

Abstract
PTEN-induced kinase 1 (PINK1) mutations are responsible for an autosomal recessive, familial form of Parkinson’s disease. PINK1 protein is a Ser/Thr kinase localized to the mitochondrial membrane and is involved in many processes including mitochondrial trafficking, mitophagy, and proteasomal function. Using a new PINK1 KO rat model we found altered brain metabolomic markers using magnetic resonance spectroscopy, identified changes in mitochondrial pathways with quantitative proteomics using sequential window acquisition of all theoretical spectra (SWATH) mass spectrometry and demonstrated mitochondrial functional alterations through measurement of oxygen consumption and acidification rates. The observed alterations included reduced creatine, a decrease in levels of complex I of the electron transport chain, and increased proton leak in the electron transport chain in PINK1 KO rat brains. In conjunction, these results demonstrate metabolomic and mitochondrial alterations occur during the asymptomatic phase of Parkinson’s disease in this model. These results indicate both potential early diagnostic markers and therapeutic pathways that can be used in PD.
Keywords: Parkinson’s disease, Mitochondria, Neurodegeneration, Proton leak
Introduction
Parkinson’s disease (PD) is a chronic, progressive neurodegenerative disorder characterized by loss of midbrain dopaminergic neurons in the substantia nigra pars compacta (SNPC) leading to a movement disorder and, ultimately, death. Research has implicated mitochondria in the pathogenesis of PD. PD patients exhibit electron transport chain (ETC) complex I deficiencies [1, 2], ROS elevation [3, 4], and mitochondrial DNA mutations [5, 6]. Several drugs including 1-methyl-4-phenylpyridinium, rotenone, and paraquat inhibit mitochondrial function producing Parkinsonian symptoms [7–10]. Finally, gene mutations can produce dysfunctional forms of proteins responsible for familial forms of PD including SNCA, PARKIN, PINK1, DJ-1, and LRRK2, and these proteins have mitochondrial functions (reviewed in [4]). These factors all suggest a mitochondrial component in the etiology of PD.
Despite the clear implication of mitochondria, studying the initiation and progression of PD has proven problematic. In general, PD models are better at mimicking the end-stages of PD rather than the progression (reviewed in [11]). Recently, the Michael J. Fox Foundation produced several rat models lacking PD-associated proteins. One model, the PINK1 knockout (PINK1 KO) rat model displays a progressive movement disorder [12]. PINK1, PTEN-induced putative kinase 1, acts as a regulator of mitochondrial quality. Localized to the mitochondrial membrane with the kinase domain facing the cytoplasm [13, 14], this protein, in addition to phosphorylating many proteins, directly interacts with other proteins to produce varied mitochondrial and cytosolic effects [15]. Interestingly, PINK1 also directly interacts with several of the other PD-associated proteins including Parkin and DJ-1 [16, 17]. Using the PINK1 KO model, it may be possible to study the progression of PD and identify novel diagnostic and therapeutic target during the asymptomatic phase of PD.
To interrogate the mitochondria-associated PD disorder, we chose to perform a longitudinal study on the PINK1 KO rat model. The metabolome, mitochondrial proteome, and mitochondrial function were assessed during the asymptomatic (4 month old) and symptomatic (9 month old) stages of PD in both the cortex and striatum of PINK1 KO and age-matched controls. Metabolomic, mitochondrial proteomic, and mitochondrial functional alterations were identified as compared to age-matched controls.
Materials and Methods
Animals
All animal experiments were conducted with PINK1 knockout and the Long Evans Hooded (LEH) control strains. Animals were 4 months or 9 months old at the time of the experiments mass spectrometry and mitochondria functional experiments. Animals for the MRS experiments were scanned longitudinally every 4 weeks starting at 10 weeks (2.5 months) of age. Animals used for MRS were used for 9 month stereology. All protocols were conducted within NIH-approved guidelines with the approval and oversight of the University of Nebraska Medical Center IACUC.
Brain isolation for stereology
Brains were rapidly harvested in accordance with the IACUC protocols at the University of Nebraska Medical Center. Six brains were taken from 9 month old animals from both the PINK1 knockout (PINK1 KO) and Long Evans Hooded (LEH) control groups. Brains were placed in Formal Fixx (Thermo Scientific, Rockford, IL) overnight (~15 hours). Brains were immersed in 30% sucrose in 0.1 M PBS overnight at 4° C. Brains were place in 70% ethanol and shipped on dry ice to the Stereology Resource Center, Inc. for stereology.
Stereology tissue processing
Serial cryostat sections (50 µm) were cut coronally through the brain stem containing the substantia nigra (SN), approximately from Bregma −4.36 mm to −6.72 mm [18]. Every 1st, 2nd and 3rd section of each series of 3 sections (interval: 150 µm) were collected separately in section storage solution (approximately 15 sections per set per brain), with free-floating sections stored at −20° C before further processing.
After inactivating the endogenous peroxidase activity with hydrogen peroxidase and washes in 0.01 M phosphate-buffered saline (PBS), sections were incubated free-floating in PBS containing the normal blocking serum, Triton X-100, and the specific tyrosine hydroxylase antibody (Abcam, Cambridge, MA) antibody for 3 days at 4°C. Subsequently, the immunoreaction product was visualized according to the avidin-biotin complex method [19] using the Vectastin elite ABC kit (Vector Lab., Burlingame, CA) and 3’, 3’-diaminobenzidine (Sigma, St. Louis, MO) as a chromogen. After thorough washes, all sections were mounted on gelatin-coated slides, and then counterstained with cresyl violet. Following dehydration in ethanol and clearing in xylene, sections were coverslipped with Permount (Fisher Scientific, Fair Lawn, NJ).
Stereology
Total volume of SNPC was quantified using the point-counting-Cavalieri approach [20]. At low power (4x objective) the SNPC was outlined on each section and the sum of area on the cut surfaces quantified by point counting. Using the average post-processing section thickness determined by automatic analysis [21], the total volume of SNPC for each brain was determined by the Cavalieri method.
