

Abstract
Third trimester-equivalent alcohol exposure causes significant deficits in hippocampal and cortical neuroplasticity, resulting in alterations to dendritic arborization, hippocampal adult neurogenesis, and performance on learning tasks. The current study investigated the impact of neonatal alcohol exposure (postnatal days 4–9, 5.25 g/kg/day) on expression of brain-derived neurotrophic factor (BDNF) and the tropomyosin-related kinase B (TrkB) receptor in the hippocampal and frontal cortex of infant Long-Evans rats. Levels of BDNF protein were increased in the hippocampus, but not frontal cortex, of alcohol-exposed rats 24 hrs after the last dose, when compared with undisturbed (but not sham-intubated) control animals. BDNF protein levels showed a trend towards increase in hippocampus of sham-intubated animals as well, suggesting an effect of the intubation procedure. TrkB protein was increased in the hippocampus of alcohol-exposed animals compared to sham-intubated pups, indicating an alcohol-specific effect on receptor expression. In addition, expression of bdnf total mRNA in alcohol-exposed and sham-intubated pups was enhanced in the hippocampus; however, there was a differential effect of alcohol and intubation stress on exon I- and IV-specific mRNA transcripts. Further, plasma corticosterone was found to be increased in both alcohol-exposed and sham-intubated pups compared to undisturbed animals. Upregulation of BDNF could potentially represent a neuroprotective mechanism activated following alcohol exposure or stress. The results suggest that alcohol exposure and stress have both overlapping and unique effects on BDNF, and highlight the need for the stress of intubation to be taken into consideration in studies that implement this route of drug delivery.
Keywords: corticosterone, cortex, FASD, neurotrophin, plasticity
Introduction
The developing brain is particularly sensitive to damage from factors such as stress and teratogenic exposure (e.g., environmental toxins, drugs of abuse, alcohol). Prenatal alcohol exposure is the leading cause of preventable intellectual disability, with an estimated occurrence of up to 5% of live births each year in the USA (May et al., 2009; CDC, 2014). Maternal drinking can result in a wide array of physical, cognitive, and behavioral abnormalities in offspring (Kodituwakku et al., 1995; Pei et al., 2011; Rasmussen et al., 2011; Franklin et al., 2008; Irner et al., 2012; Stevens et al., 2012; Mattson et al., 1996; Mattson et al., 1997; Mattson et al., 1999) and these deficits are categorized under the non-diagnostic umbrella term Fetal Alcohol Spectrum Disorders (FASD). Individuals with FASD also have a high rate of life-long secondary disabilities, such as an increased risk of drug dependence and likelihood of incarceration (CDC, 2014; Streissguth et al., 2004). The most severe form of FASD, Fetal Alcohol Syndrome, affects up to 1% of live births per year and manifests with physical aberrations including growth retardation, decreased brain weight, and craniofacial malformations (Mattson et al., 1996; Mattson et al., 2001; CDC, 2014). Caring for children with FASD can cost up to $2 million for medical and caregiver costs across the lifetime (Lupton et al., 2004; Amendah et al., 2011). Therefore, the study of the effects of alcohol exposure in utero as well as possible therapies for alcohol-induced deficits is important. Understanding the cellular and molecular changes that take place following developmental alcohol exposure is paramount in designing behavioral or pharmacological therapies to ameliorate the cognitive and behavioral deficits observed in children with FASD.
Alcohol exposure during the third trimester-equivalent (first two postnatal weeks in rodent models; Dobbing & Sands, 1979) has a significant impact on the structure and function of various brain regions forming during this time, including the hippocampus and prefrontal cortex. During this sensitive time window, known as the brain growth spurt, these brain regions undergo critical developmental changes, including massive neurogenesis, synaptogenesis, cell migration, and gliogenesis. Studies using rodent models of FASD have demonstrated that third trimester-equivalent alcohol exposure causes waves of apoptosis of post-mitotic cortical and hippocampal neurons (Ikonomidou et al., 2000; Olney et al., 2002), in addition to long-lasting reductions in hippocampal CA1 pyramidal cell number, impaired induction of CA1 long-term potentiation (LTP), decreased neuronal activation as measured by expression of the protein c-Fos, impaired survival of newly generated dentate gyrus granule cells, and alterations to mature granule cell morphology (Murawski et al., 2012; Puglia et al. 2010a, b; Hamilton et al., 2010; Hamilton et al., under revision; Whitcher et al., 2008; Tran et al., 2003; Livy et al., 2003; Klintsova et al., 2007; Gil-Mohapel et al., 2011; Hamilton et al., 2012). Targeting molecular changes immediately following alcohol exposure might prevent long-term brain damage.
One potential pathway through which developmental alcohol exposure might affect short- and long-term neuroplasticity is via alterations to levels of brain-derived neurotrophic factor (BDNF). BDNF is critical for normal cellular maturation processes, including proliferation, migration, dendritic arborization, synaptogenesis, and induction and maintenance of LTP (Chan et al., 2008; Tolwani et al., 2002; McAllister et al., 1997; Lu et al., 1999; Hall et al., 2000; Alonso et al., 2002; Heldt et al., 2007). Developmental alcohol exposure has been shown to alter BDNF protein levels in various brain regions, including the hippocampus, prefrontal and motor cortex (Heaton et al., 2000; Heaton et al., 2003; Feng et al., 2005; Barbier et al., 2008; Caldwell et al., 2008; Fattori et al., 2008); though, the nature of these effects (i.e. increases or decreases) are dependent on alcohol dose, exposure window, brain region, and timing of analysis, suggesting a developmental time-specific vulnerability. Most studies have assessed either only BDNF protein or mRNA expression, making the outcomes incomplete and comparison across studies difficult. Additionally, investigating expression of BDNF’s high-affinity tyrosine kinase receptor tropomyosin-related kinase B (TrkB; also the receptor for NT-4) in the same paradigm is necessary to better understand possible cellular mechanisms of aberrant plasticity after developmental alcohol exposure. Receptor expression or function might also be affected by developmental alcohol exposure, potentially influencing downstream signaling pathways involved in neuroplasticity and learning and memory.
Various routes of administration are used in models of developmental alcohol exposure, including intraperitoneal injection, alcohol vapor inhalation, maternal voluntary drinking paradigms, and administration via intragastric gavage of the dam or pups (reviewed in Patten et al., 2014). The suitability of each method is based on the timing, dose and pattern of alcohol exposure being modeled. The current study employs intragastric intubation of the pups from postnatal days 4–9 to mimic alcohol exposure during the third trimester of human pregnancy. Intubation allows for precise dosing of each pup based on individual weight in a physiologically valid way (via absorption through the gastrointestinal tract), while also achieving high, binge-like blood alcohol concentrations (300–400 mg/dl using our model). Since the intubation procedure is quite invasive and stressful for the pups, a sham-intubated group is used as a stress control. These animals are intubated without administration of any liquid for the same duration and number of times as the alcohol-treated pups. Previous work has shown that sham-intubation does not increase cortical and cerebellar apoptosis or cell loss in the same manner as alcohol exposure (Goodlett et al., 1998; Bonthius et al., 2001; Green et al., 2002; Tran et al., 2003) and is regarded as less stressful compared to previously used postnatal models (e.g., artificial rearing or “pup in a cup” method described in West, 1993; Dominguez & Thomas, 2008). In recent years, the role of stress in early development has been demonstrated to be detrimental on a wide variety of neuroplasticity measures, making the consideration of stress in the current intubation paradigm more essential than ever. The impact of intubation stress on subtle neuroanatomical and behavioral measures is critical for the understanding of this method and researchers’ continuing mission to develop models which most closely mimic the human condition.
