from and dependent upon the continuous regeneration of NADPH via NNT and the flow of electrons through the GSH and/or Trx redox couples. Because NNT activity is dependent on ΔΨm, electron flux through the PDHC-NNT circuit comes at the expense of energy. The greater the rate of H2O2 production by PDHC, the greater the rate of NNT activity and thus energy utilization (Figure 6).
Figure 6. Schematic of the PDHC-NNT redox circuit.

Proposed model shows the integration of PDHC with both the electron transport system (ETS) and the NNT-linked redox buffering circuit, providing a mechanism by which redox control processes regulate energy balance. P in circle represents matrix peroxidases and R in circle represents matrix reductases; V, complex V (ATP synthase).
To our knowledge, the PDHC–NNT circuit is the first intact continuously cycling redox circuit to be identified, although it is likely representative of redox circuits functioning throughout the proteome. In the context of redox biology, cells evolved to integrate redox catabolic (NAD+:NADH) and anabolic (NADP+:NADPH) processes to generate and maintain an optimal ‘redox charge’ against the default pressure of oxidation imposed by the environment. The majority of redox-sensitive thiols in the proteome are maintained in a reduced state by virtue of the thermodynamics of the reductive system dominating over the relatively slow rate of non-enzymatic protein thiol oxidation. The mid-point potentials for the NAD- and NADP-redox couples are virtually identical, which means the equilibrium constant for the reaction catalyzed by NNT:
NADH + NADP+ ↔ NAD+ + NADPH
should be ~1[26]. However, the reaction is maintained far from its theoretical equilibrium by ΔΨm which drives the reaction far to the right, generating a highly reduced NADP+:NADPH redox state. The mitochondrial and cytosolic NADP(H) redox states are maintained in equilibrium by the NADP-linked α-ketoglutarate/isocitrate redox shuttle, thus ensuring the reducing power of the mitochondria (i.e., ‘electrical charge’) is distributed and held via GSH- and Trx-dependent systems throughout the redox proteome. Analogous to the activation of ATP synthase in response to increased cellular ATPase activity, an increase in the oxidation rate of specific protein thiols anywhere in the cell will instantly draw electrons from the GSH and/or Trx redox buffering circuits, ultimately ‘pulling current’ from NADPH at the expense of NNT-mediated energy expenditure. This suggests 1) control of the redox environment is exerted and dominated by the kinetics of the reductive arm of the system rather than simply by the production of reactive oxygen species, and 2) that preservation of mitochondrial content (i.e., ‘reducing power’), particularly in the context of disease, aging, etc., is critical to sustaining a stable redox environment.
PDHC occupies a pivotal position in metabolism, raising the question as to why the complex may have evolved to produce H2O2 directly. One possibility is that the PDHC-NNT circuit is uniquely positioned in intermediary metabolism to provide a safeguard against imbalances imposed on the system. When energy supply exceeds energy demand, the increased ‘reducing pressure’ (i.e., increased NADH/NAD+, more negative reduction potential) on the ETS and therefore tendency to increase ΔΨm and NADPH/NADP+ is counterbalanced by the increase in PDHC-mediated H2O2 production and acceleration of flux through the PDHC-NNT circuit. This bilateral function protects against the potential for hyper-reduction of the proteome while simultaneously increasing the rate of energy expenditure and thereby at least partially compensating for the surplus energy. Interestingly in humans, active periods of weight gain due to excess caloric intake induce a much greater than predicted1 increase in resting energy expenditure rate, whereas active periods of weight loss due to caloric restriction induce a much lower than predicted decrease in resting energy expenditure rate [27]. The integration of redox control processes with energy utilization, as exemplified by the PDHC-NNT circuit, provides a potential mechanism for these compensatory changes in energy expenditure/efficiency that defend normal body weight [27], offering a possible new avenue for therapeutic development to treat obesity and other weight management conditions.
Acknowledgments
FUNDING
This work was supported by the United States Public Health Service grants F32 AR061946 (LAAG), R01 DK089312 (DMM), R01 DK073488 and R01 DK096907 (PDN).