At high magnification (100 x oil, n.a. 1.4 objective) the total number of tyrosine hydroxylase (TH)-positive neurons was counted using the optical fractionator method [22]. All cells with a neuronal phenotype in SNPC were counted if neuronal nuclei surrounded by TH+ cytoplasm. Dissector counting of TH-positive cells through the SNPC was repeated to a high level of sampling stringency (coefficient of error about 0.10), approximately at between 150 and 200 systematic-random locations across 8–12 sections. The mean total number of TH-positive neurons in SNPC was calculated according to the optical fractionator approach [22–24].
1H MRS Acquisitions of Parkinson’s disease metabolomic alterations
Single voxel localized spectra were acquired using point resolved spectroscopy (PRESS) [25] with outer volume suppression and high bandwidth pulses to optimize sequence performance. A 3 mm (caudal rostral) x 2 mm (anterior-posterior) x 8 mm voxel was selected in the central cerebral cortex for spectral acquisition. Spectra were acquired with a repetition time of 4 seconds, echo time of 50 ms, 576 averages, using a laboratory constructed 40 mm diameter rat brain birdcage coil on a 7 Tesla/16 cm Bruker Pharmascan (Karlsure, Germany) MRI/MRS system.
Spectroscopic processing and analyses
Spectroscopic data were processed by removal of residual water signal using the HLVSD filter. Spectra from 1H MRS data sets were curve fit in the time domain using the QUEST algorithm in jMRUI [26, 27] which fits results to a sum of individual metabolite spectra (basis set). Spectra for the basis set were acquired from phantoms using the same acquisition parameters as used in vivo. Phantoms containing either alanine, aspartate, choline, creatine, gamma-amino butyric acid, glutamate, glutamine, glycerophosphocholine, glycine, lactate, myoinositol, n-acetyl aspartate, or taurine were maintained at 38° C using a circulating water bath and buffered to pH 7.4 at physiological osmolarity. Results were normalized to the sum of all 13 metabolites as a semi-quantitative method for reporting metabolite concentrations in institutional units (I.U.).
Graphs were made in Prism (Version 6.04). A two-way repeated measures ANOVA followed by Sidak’s posthoc multiple comparison test was used to determine significance differences. Differences were found to be significantly different if .
Sequential window acquisition of all theoretical spectra mass spectrometry (SWATH-MS)
SWATH-MS is a robust methodology for quantitative mass spectrometry [28] and consists of a series of crucial steps: building a reference spectral library using data-dependent mass spectroscopic analysis, isolation of the sample and its preparation for mass spectrometry, data-independent mass spectroscopic analysis of the experimental sample performed via repetitively cycling through consecutive precursor isolation windows (swaths), processing of the mass spectroscopic data, and statistical analysis.
Data-dependent analysis for building a library
Mitochondrial protein lysates were isolated form B35, H19-7/IGF-IR, PC12, and RN33B rat cell lines of neuronal origin. The resulting protein was mixed in equal amounts, trypsin digested, quantified, and fractionated in 12 fractions by isoelectric focusing using an Agilent 3100 OffGEL Fractionator with a pH 3–10 strip in accordance with the manufacturer supplied protocols (Agilent Technologies, Santa Clara, CA). Peptides were purified using a C-18 PepClean Spin Columns (Thermo Fisher). Samples were dehydrated with a Savant ISS 110 SpeedVac Concentrator (Thermo Fisher) and resuspended in 6 µL of 0.1% formic acid for mass spectrometry. The isolation of peptides for mass spectrometry was performed twice independently. The resulting 24 fractions of peptides were analyzed by nano-LC-MS/MS in SWATH-MS mode on the 5600 TripleTOF instrument. The SWATH-MS acquisition was performed using the published protocol [28].
Additional samples were added to enrich our database for synaptic proteins. A brain from a LEH rat had synaptic mitochondria isolated as before [29]. These samples were also added to the spectral library.
Isolation of brain region specific mitochondria for SWATH-MS
Brains were rapidly isolated from 4 and 9 month old animals in both the PINK1 KO and LEH control groups. The cortex and striatum were isolated from the animals. For all mass spectrometry experiments, 4 biological replicates were used per group. After extraction, brains were immediately rinsed with ice-cold PBS to remove blood. The meninges were removed. Tissue was chopped and homogenized using a Dounce homogenizer. Brain mitochondria were isolated using a differential centrifugation kit (Mitosciences, Eugene, OR) followed by an immunomagnetic purification using a kit with TOM-22 coupled to magnetic beads (MACS Miltenyi Biotec, Auburn, CA). Mitochondria were lysed in 4% sodium dodecyl sulfate (SDS) and protein concentration was quantified using a using a Pierce 660 assay with bovine serum albumin standards (Thermo Fisher Scientific, Rockford, IL).
Sample preparation for mass spectrometry and data-independent SWATH-MS analysis
Sample preparation was conducted as before [30]. In short, proteins were digested with trypsin (Promega, Madison, WI) on a 20-µm filter (Pall Corporation, Ann Arbor, MI). Impurities were removed using a mixed-mode weak cation exchange cartridge (Waters, Milford, MA). Peptides were quantified with the aid of a Nanodrop (Thermo Fisher) in conjunction with the Scopes’ method for protein quantification [31]. For sample analysis, 2 µg of peptide was loaded into a 6 µl volume of 0.1% formic acid (Fisher Scientific).
Data-independent SWATH-MS analysis
The SWATH-MS acquisition was conducted similar to as previously described [28]. Samples of peptides from PINK1 KO and LEH rat brain mitochondrial lysates were analyzed in quadruplicate (four biological replicates per age group) using SWATH data-independent analysis (DIA). The list of peaks was generated in ProteinPilot (Version 4.5) using the Paragon algorithm (4.5.0.0) with the default settings. All of the fragment ion chromatograms were extracted and automatically integrated with PeakView (Version 2.1 Beta). The raw peak areas as reported by PeakView were used for all the quantification calculations with no data processing (neither denoising nor smoothing) of any kind applied to the extracted ion chromatograms.
To calibrate retention times, synthetic peptides (BiognoSYS; Zurich, Switzerland) were spiked-in the samples in accordance with the manufacturer’s protocol, and data was normalized to the median of common proteins in Markerview (Version 1.2.1). In accordance with previously published work [28], we selected 5 peptides and 5 transitions option for quantitative analysis by extracted-ion chromatograms (XIC) and targeted data extraction for each peptide was performed. Briefly, for each peptide the fragment ion chromatograms were extracted using the SWATH isolation window set to a width of 10 min and 50 ppm accuracy for quantification purposes in accordance with previously established protocols [28].