The current study investigates whether third trimester-equivalent binge-like alcohol exposure or intubation stress alone (PD4–9) affect BDNF and TrkB receptor protein levels in the hippocampus and frontal cortex and bdnf gene expression in the hippocampus of infant rats (PD10). Additionally, as we were interested in exploring the degree of stress caused by the intubation method and how this stress might affect experimental findings, we measured plasma corticosterone levels on PD10. This study broadens our knowledge regarding whether developmental alcohol exposure and intubation stress similarly and differentially affect BDNF production during infancy and provides important information to researchers using a similar alcohol exposure model concerning the effect on glucocorticoids.
Methods
Animals
Timed pregnant Long-Evans rat dams were obtained from Harlan Laboratories (Indianapolis, IN) and housed in standard cages (17 cm high x 145 cm long x 24 cm wide) in a 12/12 hr light cycle (lights on at 9:00 AM) upon arrival. On postnatal day (PD) 3, litters were culled to eight pups each with 6 male, 2 female when possible (achieved in 10/16 litters). On PD4, pups were randomly assigned to one of three experimental groups: suckle control (SC), sham-intubated (SI) or alcohol exposed (AE), using a split-litter design so that SI and AE animals were represented in the same litter. Following the alcohol exposure procedure, pups were left undisturbed with the dam until sacrifice (PD10). In total, 84 male rat pups were generated for the current study (16 litters). Due to the capricious nature of the TrkB Enzyme-linked Immunosorbant Assay (ELISA) kit, some tissue was lost due to kit failure, resulting in a need for increased litters of animals to be generated for this assay; thus, the specific n’s for each experimental condition used for each assay are listed on the x-axis of each figure. All procedures were carried out in accordance with NIH Animal Care Guidelines and the animal use protocol approved by University of Delaware Institutional Animal Care and Use Committee.
Neonatal Alcohol Exposure Paradigm
On PD4–9, AE pups were exposed to alcohol in a binge-like manner (5.25 g/kg/day) via intragastric intubation as described previously (Figure 1A; Klintsova et al., 2007; Helfer et al., 2009). This alcohol dose mimics heavy binge drinking during the third trimester of human pregnancy. Alcohol was administered in an 11.9% v/v milk solution in two doses, two hours apart (9:00 and 11:00 AM). On PD4, two supplemental doses of milk formula were administered, two and four hours following the second alcohol dose as a caloric supplement for the AE pups. For PD5–9, milk formula was administered only once, two hours following the second alcohol exposure. A total of 6 male AE pups died during or shortly following the intubation procedure. Sham-intubated (SI) pups did not receive any milk or alcohol solution during the intubation procedure as delivering milk was previously demonstrated to result in significant weight gain above the norm (Goodlett & Johnson, 1997). SC animals were left undisturbed with the dam apart from daily weighing (PD4–9).
Figure 1.
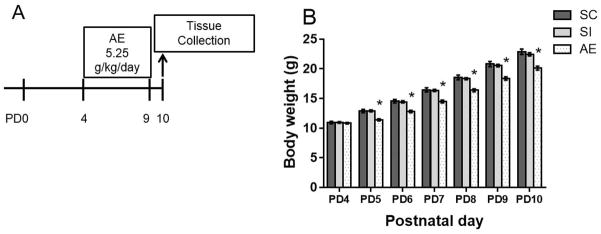
Experimental timeline and body weights. A) On PD 4-9, rats were exposed to 5.25 g/kg/day ethanol via intragastric intubation and sacrificed on PD10. AE: Alcohol-exposed; SI: Sham-intubated; SC: Suckle control. B) Average body weight ± SEM are shown for each day during the neonatal treatment period (PD4-9) as well as the day of sacrifice for alcohol-exposed (AE), sham-intubated (SI), and suckle controls (SC). AE animals weighed significantly less than SI and SC animals on PD5–10. * p < 0.05.
Blood Alcohol Concentrations (BACs)
On PD4, blood samples were obtained from AE pups via tail clip for BAC analysis. Blood was collected 90 minutes following the second alcohol exposure. Samples from the AE group were centrifuged (15,000 rpm/15 minutes) and the plasma collected and stored at −20°C. Plasma was analyzed for BAC using an Analox GL5 Alcohol Analyzer (Analox Instruments, Boston, MA). Blood samples were also taken from SI group at the same time to control for stress of the blood collection procedure, but were not analyzed for BACs.
Tissue and Plasma Collection
Animals were sacrificed via rapid decapitation on PD10 (24 hrs after the last alcohol exposure), and conditions of the animals were counterbalanced across the day to account for possible diurnal changes in corticosterone and BDNF. The brains were quickly removed and sectioned using a 1-mm brain matrix. For ELISA, hippocampus from one hemisphere (both dorsal and ventral, left/right counterbalanced) was removed from each animal, weighed, and stored at −20°C until lysation. The rest of the brain was flash -frozen on untreated slides using −20°C 2-methylbutane and stored at −80°C until processing for gene expression and ELISA assays. During sacrifice, trunk blood was collected for corticosterone assays. Samples were kept on ice until they were centrifuged at 4°C for 25 minutes, after which plasma was collected and stored at −20°C.
Gene Expression Assays
The hippocampus (both dorsal and ventral) was dissected on dry ice and homogenized. DNA/RNA were extracted (Qiagen Inc., Valencia, Calif., USA) and stored at −80°C following quantification and analysis of nucleic acid quality using spectrophotometry (NanoDrop 2000). Reverse transcription was performed using a cDNA synthesis kit (Qiagen) on RNA, and cDNA was amplified by real-time PCR (Bio-Rad CFX96) with Taqman probes (Applied Biosystems) to target bdnf total mRNA (exon IX) and exon I- and IV-specific transcripts. Tubulin was used as a reference gene based on analysis showing no differences in tubulin expression between neonatal treatment groups in these experiments. All reactions for each gene target and reference were run in triplicate. Product specificity was verified using gel electrophoresis.