Footnotes
Prediction based on thermic effect of food and change in metabolic mass (24).
REFERENCES
- 1.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Go Y-M, Jones DP. The Redox Proteome. J. Biol. Chem. 2013;288:26512–26520. doi: 10.1074/jbc.R113.464131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones DP, Go Y-M. Mapping the cysteine proteome: analysis of redox-sensing thiols. Curr. Opin. Chem. Biol. 2011;15:103–112. doi: 10.1016/j.cbpa.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012;23:142–153. doi: 10.1016/j.tem.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 6.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perevoshchikova IV, Quinlan CL, Orr AL, Gerencser AA, Brand MD. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2013;61C:298–309. doi: 10.1016/j.freeradbiomed.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watmough NJ, Frerman FE. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2010;1797:1910–1916. doi: 10.1016/j.bbabio.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambrus A, Adam-Vizi V. Molecular dynamics study of the structural basis of dysfunction and the modulation of reactive oxygen species generation by pathogenic mutants of human dihydrolipoamide dehydrogenase. Arch. Biochem. Biophys. 2013;538:145–155. doi: 10.1016/j.abb.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 11.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bunik VI, Sievers C. Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur. J. Biochem. 2002;269:5004–5015. doi: 10.1046/j.1432-1033.2002.03204.x. [DOI] [PubMed] [Google Scholar]
- 13.Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher-Wellman KH, Gilliam LA, Lin CT, Cathey BL, Lark DS, Neufer PD. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic. Biol. Med. 2013;65:1201–1208. doi: 10.1016/j.freeradbiomed.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014;289:8312–8325. doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry CG, Kane DA, Lin CT, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ, Neufer PD. Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J. 2011;437:215–222. doi: 10.1042/BJ20110366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 18.Affourtit C, Quinlan CL, Brand MD. Measurement of proton leak and electron leak in isolated mitochondria. Methods Mol. Biol. 2012;810:165–182. doi: 10.1007/978-1-61779-382-0_11. [DOI] [PubMed] [Google Scholar]
- 19.Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, Covington JD, Bajpeyi S, Ravussin E, Kraus W, Koves TR, Mynatt RL. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bunik VI. 2-Oxo acid dehydrogenase complexes in redox regulation. Eur. J. Biochem. 2003;270:1036–1042. doi: 10.1046/j.1432-1033.2003.03470.x. [DOI] [PubMed] [Google Scholar]
- 21.Hoek JB, Rydstrom J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem. J. 1988;254:1–10. doi: 10.1042/bj2540001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freeman HC, Hugill A, Dear NT, Ashcroft FM, Cox RD. Deletion of nicotinamide nucleotide transhydrogenase: a new quantitive trait locus accounting for glucose intolerance in C57BL/6J mice. Diabetes. 2006;55:2153–2156. doi: 10.2337/db06-0358. [DOI] [PubMed] [Google Scholar]
- 23.Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, Quarterman J, Freeman HC, Ashcroft FM, Cox RD. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia. 2005;48:675–686. doi: 10.1007/s00125-005-1680-z. [DOI] [PubMed] [Google Scholar]
- 24.Ronchi JA, Figueira TR, Ravagnani FG, Oliveira HC, Vercesi AE, Castilho RF. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med. 2013;63:446–456. doi: 10.1016/j.freeradbiomed.2013.05.049. [DOI] [PubMed] [Google Scholar]
- 25.Lopert P, Patel M. Nicotinamide Nucleotide Transhydrogenase (Nnt) Links the Substrate Requirement in Brain Mitochondria for Hydrogen Peroxide Removal to the Thioredoxin/Peroxiredoxin (Trx/Prx) System. J. Biol. Chem. 2014;289:15611–15620. doi: 10.1074/jbc.M113.533653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leibel RL, Rosenbaum M, Hirsch J. Changes in Energy Expenditure Resulting from Altered Body Weight. N. Engl. J. Med. 1995;332:621–628. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- 28.Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, Muoio DM. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem. 2009;284:22840–22852. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]