A Bayesian analysis was conducted using an unpaired two-condition analysis in CyberT [32, 33]. All comparisons are made against the age- and brain region-matched LEH controls. The sliding window size was set at 101 and the Bayesian confidence value was 12. The posterior probability of differential expression (PPDE) was calculated and significant changes were assigned if the p 0.05 and (α=0.05). All heat maps generated from mass spectrometry data were made in Multiple Experiment Viewer (www.tm4.org) [34].
Bioinformatic analysis
Analysis of upstream regulators was performed using Ingenuity Pathways Analysis (http://www.ingenuity.com/products/ipa) [35]. For this analysis, proteins and corresponding expression values were uploaded into the software. Based upon expression of proteins, the software was able to identify activated pathways through z-score analysis. The resulting z-score data was uploaded into Multiple Experiment Viewer and heat maps were generated.
Analysis of mitochondrial function
Mitochondrial function was analyzed with a Seahorse XF 24 analyzer based on the protocol of Rogers with minor alterations [36]. The amount of mitochondria used for the assay was optimized before the experiment for both cortical (10 µg) and striatal (10 µg) mitochondria. Additionally, we optimized the amount of ADP to be used to calculate State 3 respiration (4 µM). In Seahorse experiments, 3 animals were used per group for mitochondrial coupling and 3 animals were used per group for mitochondrial flux assays. Each biological replicate had 4 technical replicates for the experiments. Statistical significance was determined using a repeated measures two-way ANOVA followed by Sidak’s post-hoc test with α=0.05. Graphs were generated in Prism.
For all assays mitochondria were isolated by differential centrifugation, described above, and quantified. Equal amounts of mitochondria were then loaded onto Seahorse plates, centrifuged for 15 min, followed by 5 min incubation at 37° C, and loaded onto the Seahorse XF24. For the coupling assay, mitochondria were subjected to subsequent injections of ADP, oligomycin, FCCP and antimycin A while measuring the oxygen consumption rate (OCR) in the presence of succinate and rotenone. OCR is an indirect measurement of ATP production as higher oxygen consumption correlates with increased ATP production. Treatments of mitochondria with ADP, oligomycin, FCCP and antimycin A mimic various respiratory states. State 2, or basal respiration, is the amount of respiration at rest or in the presence of no treatment. State 3 respiration is the ADP-stimulated respiration in the presence of saturating substrate or during ADP treatment. State 4o is the oxygen consumption in the absence of oxidative phosphorylation as oligomycin binding inhibits ADP consumption at complex 4. State 3u is the maximal uncoupled respiratory state of the mitochondria as denoted by treatment with FCCP (for review of respiratory states see [37]). Antimycin A treatment mimics the absence of respiration by blocking complex III of the ETC.
To measure electron flux, after a baseline measurement, mitochondria are exposed to subsequent treatments of rotenone, succinate, antimycin A and ascorbic acid/tetramethylphenylenediamine (ASC/TMPD). These treatments inhibit certain electron transport subunits allowing us to measure the function of the complex I, complex II and complex IV when each complex act as the rate-limiting complex by providing complex-appropriate substrates in excess such as pyruvate and malate (basal), succinate (complex II), or ASC/TMPD (complex IV) treatment respectively after inhibition of other complexes by rotenone (complex I) or antimycin A (complex III).
Results
Assessment of loss of midbrain dopaminergic neurons
A major limitation of PD genetic models is most models do not recapitulate the progressive neurodegenerative hallmarks of the disease such as the progressive movement disorder or the loss of midbrain dopaminergic neurons in the substantia nigra (reviewed in [11]). As such, the relevance of the findings in mouse models to human PD is questionable. To circumvent this issue, a novel rat model that is deficient in the PINK1 protein and presents a progressive movement disorder was obtained [12].
To confirm the loss of midbrain dopaminergic neurons in the substantia nigra, we performed stereology of tyrosine hydroxylase positive neurons in wild-type Long-Evans Hardy (LEH) and PINK1 KO (produced on the LEH background) animals at 9 months of age (Fig. 1A). Dopaminergic neurons express high levels of tyrosine hydroxylase because this enzyme catalyzes the final and rate-limiting reaction of dopamine synthesis. Based on tyrosine hydroxylase staining, the number of dopaminergic neurons in the SNPC was decreased in PINK1 KO rats (Fig. 1B). A corresponding decrease in the size of the SNPC was also detected (Fig. 1C). These data demonstrate that the PINK1 knockout rat model recapitulates the midbrain dopaminergic cell death characteristic of PD.
Fig. 1.

Analysis of nigral dopaminergic loss in the PINK1 KO animals. Brain tissue was isolated, sectioned, and stained for tyrosine hydroxylase (TH) (A). The number of TH-positive neurons (B) and volume (C) were quantified for the substantia nigra pars compacta. *: p≤0.05 on a t-test. n=6 animals for both groups.
Non-invasive identification of metabolic changes due to loss of PINK1
Identifying early diagnostic markers in PD has been problematic, as patients do not present before loss of dopaminergic neurons. With the advent of the PINK1 knockout rat model to simulate the disease progression of PD, marker identification is feasible. Non-invasive magnetic resonance spectroscopy (MRS) was utilized to determine the presence of metabolomic alterations in 13 common brain metabolites in the cortex and striatum of PINK1 knockout and LEH control animals longitudinally. These metabolites are produced in the mitochondria or interact with mitochondrial products (Fig. 2A and 2B). As a result, identification of an altered metabolite not only indicates a possible PD diagnostic marker but also indicates altered PD-associated mitochondrial function.
Fig. 2.

Mitochondrial interaction of metabolites. Figure depicting metabolites originating from mitochondrial processes (A). Conversely, metabolites may be transported into the mitochondria or interact with mitochondrial products (B). OMM: Outer mitochondrial membrane; IMM: Inner mitochondrial membrane.