ELISA Assays
Hippocampal and cortical tissue was homogenized in 4X cell lysis buffer (20mM Tris-HCL [pH 7.5], 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1mM β-glycerophosphate, 1 mM Na3VO4, 1μg/ml leupeptin; Cell Signaling Technology) and 1X protease and phosphatase inhibitor cocktail (ThermoScientific). The final concentrations of the homogenized tissue were 1:8 for hippocampus and 1:7 for cortex. The lysate was then centrifuged (25 min at 15,000 rpm at 4°C) and the supernatant collected for analysis. Kits used for each analysis were Chemikine BDNF Sandwich ELISA kit (Millipore, Billerica, MA), Rat Neurotrophic Tyrosine Kinase Receptor Type 2 ELISA kit (NovateinBio, Woburn, MA), and Corticosterone ELISA kit (Enzo Life Sciences, Farmingdale, NY). Total brain protein was detected with Coomassie Protein Assay (ThermoScientific). Tissue and plasma were diluted and processed according to the manufacturer’s specifications and suggestions for each kit. All plates were read using a spectrophotometer (Tecan Infinite F50). Data were expressed as pg target protein per mg of total brain protein for tissue and pg corticosterone per mL for plasma. Results were then combined as percent of control between plates.
Statistical Analyses
Weights for each day of dosing (PD4-9) and day of tissue harvest (PD10) were averaged across neonatal condition for each day and analyzed using a repeated-measures analysis of variance (ANOVA) followed by post hoc tests when appropriate. For blood alcohol concentrations, average PD4 BACs for each animal (two or three analyses run per animal) ± standard error of the mean (SEM) are reported as mg/dl. For BDNF and TrkB protein data, pg target protein/mg total brain protein data were transformed into percent of control between plates to control for differences in the ELISA kit standards. Inconsistent optical density values (>30% difference between duplicate wells) were removed from the analyses. Differences in BDNF and TrkB protein data were analyzed using one-way ANOVAs with appropriate post hoc tests run as needed (Tukey’s). For gene expression data, the comparative Ct method was used to obtain the relative fold change of experimental (AE or SI) versus the average of controls (SC) per plate (Livak & Schmittgen, 2001). Comparisons between the experimental groups were performed using unpaired t-tests and comparisons between the experimental (AE and SI) and the control group (SC) were analyzed with a Wilcox Signed Rank Test (hypothetical value set to 1.0). A mean value of 1 would indicate no change in transcript level in comparison to the suckle control group. For plasma corticosterone data, an independent-samples Kruskal-Wallis test was run to account for unequal variance between groups with post hoc pairwise comparisons. Differences were considered to be statistically significant at p < 0.05 and nonsignificant trends at p < 0.1 are also reported. For all studies, outliers were identified using Grubb’s test. All statistical analyses were run using Prism 6 software (GraphPad Inc.).
Results
Weights
All neonatal treatment groups gained weight throughout the treatment period. A two-way ANOVA revealed a significant interaction between postnatal day and neonatal treatment (F(12, 567) = 2.413, p < 0.01; Figure 1B). In addition, there were main effects of both postnatal day (F(6,567) = 556.3, p < 0.001) and neonatal treatment (F(2, 567)= 78.66, p < 0.001). While animals from all neonatal treatments did not differ in weight on PD4 (means ± SD: SC: 10.9g ±1.2, SI: 10.9g±0.7, AE: 10.8g±0.7), Tukey’s HSD post hoc test revealed that AE animals weigh significantly less starting on PD5 (p < 0.05, Figure 1B). By PD10, AE animals weighed an average of 2 g less compared to both SC and SI (SC: 22.9g±2.7, SI: 22.4g±1.4, AE: 20.1g±1.7). Similar decreases in AE weights have been reported using this model of alcohol exposure. The effects on weight are temporally limited, as we have shown in other studies that AE animals do not differ in weight from SC and SI rats when assessed on PD30 (Hamilton et al., 2012; Boschen et al., 2014). However, the role of under-nutrition should not be discounted when interpreting the results of the current study.
BACs
AE animals had an average BAC concentration of 366.4 mg/dl (± 84.1 SD) on PD4–90 min following the second alcohol dose. This value is consistent with previous work published from our lab using the same alcohol exposure paradigm (Hamilton et al., 2012, Boschen et al., 2014).
BDNF and TrkB protein - hippocampus and cortex
BDNF protein levels were measured from PD10 hippocampal and frontal cortex tissue. For hippocampus, a one-way ANOVA revealed a main effect of neonatal condition (F(2,36) = 4.833, p = 0.014, Figure 2A). Post hoc tests (Tukey’s HSD) showed that AE pups had significantly increased BDNF protein compared to the SC group (p = 0.016), while SI exhibited a trending effect vs. SC (p = 0.074) but did not differ significantly from AE pups (p = 0.9). No effect of neonatal treatment was found in the frontal cortex (Figure 2B, p = n.s.).
Figure 2.

BDNF and TrkB protein in the hippocampus and cortex on PD10. A) Alcohol-exposed (AE) animals had significantly elevated BDNF protein compared to suckle control (SC) animals. Sham-intubated (SI) animals showed a trending increase in BDNF protein compared to the SC group (p = 0.074) but were not significantly different from AE pups. B) No effect of neonatal condition was found in frontal cortex. C) AE animals had increased TrkB protein in the hippocampus relative to SI animals, but not SC pups. No change was found in frontal cortex (D). * p < 0.05. Data is shown as mean percent of control ± SEM.
TrkB protein levels were also measured from PD10 hippocampal and frontal cortex tissue. For hippocampus, a one-way ANOVA revealed a main effect of neonatal condition (F(2,34) = 4.875, p = 0.014, Figure 2C) showing that AE pups had significantly higher TrkB protein levels compared to SI animals (p = 0.01), but not from SC animals (p = 0.12). Similar to the BDNF protein results, there was no effect of neonatal treatment on TrkB protein in the frontal cortex (Figure 2D, p = n.s.).
BDNF gene expression – hippocampus
Total bdnf gene expression (by targeting all exon-IX containing transcripts) was measured in the hippocampus on PD10. Gene expression assays were only conducted using hippocampal tissue due to the lack of significant effects of neonatal treatment in frontal cortex on BDNF or TrkB protein. Total bdnf gene expression was increased in both the AE and SI groups compared to the SC group on PD10 (p = 0.019 and 0.034 for AE and SI vs. SC, respectively; Figure 3A), and a t-test revealed that the AE and SI groups did not differ from one another (p = 0.28). We also examined exon I- and IV-specific transcripts as expression of these mRNAs are vulnerable to early-life experiences (Sathanoori et al., 2004; Nair et al., 2011). For exon I-specific transcripts, only AE pups had significantly increased gene expression compared to SC pups (p = 0.027; Figure 3B), but did not significantly differ from SI (p = 0.29). For exon IV-specific transcripts, gene expression was significantly increased in AE animals (p = 0.027; Figure 3C) and there was a trending effect found in the SI group (p = 0.09). AE and SI groups did not differ from one another (p = 0.90).
Figure 3.
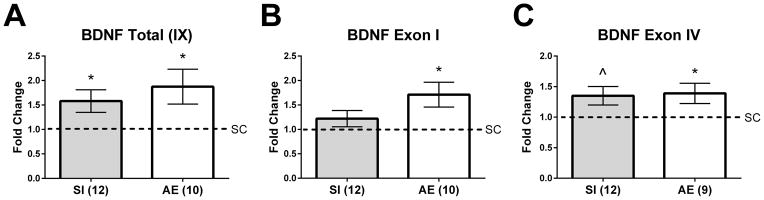
BDNF gene expression in the PD10 hippocampus. A) Alcohol-exposed (AE) and sham-intubated (SI) animals exhibited an increase in total bdnf gene expression in the hippocampus. B) and C) Exon I- and IV-specific transcripts were significantly elevated in AE animals. A trending increase was also found for exon IV-specific transcript gene expression in SI pups relative to controls (p = 0.09, indicated as ^ on graph). * p < 0.05. Data is expressed as a fold change from the suckle control (SC) group (shown as 1 on graphs).
Corticosterone - total corticosterone and correlation
Corticosterone was measured from PD10 plasma to assess the role that stress might play in the current results. Trunk blood was collected at the time of sacrifice for all generated animals, making it possible to analyze corticosterone levels for all the cohorts generated for these experiments. An independent-samples Kruskal-Wallis test revealed a main effect of neonatal treatment (χ2(2) = 13.576, p < 0.001; Figure 4), with AE and SI animals showing significantly increased corticosterone levels compared to the SC group (p < 0.001 and p = 0.045, respectively) but not differing from one another (p = 0.924). These results support the use of sham intubation as a stress control for the alcohol dosing procedure. Corticosterone levels have a diurnal pattern, with levels increasing throughout the progression of the light cycle (Ader, 1969; Dauchy et al., 2010). To account for these variations, corticosterone levels were correlated with time of day (specifically, minutes since lights were turned on for the day: 9 AM). Both the SC and SI pups showed significant correlations between time of day and corticosterone levels (SC: Pearson’s r = 0.5716, p < 0.01; SI: Pearson’s r = 0.5582, p < 0.05; Figure 5), while AE pups did not show a correlation (Pearson’s r = −0.1774, p = n.s.), suggesting a disrupted diurnal corticosterone rhythm in the AE group.
Figure 4.

Plasma corticosterone on PD10. Both alcohol-exposed (AE) and sham-intubated (SI) animals showed an increase in corticosterone compared to the suckle control (SC) group. * p < 0.05; ** p < 0.01. SI and AE did not statistically differ from one another. Data is shown as mean percent of control ± SEM.
Figure 5.

Plasma corticosterone and time of sacrifice correlations. Significant correlations between time of day (minutes since lights on at 9 AM) and corticosterone levels were observed in suckle control (SC) and sham-intubated (SI) pups, but there was no correlation observed in alcohol-exposed (AE) animals. Pearson’s r value given for each correlation. SC and SI: p < 0.01; AE: p = n.s.
Discussion
This study has shown that third trimester-equivalent binge-like alcohol exposure (AE) and sham intubation (SI) both influence expression of brain-derived neurotrophic factor (BDNF) in the neonatal brain. However, some measures, such as receptor and gene expression, were affected by alcohol exposure alone. Specifically, both AE and SI groups showed increases of BDNF protein expression in the hippocampus, but not the frontal cortex, of PD10 rats. TrkB receptor expression in the hippocampus was only increased in the AE animals on PD10 compared to the SI pups, suggesting that the increase in TrkB receptors is specific to AE animals at this time point. Supporting the observed increases in BDNF protein in these two groups, both AE and SI pups showed significant increases in bdnf total gene expression in the hippocampus. Interestingly, while both groups also showed increases in exon IV-specific transcripts (increase was p = 0.09 in SI vs. SC), increases in exon I-driven gene expression were specific to AE pups, suggesting a differential effect of alcohol exposure on exon-specific bdnf gene transcription. To our knowledge, these results are the first to examine BDNF mRNA, protein, and TrkB receptor levels using the same model of alcohol exposure. Analysis of plasma corticosterone levels showed that both the AE and SI groups have significant increased levels of corticosterone compared to the SC group. Overall, these findings suggest that exposure to alcohol or to stress from experimental manipulation during development alters BDNF and receptor levels in the hippocampus.
Expression of the bdnf gene is regulated via transcription starting at one of at least nine promoter regions (Timmusk et al., 1993; Timmusk et al., 2005; Aid et al., 2007; Liu et al., 2007; Pruunsild et al., 2007). Consistent with the protein data (Figure 2A), the current study found that both postnatal alcohol exposure and sham intubation increased total bdnf gene expression (Figure 3A). Interestingly, exon I-specific transcription was not increased to the same degree in SI pups as compared to AE animals (exon IV-specific expression was also not statistically significant from the SC group at p = 0.09), suggesting that exon I did not contribute as much to the overall increases in bdnf gene expression in the SI group compared to the AE condition. While both the AE and SI groups exhibit increased BDNF production, the transcripts through which the protein is generated differs between the conditions. Both exons I and IV have been shown to regulate experience-dependent BDNF release, and transcription can be differentially affected based on the type of stressor, brain region, and age of the animal (Metsis et al., 1993; Oliff et al., 1998; Nair et al., 2007; Suri et al., 2013). The gene expression increase in SI animals is consistent with previous literature reporting transient increases in hippocampal bdnf mRNA following maternal deprivation stress (Roceri et al., 2004).
The findings of increased BDNF protein in the hippocampus following PD4-9 alcohol exposure are consistent with previous literature investigating the effects of developmental alcohol exposure on BDNF and other neurotrophins in various regions of the brain. Both pre- and postnatal alcohol exposure paradigms have been found to increase hippocampal NGF (Angelucci et al., 1997; Heaton et al., 2000). Heaton and colleagues (2000) reported increased BDNF protein in the hippocampus on PD10 immediately following cessation of a week-long alcohol exposure paradigm. Interestingly, previous work with postnatal models has suggested a temporally-based fluctuation in cortical BDNF protein levels during the hours following a single alcohol exposure, with BDNF increasing immediately, decreasing after two hours, and then increasing again after 12 hours (Heaton et al., 2003). Temporally-specific alterations to BDNF levels following developmental alcohol exposure, could shed light on the functional significance of BDNF at each time point, particularly when taken together with levels of other pro-apoptotic and pro-survival molecules. More tissue collection time points are needed to fully address this question. If BDNF levels do change based over time since the exposure, it would imply that in our model, BDNF levels return to baseline within 24 hours in the cortex but remain elevated in the hippocampus.
BDNF and its specific receptor TrkB are critical for proper neural development and have distinct developmental timeline of expression, with increasing expression following the maturation of brain structures: the earlier maturing rodent brain regions (brainstem, midbrain) start to demonstrate the higher levels of expression during the first postnatal days (e.g., Maisonpierre et al., 1990, Fryer et al., 1996). BDNF mediates neuronal differentiation during development (Patapoutian & Reichard, 2001) and is required for the terminal differentiation of new neurons (Chan et al., 2008). Balanced expression of BDNF during critical periods in development is essential for synaptic maturation as BDNF potentiates excitatory and attenuates inhibitory synaptic transmission by acting on presynaptic terminals (Carvalho et al., 2008; Gao et al., 2014) and postsynaptic receptors (Tanaka et al., 1997; Brunig et al., 2001). BDNF may also play a vital role in mediating processes associating early life environment with brain development and behavior (Branchi et al., 2006a,b; Branchi et al., 2013). Thus, alterations to BDNF and TrkB signaling during development could result in long-lasting neuroplastic consequences. Furthermore, aberrant BDNF signaling could represent either a neuroprotective mechanism or a sign of dysregulation within the neurotrophic factor “push-pull” synchrony (McAllister et al., 1997).