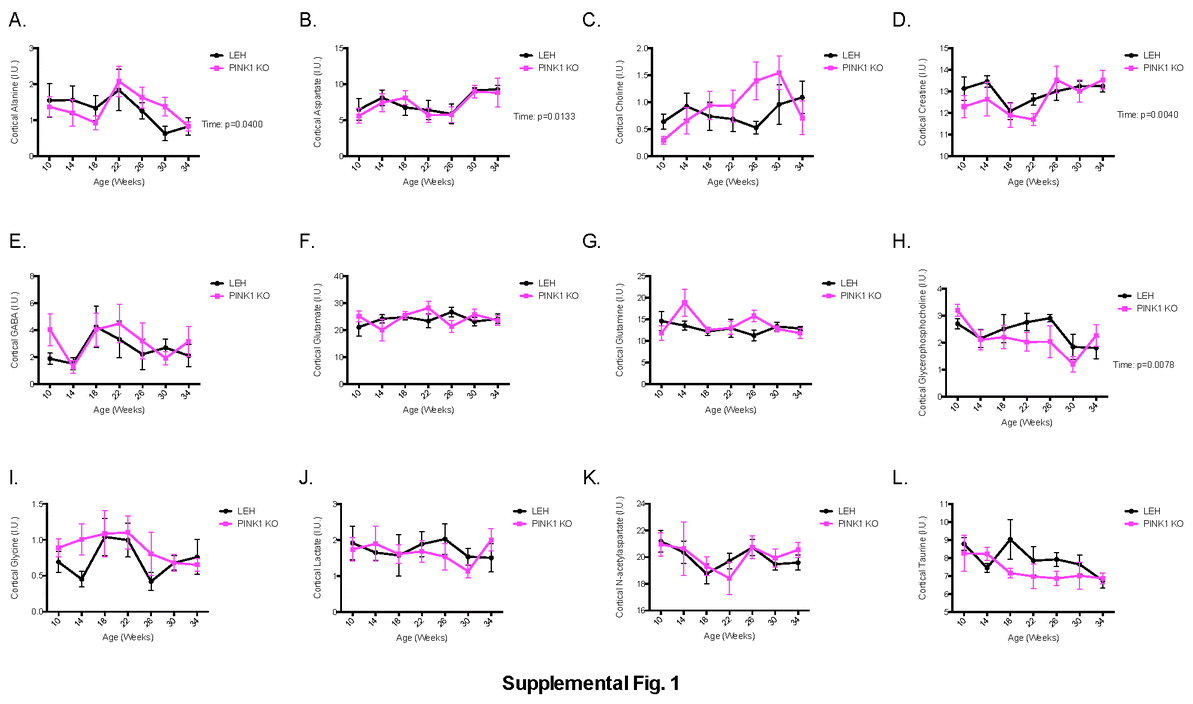
In the cortex, the majority of the metabolites were unchanged. Alanine, aspartate, creatine and glycerophosphocoline change with time, but not genotype, with p-values of 0.04, 0.01, 0.004 and 0.008 respectively (Supplemental Fig. 1). Only one metabolite was found to be altered in the cortex based on genotype. Cortical myoinositol was found to be significantly lower in the PINK1 KO animals (p= 0.03), but no time point was altered enough to pass Sidak’s post-hoc comparison test (Fig. 3A).
Fig. 3.

Metabolomic alterations produced by PINK1-deficiency. Metabolites were measured in the cortex and striatum using magnetic resonance spectroscopy (MRS) in vivo. Measurements were taken at 10 weeks (2.5 months) of age every 4 weeks until 34 weeks (8.5 months) of age. The measurements were expressed as a percentage of the total metabolite concentration (I.U.). Statistical significance was determined by a repeated measures two-way ANOVA. Sidak’s post-hoc comparison test was used to determine difference at any given time point. Listed p-values correspond to p-values generated by ANOVA. *: p≤0.05 on Sidak’s post-hoc comparison test. n=6 for cortical LEH and PINK1 KO animals, respectively. n=6 and n=5 for striatal LEH and PINK1 KO animals, respectively.
In the striatum, the majority of metabolites were also unchanged. Alanine, creatine, lactate and myoinositol changed with time with p-values of 0.0004, 0.03, 0.003, and 0.001 respectively (Supplemental Fig. 2), but not with genotype. Three metabolites were found to be significantly altered based on genotype in the striatum. Striatal aspartate was found to be increased in the PINK1 KO animals (p=0.05) with the 26 week time point passing the post-hoc comparison test (p=0.03) (Fig. 3B). However for aspartate there was an interaction between genotype and time (p=0.01) making interpretation of these data difficult. Striatal taurine was significantly decreased in the PINK1 KO animals (p=0.02) with the 18 week time point being significantly different after multiple comparison testing (p=0.01) (Fig. 3C). Striatal creatine was found to be significantly lowered in the PINK1 KO animals (p=0.0003) with the 10 week time point passing Sidak’s comparison test (p=0.05) (Fig. 3D).
Mechanistic pathways affected by loss of PINK1
Although our analysis indicated metabolomics alterations in the PINK1 KO rats, the mechanisms producing these changes remain elusive. Identification of the mechanism behind the metabolic alterations could potential reveal new PD therapeutic targets. Therefore, we performed a mass spectrometry experiment to interrogate mitochondrial proteomic alterations caused by PINK1-deficiency in 4 and 9 month old rats.
Analysis of the ETC complex subunits revealed an almost ubiquitous decrease in the levels of subunits identified (Fig. 4) regardless of age or brain area. The observed decrease in complex I subunits was more extreme in the striatum, compared to the cortex, of PINK1 KO rats regardless of age. Complexes III, IV, and V had a general decrease in ETC subunit expression with the PINK1 KO striatal mitochondria having a more severe depression of protein expression than the PINK1 KO cortex as compared to controls (Fig. 4). Complex II subunit levels were unchanged regardless of genotype or age.
Fig. 4.

Heat maps of the electron transport chains complex subunit expression in the PINK1 KO rat model. Subunit levels were measured through SWATH mass spectrometry. n=4 biological replicates for each measure. Data are displayed as a Log2 ratio of the averages.