The simultaneous increase in BDNF and TrkB protein in the hippocampus following postnatal alcohol exposure might represent a compensatory protective mechanism in which the brain responds to the damaging effects of alcohol through upregulation of growth factors. Elevated BDNF levels in hippocampus of sham-intubated animals could represent protective response to stress of intubation, however this increase was not accompanied by an upregulation in TrkB receptor. Increased release of neurotrophins have been reported following other types of brain trauma, including experimental models of traumatic brain injury and seizures (Dekosky et al., 1994; Hicks et al., 1997; Rudge et al., 1998). Postnatal alcohol exposure has also been shown to transiently increase BDNF protein levels in the hippocampus immediately following the last alcohol exposure (Heaton et al., 2000). The current results show that postnatal alcohol exposure also increases TrkB receptor expression and that upregulated levels of BDNF and TrkB persist for at least 24 following the last ethanol administration. As neurotrophins play a critical role in neuronal proliferation and survival and induce dendritic reorganization, upregulation of these proteins could potentially offset alcohol-induced neurotoxic and apoptotic effects. Some evidence supports a role for BDNF in neuroprotection (Heaton et al., 1994; Mitchell et al., 1999; Nagahara & Tuszynski, 2011), though exogenously administered BDNF does not necessarily result in therapeutic effects (Rudge et al., 1998; Nagahara & Tuszynski, 2011). The lack of upregulation of BDNF and TrkB in the frontal cortex as compared to the hippocampus could indicate differential vulnerabilities of these two brain structures to alcohol-induced insult. However, any deviation from normal levels of BDNF could result in detrimental effects, as many neurotrophic factors and other growth hormones act in synchrony and levels are precisely developmentally-timed. Prolonged overexpression of neurotrophins, such as BDNF, could result in aberrant synaptic connectivity (Miller & Al-Rabiai, 1994), dendritic arborization, or even increased apoptosis via activation of the p75 receptor (Casaccia-Bonnefil et al., 1996). While not systematically studied in the current experiment, the potential role of withdrawal effects should also be taken into consideration with the current results, given that animals were sacrificed 24 hours following the last alcohol exposure and physiological signs and symptoms of withdrawal start as soon as six hours following alcohol in adult animals (Becker, 2000). Alcohol withdrawal effects are thought to be in part due to neuronal excitotoxicity following a compensatory upregulation of NMDA receptors following exposure (Young et al., 2010, Idrus et al., 2014), which might contribute “rebound hyperexcitability” in the hippocampus of human alcoholics (described in Becker, 2000). CNS hyperexcitability could play a role in the observed increase in BDNF and TrkB in AE pups in the current study.
The present study found that both AE and SI animals displayed increased levels of plasma corticosterone compared to undisturbed animals and AE pups had disrupted diurnal corticosterone patterns. The PD10 age point falls within the “stress hyporesponsive” period, which occurs during the first two weeks of postnatal life and is characterized by low baseline glucocorticoids plasma levels, attenuated hormonal responses to acute stress, impaired negative feedback of circulating corticosterone, and a 3x longer half-life of corticosterone (Lupien et al., 2009; Sapolsky & Meaney, 1986). In the current study, two main factors could have resulted in the observed increase in corticosterone levels in the AE and SI pups: binge-like alcohol exposure and the stress of the intubation paradigm. While limited literature has examined the short-term impact of postnatal alcohol exposure on plasma corticosterone, neonatal rats prenatally exposed to alcohol are known to exhibit elevated basal levels of corticosterone in plasma and brain and exhibit a blunted stress response (Weinberg, 1989; Taylor et al., 1986). Furthermore, prenatally alcohol-exposed animals are hyper-responsive to stress later in life, which can manifest as hypersecretion of adrenocorticotropic hormone (ACTH) and corticosterone or delayed return to basal levels after acute stressors (Kim et al., 1999). Early-life stress, such as experiencing the intubation procedure, also affects the stress response in neonatal rats, either through short-term increases in corticosterone and ACTH or long-term alterations to the hypothalamic-pituitary-adrenal (HPA) axis (Walker et al., 1991; Weinstock et al., 2009). While short durations of separation or handling (<15 min) speed recovery of corticosterone following a later stressful event (Ader, 1970), prolonged separation increases and extends stress-induced elevations of circulating corticosterone (Levine et al., 1991; Gilles et al., 1996; McCormick et al., 1998). The timing of the increase in corticosterone levels is characteristic of an impaired effective termination mechanism negative feedback system. Thus, early exposures to stressors, such as the intubation procedure, might prime the HPA axis to respond more strongly to minor stressors later in life, such as brief maternal separation or siblings being removed from the home cage (Gilles et al., 1996; McCormick et al., 1998). The close temporal proximity between the last intubations and sacrifice suggests that stress plays an important role in the interpretation of the results.
In summary, third trimester-equivalent binge-like alcohol exposure and intubation stress effect neuroplasticity in overlapping and distinct ways. Alcohol exposure increases BDNF and TrkB receptor protein in the hippocampus, but not in frontal cortex, 24 hours following the final alcohol exposure (PD10). Sham intubation results in a trending increase in BDNF protein in the hippocampus, but does not affect TrkB protein expression. Bdnf total gene expression is also increased at this time point in both AE and SI pups. The increase in total gene expression in SI and AE animals might be driven through different promoter regions, as AE animals showed an increase in exon I-specific transcripts while SI animals did not. These data suggest a reactive increase in BDNF following exposure to a teratogen or intubation stress, potentially as a neuroprotective mechanism. The temporal pattern of the BDNF and TrkB upregulation needs to be further investigated. Basal plasma corticosterone was also elevated in AE and SI animals and the diurnal cycling pattern seems to be disrupted in the AE group. The long-term impact of developmental alcohol exposure on stress hormone function, both at basal levels and in response to novel stressors, remains to be elucidated. Continual efforts to understand and refine animal models of human disorders is a critical scientific process. While the intragastric intubation method of alcohol administration is a valid model of FASD on many measures, it’s more subtle effects on cells were not originally known and it seems that some neuroplastic measures may be impacted by stress alone using this technique. The results of the current study were unexpected, but highlight that the potential effects of intubation stress should not be ignored – in fact, the opposite: future studies should investigate how stress might play a role in the observed neuroplastic or behavioral changes so the intubation procedure can be altered to reduce stress effects. Overall, these data give important insight into how the brain responds to teratogenic insult in the short-term, as well as critical information regarding how this model of alcohol exposure affects the HPA axis and stress hormone production.
Highlights.
We use a model of third trimester-equivalent binge alcohol exposure in rats.
We investigated levels of BDNF and TrkB in the neonatal hippocampus and cortex.
Neonatal alcohol increased bdnf exon I mRNA and TrkB protein in the hippocampus.
Alcohol and intubation raised hippocampal BDNF protein and total and exon IV mRNA.
Plasma corticosterone was increased in alcohol-exposed and sham-intubated animals.
Acknowledgments
The authors would like to thank Jennifer Blaze, Tiffany Doherty, Shaqran Shareeq, and Zubin Hussain for their assistance with the gene expression assays, spectrophotometry, and gel electrophoresis, and all undergraduate research assistants for help with animal generation and care. This work was supported by the National Institutes of Health/NIGMS COBRE: The Delaware Center for Neuroscience Research 1P20GM103653 – 01A1 to AYK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ader R. Early experiences accelerate maturation of the 24-hour adrenocortical rhythm. Science. 1969;163(3872):1225–1226. doi: 10.1126/science.163.3872.1225. [DOI] [PubMed] [Google Scholar]
- Ader R. The effects of early experience on the adrenocortical response to different magnitudes of stimulation. Physiol Behav. 1970;5(8):837–839. doi: 10.1016/0031-9384(70)90168-x. [DOI] [PubMed] [Google Scholar]
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85(3):525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Vianna MR, Depino AM, Mello e Souza T, Pereira P, Szapiro G, Viola H, Pitossi F, Izquierdo I, Medina JH. BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus. 2002;12(4):551–560. doi: 10.1002/hipo.10035. [DOI] [PubMed] [Google Scholar]
- Amendah DD, Grosse SD, Bertrand J. Medical expenditures of children in the United States with fetal alcohol syndrome. Neurotoxicol Teratol. 2011;33(2):322–324. doi: 10.1016/j.ntt.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Angelucci F, Cimino M, Balduini W, Piltillo L, Aloe L. Prenatal exposure to ethanol causes differential effects in nerve growth factor and its receptor in the basal forebrain of preweaning and adult rats. J Neural Transplant Plast. 1997;6(2):63–71. doi: 10.1155/NP.1997.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier E, Pierrefiche O, Vaudry D, Vaudry H, Daoust M, Naassila M. Long-term alterations in vulnerability to addiction to drugs of abuse and in brain gene expression after early life ethanol exposure. Neuropharmacology. 2008;55(7):1199–1211. doi: 10.1016/j.neuropharm.2008.07.030. [DOI] [PubMed] [Google Scholar]
- Becker HC. Animal models of alcohol withdrawal. Alcohol Res Health. 2000;24(2):105–113. [PMC free article] [PubMed] [Google Scholar]
- Bonthius DJ, Woodhouse J, Bonthius NE, Taggard DA, Lothman EW. Reduced seizure threshold and hippocampal cell loss in rats exposed to alcohol during the brain growth spurt. Alcohol Clin Exp Res. 2001;25(1):70–82. [PubMed] [Google Scholar]
- Boschen KE, Hamilton GF, Delorme JE, Klintsova AY. Activity and social behavior in a complex environment in rats neonatally exposed to alcohol. Alcohol. 2014;48(6):533–541. doi: 10.1016/j.alcohol.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchi I, Curley JP, D’Andrea I, Cirulli F, Champagne FA, Alleva E. Early interactions with mother and peers independently build adult social skills and shape BDNF and oxytocin receptor brain levels. Psychoneuroendocrinology. 2013;38(4):522–532. doi: 10.1016/j.psyneuen.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchi I, D’Andrea I, Fiore M, Di Fausto V, Aloe L, Alleva E. Early social enrichment shapes social behavior and nerve growth factor and brain-derived neurotrophic factor levels in the adult mouse brain. Biol Psychiatry. 2006;60(7):690–696. doi: 10.1016/j.biopsych.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Branchi I, D’Andrea I, Sietzema J, Fiore M, Di Fausto V, Aloe L, Alleva E. Early social enrichment augments adult hippocampal BDNF levels and survival of BrdU-positive cells while increasing anxiety- and “depression” like behavior. J Neurosci Res. 2006;83(6):965–973. doi: 10.1002/jnr.20789. [DOI] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur J Neurosci. 2001;13(7):1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Caldwell KK, Sheema S, Paz RD, Samudio-Ruiz SL, Laughlin MH, Spence NE, Roehlk MJ, Alcon SN, Allan AM. Fetal alcohol spectrum disorder-associated depression: evidence for reductions in the levels of brain-derived neurotrophic factor in a mouse model. Pharmacol Biochem Behav. 2008;90(4):614–624. doi: 10.1016/j.pbb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AL, Caldeira MV, Santos SD, Duarte CB. Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br J Pharmacol. 2008;153(Suppl 1):S310–324. doi: 10.1038/sj.bjp.0707509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383(6602):716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- CDC. Fetal Alcohol Spectrum Disorders (FASDs) 2014 [Google Scholar]
- Chan JP, Cordeira J, Calderon GA, Iyer LK, Rios M. Depletion of central BDNF in mice impedes terminal differentiation of new granule neurons in the adult hippocampus. Mol Cell Neurosci. 2008;39(3):372–383. doi: 10.1016/j.mcn.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauchy RT, Dauchy EM, Tirrell RP, Hill CR, Davidson LK, Greene MW, Tirrell PC, Wu J, Sauer LA, Blask DE. Dark-phase light contamination disrupts circadian rhythms in plasma measures of endocrine physiology and metabolism in rats. Comp Med. 2010;60(5):348–356. [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Goss JR, Miller PD, Styren SD, Kochanek PM, Marion D. Upregulation of nerve growth factor following cortical trauma. Exp Neurol. 1994;130(2):173–177. doi: 10.1006/exnr.1994.1196. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3(1):79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Dominguez HD, Thomas JD. Artificial rearing. Methods Mol Biol. 2008;447:85–100. doi: 10.1007/978-1-59745-242-7_7. [DOI] [PubMed] [Google Scholar]
- Fattori V, Abe S, Kobayashi K, Costa LG, Tsuji R. Effects of postnatal ethanol exposure on neurotrophic factors and signal transduction pathways in rat brain. J Appl Toxicol. 2008;28(3):370–376. doi: 10.1002/jat.1288. [DOI] [PubMed] [Google Scholar]
- Feng MJ, Yan SE, Yan QS. Effects of prenatal alcohol exposure on brain-derived neurotrophic factor and its receptor tyrosine kinase B in offspring. Brain Res. 2005;1042(2):125–132. doi: 10.1016/j.brainres.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Franklin L, Deitz J, Jirikowic T, Astley S. Children with fetal alcohol spectrum disorders: problem behaviors and sensory processing. Am J Occup Ther. 2008;62(3):265–273. doi: 10.5014/ajot.62.3.265. [DOI] [PubMed] [Google Scholar]