To assess whether electron flow into the ETC was altered and because many metabolites stem from glycolytic processes, we investigated protein expression of glycolytic proteins (Fig. 5A). A total of 27 enzymes involved in glycolysis were identified. Unlike the ETC subunit levels, the directionality of altered protein levels varied with brain region and time. The glycolytic protein levels in the 4 month old cortex and striatum were very similar to controls. In the 9 month old samples, the cortical glycolytic protein levels were similar to controls. However, the striatal glycolytic protein levels were, in general, elevated, suggesting increased glycolysis. Interestingly, one protein, ALDOA, was consistently expressed at lower levels in PINK1 KO mitochondria regardless of age or brain region. ALDOA was decreased 2.29-fold, 2.60-fold, 1.79-fold and 1.98-fold in the 4 month old cortex, 4 month old striatum, 9 month old cortex, and 9 month old striatum respectively, as compared to controls.
Fig. 5.

Heat maps depicting glycolysis, dynamics/trafficking, and fatty-acid metabolism proteins in the PINK1 KO rat model. Subunit levels were measured through SWATH mass spectrometry. n=4 biological replicates for each measure. Data are displayed as a Log2 ratio of the averages.
In addition to regulating mitochondrial properties by protein levels of ETC subunits and glycolytic enzymes, cells regulate function by localization of mitochondria within the cell [38]. To assess whether proteins controlling mitochondrial localization were altered, proteins involved in mitochondrial dynamics and trafficking were interrogated (Fig. 5B). A total of 19 known trafficking/dynamics proteins were identified. The main fission protein DRP1 was increased in the striatum while the main fusion protein, MFN2, was decreased in the striatum. Together, these results suggest increased fission and thus mitochondrial fragmentation. In addition, mitochondrial adaptor proteins such as KLC1 (2.56-fold), SH3LG1 (1.71-fold), SH3GL2 (1.66-fold), SH3GLB1 (1.41-fold), and SH3GLB2 (1.47-fold) in addition to the trafficking proteins DNM1 (1.51) and MIRO1 (1.34-fold) were increased in the 4 month old PINK1 KO striatum as compared to controls. These results suggest mitochondrial trafficking and dynamics are altered in PINK1 animal striatum at 4 months of age.
The mitochondrion is also the primary locale for fatty acid oxidation. We identified 25 proteins involved in fatty-acid metabolism (Fig. 5C). There was a ubiquitous decrease of dehydrogenases in PINK1 KOs, such as ACADL, ACADM, ACADS, ACADVL, HADH, HADHA, and IVD regardless of brain region or age. Correspondingly, the proteins involved with transporting the electrons generated from the dehydrogenases to the ETC (ETFA and ETFB) were decreased in PINK1 KOs regardless of brain region or age. A decrease in electron passage to the ETC from this process in PINK1 KO animals may be a contributing factor to the development of PD symptoms. Two proteins deserve special mention: ACAA2 and ETFB. ACAA2 catalyzes the final step of β-oxidation while ETFB transfers electrons from dehydrogenases involved in β-oxidation to the ETC. Both proteins were ubiquitously decreased in all samples and significantly so in the 4 month old in PINK1 KO cortex and striatum (Supplemental Table 1).
Based upon the MRS data indicating lower levels of creatine in the striatum, we investigated enzymes known to regulate the levels of this metabolite. The principle enzyme response catalyzing the rate-limiting reaction in creatine synthesis, GATM, was depressed in the striatum of PINK1 KO animals, suggesting deficient creatine production may be responsible for the low levels of creatine observed in PINK1 KO animal striatum (Supplemental Table 2).
Also of interest are the proteins with significantly altered levels in both the striatal and cortical mitochondria of PINK1 KO animals as compared to control animals (Supplemental Table 1). These consistent protein alterations suggest the PINK1 protein is specifically important in regulating levels regardless of the functional changes. There were a number of proteins identified in 4 month old animals as altered in the PINK1 KO cortex and striatum including ACAA2, ALDOA, ATX10, ETFB, and SPRY4. As stated earlier, ACAA2 and ETFB are involved in β-oxidation of fatty-acids (Fig. 5C) and display similar levels in the cortex and striatum. ALDOA is a glycolytic protein (Fig. 5A) and is ubiquitously expressed. ATX10, however, is a protein related to cerebrospinal ataxia and the levels are altered, decreased in the cortex but increased in the striatum of 4 month old PINK1 KO animals. SPRY4, an inhibitor of the mitogen-activated protein kinase signaling pathway, was decreased in 4 month old striatum and cortex. Surprisingly, no proteins were found to be significantly altered in the cortex of 9 month old animals. Further, as animals aged, the number of significantly changed proteins decreased in both brain regions.
Despite the amount of data gained from mitochondrial proteomics, most mitochondrial proteins are nuclear encoded, and their levels controlled by nuclear and cytoplasmic protein pathways, which were not investigated here. To identify pathways regulating the observed processes, Ingenuity Pathways Analysis was utilized. The pathways identified as regulating the observed processes were involved in cellular developmental signaling, mitochondrial proliferation, receptor signaling, ROS processes or cellular stress (Fig. 6).
Fig. 6.

IPA predicted activated and deactivated pathways in the PINK1 KO rat model. Data and corresponding expression values were uploaded into the IPA prediction algorithm. Z-score activation was generated and is depicted through the color scale. Upstream regulators were sorted by the cellular processes that each influences. Activation numbers depict level of activation of the PINK1 KO as compared to the LEH control. n=4 for all groups in the analysis.
The pathways involved in cellular developmental signaling (BDNF, NRG1 and PHF21A) displayed a similar pattern (Fig. 6). The developmental signaling pathways were decreased in 4 month old PINK1 KO cortex but increased in the 4 month striatum as well as the 9 month cortex and striatum. The pathways regulating mitochondrial proliferation (PGC1A, PGC1B and TFAM) were increased in the 4 month cortex but decreased in the 4 month striatum. The proliferative pathways remained low in the 9 month cortex and striatum in general. Taken together, these results suggest a low level of mitochondrial biogenesis occurring in PINK1 KO brains as opposed to controls.