- Gao M, Maynard KR, Chokshi V, Song L, Jacobs C, Wang H, Tran T, Martinowich K, Lee HK. Rebound potentiation of inhibition in juvenile visual cortex requires vision-induced BDNF expression. J Neurosci. 2014;34(32):10770–10779. doi: 10.1523/JNEUROSCI.5454-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles EE, Schultz L, Baram TZ. Abnormal corticosterone regulation in an immature rat model of continuous chronic stress. Pediatr Neurol. 1996;15(2):114–119. doi: 10.1016/0887-8994(96)00153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Mohapel J, Boehme F, Patten A, Cox A, Kainer L, Giles E, Brocardo PS, Christie BR. Altered adult hippocampal neuronal maturation in a rat model of fetal alcohol syndrome. Brain Res. 2011;1384:29–41. doi: 10.1016/j.brainres.2011.01.116. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Pearlman AD, Lundahl KR. Binge neonatal alcohol intubations induce dose-dependent loss of Purkinje cells. Neurotoxicol Teratol. 1998;20(3):285–292. doi: 10.1016/s0892-0362(97)00102-5. [DOI] [PubMed] [Google Scholar]
- Green JT, Tran T, Steinmetz JE, Goodlett CR. Neonatal ethanol produces cerebellar deep nuclear cell loss and correlated disruption of eyeblink conditioning in adult rats. Brain Res. 2002;956(2):302–311. doi: 10.1016/s0006-8993(02)03561-8. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3(6):533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- Hamilton GF, Boschen KE, Goodlett CR, Greenough WT, Klintsova AY. Housing in environmental complexity following wheel running augments survival of newly generated hippocampal neurons in a rat model of binge alcohol exposure during the third trimester equivalent. Alcohol Clin Exp Res. 2012;36(7):1196–1204. doi: 10.1111/j.1530-0277.2011.01726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton GF, Whitcher LT, Klintsova AY. Postnatal binge-like alcohol exposure decreases dendritic complexity while increasing the density of mature spines in mPFC Layer II/III pyramidal neurons. Synapse. 2010;64(2):127–135. doi: 10.1002/syn.20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton MB, Mitchell JJ, Paiva M, Walker DW. Ethanol-induced alterations in the expression of neurotrophic factors in the developing rat central nervous system. Brain Res Dev Brain Res. 2000;121(1):97–107. doi: 10.1016/s0165-3806(00)00032-8. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Paiva M, Madorsky I, Shaw G. Ethanol effects on neonatal rat cortex: comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Brain Res Dev Brain Res. 2003;145(2):249–262. doi: 10.1016/j.devbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Paiva M, Swanson DJ, Walker DW. Responsiveness of cultured septal and hippocampal neurons to ethanol and neurotrophic substances. J Neurosci Res. 1994;39(3):305–318. doi: 10.1002/jnr.490390308. [DOI] [PubMed] [Google Scholar]
- Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12(7):656–670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfer JL, Goodlett CR, Greenough WT, Klintsova AY. The effects of exercise on adolescent hippocampal neurogenesis in a rat model of binge alcohol exposure during the brain growth spurt. Brain Res. 2009;1294:1–11. doi: 10.1016/j.brainres.2009.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks RR, Numan S, Dhillon HS, Prasad MR, Seroogy KB. Alterations in BDNF and NT-3 mRNAs in rat hippocampus after experimental brain trauma. Brain Res Mol Brain Res. 1997;48(2):401–406. doi: 10.1016/s0169-328x(97)00158-7. [DOI] [PubMed] [Google Scholar]
- Idrus NM, McGough NN, Riley EP, Thomas JD. Administration of memantine during withdrawal mitigates overactivity and spatial learning impairments associated with neonatal alcohol exposure in rats. Alcohol Clin Exp Res. 2014;38(2):529–537. doi: 10.1111/acer.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287(5455):1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Irner TB, Teasdale TW, Olofsson M. Cognitive and social development in preschool children born to women using substances. J Addict Dis. 2012;31(1):29–44. doi: 10.1080/10550887.2011.642766. [DOI] [PubMed] [Google Scholar]
- Kim CK, Giberson PK, Yu W, Zoeller RT, Weinberg J. Effects of prenatal ethanol exposure on hypothalamic-pituitary-adrenal responses to chronic cold stress in rats. Alcohol Clin Exp Res. 1999;23(2):301–310. [PubMed] [Google Scholar]
- Klintsova AY, Helfer JL, Calizo LH, Dong WK, Goodlett CR, Greenough WT. Persistent impairment of hippocampal neurogenesis in young adult rats following early postnatal alcohol exposure. Alcohol Clin Exp Res. 2007;31(12):2073–2082. doi: 10.1111/j.1530-0277.2007.00528.x. [DOI] [PubMed] [Google Scholar]
- Kodituwakku PW, Handmaker NS, Cutler SK, Weathersby EK, Handmaker SD. Specific impairments in self-regulation in children exposed to alcohol prenatally. Alcohol Clin Exp Res. 1995;19(6):1558–1564. doi: 10.1111/j.1530-0277.1995.tb01024.x. [DOI] [PubMed] [Google Scholar]
- Levine S, Huchton DM, Wiener SG, Rosenfeld P. Time course of the effect of maternal deprivation on the hypothalamic-pituitary-adrenal axis in the infant rat. Dev Psychobiol. 1991;24(8):547–558. doi: 10.1002/dev.420240803. [DOI] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067(1):1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol. 2003;25(4):447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. 1999;58(1):76–87. [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10(6):434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Lupton C, Burd L, Harwood R. Cost of fetal alcohol spectrum disorders. Am J Med Genet C Semin Med Genet. 2004;127c(1):42–50. doi: 10.1002/ajmg.c.30015. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5(4):501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Goodman AM, Caine C, Delis DC, Riley EP. Executive functioning in children with heavy prenatal alcohol exposure. Alcohol Clin Exp Res. 1999;23(11):1808–1815. [PubMed] [Google Scholar]
- Mattson SN, Riley EP, Delis DC, Stern C, Jones KL. Verbal learning and memory in children with fetal alcohol syndrome. Alcohol Clin Exp Res. 1996;20(5):810–816. doi: 10.1111/j.1530-0277.1996.tb05256.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Riley EP, Gramling L, Delis DC, Jones KL. Heavy prenatal alcohol exposure with or without physical features of fetal alcohol syndrome leads to IQ deficits. J Pediatr. 1997;131(5):718–721. doi: 10.1016/s0022-3476(97)70099-4. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Schoenfeld AM, Riley EP. Teratogenic effects of alcohol on brain and behavior. Alcohol Res Health. 2001;25(3):185–191. [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M, Hoyme HE. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev Disabil Res Rev. 2009;15(3):176–192. doi: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18(5):767–778. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Kehoe P, Kovacs S. Corticosterone release in response to repeated, short episodes of neonatal isolation: evidence of sensitization. Int J Dev Neurosci. 1998;16(3–4):175–185. doi: 10.1016/s0736-5748(98)00026-4. [DOI] [PubMed] [Google Scholar]