Several receptor pathways were also determined to be activated. Either the pathway activation remained elevated in all samples as compared to controls as in the case of ADORA2A or NMDAR; or the pathways were activated in the 4 month old cortex but deactivated in all other samples as in the case of IGF1R or INSR. The ROS processes pathways had one of two patterns. Either the pathway was decreased in the 4 month cortex and elevated in other time points (ARNT or HIF1B, HIF1A or NFE2L2) or the pathway was constitutively depressed (GPX1). Finally, the regulatory pathway for autophagy, mTOR, was decreased in the cortex of both 4 month and 9 month samples but increased in the striatum regardless of age. XBP1, the main regulatory pathway for the unfolded protein response, was decreased in the 4 month old samples but increased in the 9 month old samples.
Mitochondrial coupling
The mitochondrial respiratory states were interrogated in from cortical and striatal PINK1 KO and LEH control brain mitochondria of 4 and 9 month old animals. Alterations in the mitochondrial states were detected based upon genotype in the striatum with the knockout animal displaying a higher OCR in the 9 month old animals (p=0.03) (Fig. 7D). State 3 respiration and state 3u respiration in the striatum were significantly higher (p=0.0340 and p=0.0079 respectively on Sidak’s posthoc comparison test) with the measurements for state 3 being 684 ± 175 pmol/min (mean ± SD) and 968 ± 167 pmol/min for 9 month old LEH and PINK1 KO animals respectively while state 3u respiration measured pmol/min and 1004 ± 198 for the 9 month old LEH and PINK1 KO animals respectively. No significantly different respiratory states were measured in the 9 month old cortex following Sidak’s posthoc comparison test (Fig. 7B). Additionally, although no mitochondrial alterations were detected in 4 month old PINK1 KO mitochondria, the 4 month animal results demonstrated a similar trend as the PINK1 KO rat mitochondria from 9 month old animals (Fig. 7A and 7C).
Fig. 7.

Mitochondrial functional coupling in the PINK1 KO rats. Mitochondrial oxygen consumption rate was determined using a Seahorse XF24 analyzer. For coupling (A, B, C, &D), mitochondria were administered subsequent injections of ADP, Oligomycin, FCCP, and Antimycin A in the presence of succinate and rotenone. The cortex and striatum were interrogated at 4 months (A & C, respectively) and 9 months (B & D, respectively). Based upon the coupling, the respiratory control ratios (RCR) of the cortex and striatum for 4 month (E) and 9 month (F) old animals were calculated (RCR=OCRFCCP/OCROligomycin). Additionally, mitochondrial proton leak was calculated for the cortex and striatum of 4 month old (G) and 9 month old (H) animals (Proton leak = OCROligomycin-OCRAntimycin A). Listed p-values correspond to p-values generated by two-way repeated measures ANOVA. *: p≤0.05 on Sidak’s post-hoc comparison test. For all experiments, n=3 biological replicates.
The respiratory control ratio (RCR), a measure of mitochondrial coupling, was also assessed (RCR = State3u/State4o = OCRFCCP/OCROligomycin). The RCR is frequently used as a measure of the degree of mitochondrial coupling of oxygen consumption to electron flow [39]. In general, a higher RCR suggests superior mitochondrial function. No alteration was detected in RCR in the PINK1 KO rats at either 4 or 9 months as compared to controls (Fig. 7E and 7F).
Proton leak was also interrogated. Proton leak is a measure of the depletion of the proton motive force in the absence of oxidative phosphorylation (Proton leak = State4o-Antimycin = OCROligomycin-OCRAntimycin A [40]). Proton leak was altered in 4 (p=0.0348) and 9 (p=0.0470) month old animals based upon genotype, with PINK1 KO increased relative to wild-type LEH (Fig. 7G and 7H). These results suggest depletion of the proton motive force, hindering energy generation, is a consequence of PINK1-deficiency.
Mitochondrial Flux
Because mitochondrial respiratory states are dependent on the functionality of the mitochondrial complexes, the function of individual mitochondrial complexes were interrogated. For this experiment, mitochondrial OCR was measured by the Seahorse XF24 analyzer for both the PINK1 KO and LEH animals of 4 and 9 months of age.
Striatal mitochondrial complex I, II and IV were found to have significantly increased capacity for electron flux in the 9 month old PINK1 KO animals (Fig. 8D). Complex I function (basal) was measured at 594 ± 142 pmol/min (mean ± SD) in the LEH animals, and significantly elevated (p<0.0001) at 1129 ± 103 pmol/min in the PINK1 KO animals (Fig. 8D). Complex II function (succinate) was measured at 425 ± 55 pmol/min in the LEH animals and significantly elevated (p<0.0001) at 892 ± 133 pmol/min in the PINK1 KO animals (Fig. 8D). Similarly complex IV respiration was measured at 485 ± 61 pmol/min in the LEH animals and significantly elevated (p<0.0001) at 1123 ± 178 pmol/min in the PINK1 KO animals (Fig. 8D). No alterations were found between the 9 month old LEH and PINK1 KO cortical mitochondria (Fig. 8B) or between the 4 month old LEH and PINK1 KO cortical (Fig. 8A) or striatal (Fig. 8C) mitochondria. However, the mitochondria of PINK1 KO 4 month old animals displayed a trend similar to the mitochondria of 9 month old PINK1 KO animals, perhaps preceding the significant increase.
Fig. 8.

Assessment of electron transport chain complex function in PINK1 KO animals. Mitochondrial assessment of the electron transport chain complexes in the cortex and striatum as of 4 month old (A & C, respectively) and 9 month old (B & D, respectively) as assessed by oxygen consumption rate (OCR) and measured by a Seahorse XF24 analyzer. Mitochondrial were treated to subsequent injections of Rotenone, Succinate, Antimycin A and TMPD/Asc while measuring OCR. Statistical significance was assessed by a repeated-measures two-way ANOVA followed by Sidak’s post-hoc test. *: p≤0.05 on Sidak’s post-hoc comparison test. For all experiments, n=3 biological replicates.
Discussion
Metabolomic, mitochondria proteomic, and mitochondrial functional alterations were detected in a PINK1 KO rat model before the characteristic loss of dopaminergic neurons. Using magnetic resonance spectroscopy, we detected alterations in myoinositol in the cortex, and aspartate, taurine and creatine in the striatum. Mitochondrial proteomic alterations that coincide with metabolic alterations as well as novel alterations were detected. Mitochondrial functional assays revealed altered coupling and flux in the striatum at the 9 month time point. Additionally, we detected mitochondrial proton leak was elevated in PINK1 KO animals regardless of age or brain region measured. While these results suggest mitochondrial properties are altered well in advance of the presentation of PD symptoms and current methodologies may be able to predict whether a person will develop PD, these experiments were conducted in PINK1 KO rats and further work is needed to determine the translatability of these data to idiopathic PD patients.