- Metsis M, Timmusk T, Arenas E, Persson H. Differential usage of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc Natl Acad Sci U S A. 1993;90(19):8802–8806. doi: 10.1073/pnas.90.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, al-Rabiai S. Effects of prenatal exposure to ethanol on the number of axons in the pyramidal tract of the rat. Alcohol Clin Exp Res. 1994;18(2):346–354. doi: 10.1111/j.1530-0277.1994.tb00024.x. [DOI] [PubMed] [Google Scholar]
- Mitchell JJ, Paiva M, Walker DW, Heaton MB. BDNF and NGF afford in vitro neuroprotection against ethanol combined with acute ischemia and chronic hypoglycemia. Dev Neurosci. 1999;21(1):68–75. doi: 10.1159/000017368. [DOI] [PubMed] [Google Scholar]
- Murawski NJ, Klintsova AY, Stanton ME. Neonatal alcohol exposure and the hippocampus in developing male rats: effects on behaviorally induced CA1 c-Fos expression, CA1 pyramidal cell number, and contextual fear conditioning. Neuroscience. 2012;206:89–99. doi: 10.1016/j.neuroscience.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10(3):209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- Nair A, Vadodaria KC, Banerjee SB, Benekareddy M, Dias BG, Duman RS, Vaidya VA. Stressor-specific regulation of distinct brain-derived neurotrophic factor transcripts and cyclic AMP response element-binding protein expression in the postnatal and adult rat hippocampus. Neuropsychopharmacology. 2007;32(7):1504–1519. doi: 10.1038/sj.npp.1301276. [DOI] [PubMed] [Google Scholar]
- Oliff HS, Berchtold NC, Isackson P, Cotman CW. Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Brain Res Mol Brain Res. 1998;61(1–2):147–153. doi: 10.1016/s0169-328x(98)00222-8. [DOI] [PubMed] [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002;133(2):115–126. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11(3):272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Patten AR, Fontaine CJ, Christie BR. A comparison of the different animal models of fetal alcohol spectrum disorders and their use in studying complex behaviors. Front Pediatr. 2014;2:93. doi: 10.3389/fped.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Job J, Kully-Martens K, Rasmussen C. Executive function and memory in children with Fetal Alcohol Spectrum Disorder. Child Neuropsychol. 2011;17(3):290–309. doi: 10.1080/09297049.2010.544650. [DOI] [PubMed] [Google Scholar]
- Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T. Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics. 2007;90(3):397–406. doi: 10.1016/j.ygeno.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglia MP, Valenzuela CF. Ethanol acutely inhibits ionotropic glutamate receptor-mediated responses and long-term potentiation in the developing CA1 hippocampus. Alcohol Clin Exp Res. 2010;34(4):594–606. doi: 10.1111/j.1530-0277.2009.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglia MP, Valenzuela CF. Repeated third trimester-equivalent ethanol exposure inhibits long-term potentiation in the hippocampal CA1 region of neonatal rats. Alcohol. 2010;44(3):283–290. doi: 10.1016/j.alcohol.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen C, Soleimani M, Pei J. Executive functioning and working memory deficits on the CANTAB among children with prenatal alcohol exposure. J Popul Ther Clin Pharmacol. 2011;18(1):e44–53. [PubMed] [Google Scholar]
- Roceri M, Cirulli F, Pessina C, Peretto P, Racagni G, Riva MA. Postnatal repeated maternal deprivation produces age-dependent changes of brain-derived neurotrophic factor expression in selected rat brain regions. Biol Psychiatry. 2004;55(7):708–714. doi: 10.1016/j.biopsych.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, Anderson K, Lindsay RM, Wiegand SJ. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp Neurol. 1998;149(2):398–410. doi: 10.1006/exnr.1997.6737. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Meaney MJ. Maturation of the adrenocortical stress response: neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res. 1986;396(1):64–76. doi: 10.1016/s0006-8993(86)80190-1. [DOI] [PubMed] [Google Scholar]
- Stevens SA, Majors D, Rovet J, Koren G, Fantus E, Nulman I, Desrocher M. Social problem solving in children with fetal alcohol spectrum disorders. J Popul Ther Clin Pharmacol. 2012;19(1):e99–110. [PubMed] [Google Scholar]
- Streissguth AP, Bookstein FL, Barr HM, Sampson PD, O’Malley K, Young JK. Risk factors for adverse life outcomes in fetal alcohol syndrome and fetal alcohol effects. J Dev Behav Pediatr. 2004;25(4):228–238. doi: 10.1097/00004703-200408000-00002. [DOI] [PubMed] [Google Scholar]
- Suri D, Veenit V, Sarkar A, Thiagarajan D, Kumar A, Nestler EJ, Galande S, Vaidya VA. Early stress evokes age-dependent biphasic changes in hippocampal neurogenesis, BDNF expression, and cognition. Biol Psychiatry. 2013;73(7):658–666. doi: 10.1016/j.biopsych.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17(9):2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AN, Branch BJ, Nelson LR, Lane LA, Poland RE. Prenatal ethanol and ontogeny of pituitary-adrenal responses to ethanol and morphine. Alcohol. 1986;3(4):255–259. doi: 10.1016/0741-8329(86)90034-0. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10(3):475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Tolwani RJ, Buckmaster PS, Varma S, Cosgaya JM, Wu Y, Suri C, Shooter EM. BDNF overexpression increases dendrite complexity in hippocampal dentate gyrus. Neuroscience. 2002;114(3):795–805. doi: 10.1016/s0306-4522(02)00301-9. [DOI] [PubMed] [Google Scholar]
- Tran TD, Kelly SJ. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicol Teratol. 2003;25(5):519–528. doi: 10.1016/s0892-0362(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Walker CD, Scribner KA, Cascio CS, Dallman MF. The pituitary-adrenocortical system of neonatal rats is responsive to stress throughout development in a time-dependent and stressor-specific fashion. Endocrinology. 1991;128(3):1385–1395. doi: 10.1210/endo-128-3-1385. [DOI] [PubMed] [Google Scholar]
- Weinberg J. Prenatal ethanol exposure alters adrenocortical development of offspring. Alcohol Clin Exp Res. 1989;13(1):73–83. doi: 10.1111/j.1530-0277.1989.tb00287.x. [DOI] [PubMed] [Google Scholar]
- Weinstock M. Stress - From Molecules to Behavior. Wiley-VCH Verlag GmbH & Co KGaA; 2009. Contribution of Early Life Stress to Anxiety Disorder; pp. 189–205. [Google Scholar]
- West JR. Use of pup in a cup model to study brain development. J Nutr. 1993;123(2 Suppl):382–385. doi: 10.1093/jn/123.suppl_2.382. [DOI] [PubMed] [Google Scholar]
- Whitcher LT, Klintsova AY. Postnatal binge-like alcohol exposure reduces spine density without affecting dendritic morphology in rat mPFC. Synapse. 2008;62(8):566–573. doi: 10.1002/syn.20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BW, Sengelaub DR, Steinmetz JE. MK-801 administration during neonatal ethanol withdrawal attenuates interpositus cell loss and juvenile eyeblink conditioning deficits. Alcohol. 2010;44(4):359–369. doi: 10.1016/j.alcohol.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]