Through these experiments, metabolic alterations were identified as possible diagnostic markers for early PD. The three molecules altered in the striatum (aspartate, creatine, and taurine) are important for three reasons: (1) these metabolites impact mitochondria suggesting mitochondria are important to the pathological progression of PD, (2) these differences were observed in the striatum which receives heavy innervation from the substantia nigra, and (3) these metabolomics alterations were present before the loss of midbrain dopaminergic neurons and the corresponding movement disorder.
Aspartate is an organic acid and critical for transferring high energy phosphate groups from the mitochondria to the cytoplasm in the malate-aspartate shuttle. In this study, aspartate levels were determined to be elevated in the striatum of PINK1 KO animals (Fig. 3B). These results could be indicative of an altered malate-aspartate shuttle that would lead to altered energy transfer throughout the cell. Additionally, aspartate could also lead to increased stimulation of NMDAR [41]. These results are consistent with proteomic suggesting the NMDAR pathway is activated in 4 and 9 month striatum of PINK1 KO animals (Fig. 6). We found the NMDAR pathway increasingly activated in the striatum of PINK1 KO animals regardless of age as compared to controls. However, no changes were observed in the cortical NMDAR pathway activation. The elevated aspartate levels in the striatum may provide the mechanism for this increased striatal NMDAR activation.
Taurine was also found to be significantly lower in PINK1 KO animals. Taurine is necessary for proper nervous system function [42, 43]. Specifically, taurine is critical for many biological processes including long-term potentiation [44], calcium homeostasis [45], and neuroprotection against excitotoxicity [46]. Further, the importance of taurine to the mitochondria has been realized [47]. In the mitochondria, taurine has been suggested to influence oxidative stress [48] and the buffering capacity of mitochondria [49]. CSF levels of PD patients was found to have decreased taurine levels [50] further supporting our findings.
Interestingly, alteration of acidity of the mitochondrial matrix would directly alter the function of the dehydrogenases involved in fatty-acid metabolism [51]. As noted earlier, there was a fairly ubiquitous decrease in proteins involved fatty-acid metabolism in the striatum of PINK1 KO animals at 4 months of age. The observed taurine alterations could be a contributing factor in the observed depression. Taurine levels alter the matrix pH. The dehydrogenases become inactive and as such are recycled faster than they are produced. By truncating the electron transfer potential from fatty-acid metabolism to the ETC, a source of cellular energy would be removed leading to decreased energy levels. The consistent depression of proteins levels for fatty-acid metabolism proteins in the cortex and striatum suggest the role of fatty-acid metabolism deserves more attention in regards to the PD pathology.
Metabolic alterations were also observed in a major energy metabolite, creatine. Creatine is an organic acid containing high energy phosphate bonds and serves to provide an addition energy source to cells. Additionally, creatine has antioxidant properties. Preliminary studies have demonstrated it to be neuroprotective for PD patients [52–54], but a different placebo-controlled study showed creatine had no effect on PD scores, dopamine transporter imaging, or non-motor symptoms of PD [55]. While the exact mechanism of creatine neuroprotection remains unclear, the ATP energy pool is tightly coupled to the creatine kinase system suggesting creatine supplementation may alter mitochondrial properties [56].
Creatine was found to be decreased in the striatum of PINK1 KO animals. Interestingly, cortical creatine levels did not vary between PINK1 KO and LEH control animals. The mitochondrial proteomic data are in agreement with the metabolomic data. The enzyme catalyzing the rate-limiting step of creatine production, GATM (glycine amidinotransferase, mitochondrial), was depressed in PINK1 KO as compared to LEH striatal mitochondria (1.43-fold decrease) at 4 months of age, but no change was observed in PINK1 KO as compared to LEH cortical mitochondria (only a 1.01-fold decrease) (Supplemental Table 2). The depression of GATM levels would result in deficient creatine production and could explain the deficient creatine levels in the PINK1 KO striatum. These data suggest creatine levels are directly attributable to the mitochondrial proteomic alterations.
Analysis of the mitochondrial proteome revealed the deficiency of complex I subunits in PINK1 KO rats regardless of age and brain region (Fig. 4). In our studies, we identified an almost ubiquitous decrease of complex I subunits of the ETC. For the 4 and 9 month time points, the decrease in complex I subunits was more severe in the striatum. These results suggest the mitochondrial effects of PINK1-deficiency may disproportionately affect the striatum. Given that the pathology of PD alters striatal function early during the disease pathogenesis [57], these findings are not surprising. However, it is surprising that the decrease in ETC subunits was not unique to complex I. Complex III, IV and IV also displayed a general decrease in subunit expression. Whether this finding is a result of the decrease in complex I is unknown but more work is necessary to clarify this issue.
In our experiment, the levels of glycolytic enzymes associated with the mitochondria were altered. By increasing the levels of these enzymes associated with mitochondria, the cells can increase glycolytic flux into the ETC [58]. Further, previous work has demonstrated that glycolytic enzymes can alter the electron flux into the mitochondria [58] and has demonstrated PINK1 KO mice have increased glycolysis in neurons and myocytes [59]. Our data seems to support this finding. The rate-limiting step in this pathway the conversion of fructose-6-phosphate to fructose-1,6-biphosphate catalyzed by phosphofructokinases (PFKL and PFKP). In this experiment, PFKL and PFKP were found to be increased in PINK1 KO cortex and striatum. Also, these enzymes increased in expression with time. These observations suggest these cells increase glycolysis as a compensatory mechanism to compensate for increased energy demands or decreased ETC flux.
It is important to note, however, not all the glycolytic enzymes levels were increased. However, the decrease in certain enzymes may be a defense mechanism of the cell. By decreasing certain enzymes and increasing others, pools of metabolic intermediates could be increased and decreased. Previous work has demonstrated that certain glycolysis intermediates are neuroprotective [60] suggesting the cell may be inflating certain pools of intermediates as a neuroprotective stress response. In regards to our data, this characteristic of the glycolytic cycle is most pertinent to ALDOA. ALDOA is a glycolytic enzyme responsible for converting fructose-1,6-biphosphate to glyceraldehyde-3-phosphate and dihydroxyacetone phosphate. Previous work has demonstrated ALDOA is heavily oxidized in PD patients even more so than in patients with Lewy body dementia [61]. Heavy oxidation of ALDOA leads to increased degradation and lower levels consistent with the findings we presented in the 4 and 9 month old animals (Fig. 5A). Low levels of ALDOA would lead to a surplus of fructose-1,6-biphosphate which has been demonstrated to be neuroprotective [60]. Hence, low level of ALDOA is likely a neuroprotective mechanism operating in response to PINK1-deficiency.
To assess if mitochondrial localization properties may be altered, proteins involved in mitochondrial dynamics and trafficking were interrogated. The increase in adaptor proteins in conjunction with motor proteins suggest altered mitochondrial distribution in cells. The altered distribution of mitochondria would alter synaptic signaling [62–64] and calcium signaling [38]. Additionally, DRP1 was increased but only in the striatum of PINK1 KO animals at both 4 and 9 months of age. These results suggest there is mitochondrial fragmentation that would limit the cellular ability to handle cellular insults and detrimentally affect ATP production [65]. Interestingly, trafficking/dynamic protein levels were similar to control levels in the cortex of PINK1 KO animals regardless of age. Together these results suggest altered mitochondrial trafficking is altered in the striatum of PINK1 KO animals and likely produces signaling abnormalities in the striatum.
Bioinformatic analysis of the proteomic studies revealed the PINK1-deficiency caused increased developmental signaling, decreased mitochondrial proliferation, altered signaling, increased ROS signaling and altered stress pathways in the striatum of 4 month old PINK1 KO animals (Fig. 6). Surprisingly, the mitochondrial (PGC1A, PG1B and TFAM) and cellular (IGF1R and INSR) growth pathways were deactivated in the 4 month old striatum and 9 month old cortex and striatum but activated in the 4 month cortex. These results suggest at 4 months of age, the cortex may be successfully combating the inevitable decline related to PD but the striatum has already succumbed to PINK1-deficiency. The ROS pathways (ARNT, HIF1A, and NFE2L2) seem to confirm these findings. These pathways are important for reigning in aberrant ROS production and, in general, these pathways are altered to minimize ROS in the 4 month old striatum. Additionally, the mTOR pathway, which controls autophagy, is demonstrated to be increased only in the striatum, and the autophagy pathway has recently been identified as an antioxidant pathway [66]. Together these results suggest there is additional stress on the striatum at 4 months of age. These results would be consistent with current knowledge on the progression of PD.
Additionally, the prediction of ADORA2A activation is interesting. ADORA2A is the adenosine A2 receptor and previous work has demonstrated caffeine blocks this receptor to inhibit MPTP-induced PD-like injuries [67–69]. Additionally, polymorphisms in the ADORA2A gene have been demonstrated to reduce the risk of developing PD [70]. Our finding of ubiquitous activation of the ADORA pathway regardless of age or brain region (Fig. 6) suggests PINK1-deficiency is responsible for activating this pathway. It also confirms that blocking the ADORA2A pathway is directly inhibiting PD progression.
To determine whether these changes correlated with mitochondrial abnormalities, mitochondrial function was assessed using a Seahorse XF24 analyzer. Genotypic differences in respiratory state of 9 month old animals were observed in PINK1 KO rat mitochondria from the striatum but not the cortex. PINK1 KO mitochondria from 9 month old rats have altered mitochondrial respiratory states and ETC subunit efficiency as analyzed by the coupling assay (Fig. 7B and 7D) and the flux assay (Fig. 8B and 8D) respectively. No respiratory state alterations were detected in 4 month old PINK1 KO mitochondria regardless of origin (Fig. 7A, 7C, 8A and 8C). However, similar trends were observed in the 4 month old PINK1 KO rats. Despite observed differences in coupling and flux, no changes were observed in RCR (Fig. 7E and 7F) suggesting that although the mitochondria are functioning differently, the mitochondria are still functionally intact.
In general, the mitochondria of PINK1 KO rats displayed increased oxygen consumption. These results, while initially confounding, are consistent with research on mitochondria from PINK1 PD patients. Neuronal cells derived from PD patient fibroblast-derived induced pluripotent stem cells (iPSCs) have mitochondrial properties remarkably similar to the PINK1 KO rat brain mitochondria [40]. In these experiments, iPSC-derived neurons with a Q458X PINK1 mutation had increased oxygen consumption rates.
Additionally, proton leak was increased in the PINK1 KO rats (Fig. 7G and 7H). Such a finding is important as it suggests the PINK1 KO rat has increased ROS generation. These findings are consistent with what would be expected in PD patients [4]. Additionally, a previous experiment demonstrated that mitochondria derived from a patient with Q458X PINK1 mutation have increased proton leak [40]. While proton leak may serve as a protective mechanism [71], the prolonged elevation of proton leak observed in this model likely indicates mitochondrial dysfunction as elevated reactive oxygen species (ROS) have been demonstrated to increase proton leak [72]. Increased proton leak, in turn, would decrease ROS [73]. Regardless, the increased proton leak in the striatum would dissipate the cellular capacity to generate ATP and may explain why neurons responding to dopamine display heightened sensitivity.
Through this work, we have identified possible early stage diagnostic markers, early stage altered pathways, and mitochondrial functional abnormalities. These results are critical because they indicate known late-stage PD abnormalities such as elevated proton leak and depressed taurine levels are present during the asymptomatic PD stages. Using this research, we may be able to target early processes pre-movement abnormalities for early diagnosis and allow early interventions to halt the progression of PD.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
We would like to thank the Proteomics Core Facility members at the University of Nebraska Medical Center, under the direction of Dr. Pawel Ciborowski, for all their support and aid in the proteomics experiments.
Funding Sources.
This work was funded by the National Institute of Health (NIH) MH073490 and NIH MH062261.
Footnotes
Online supplemental material.
Supplemental information includes a complete listing of all metabolite measurements from the cortex (Supplemental Fig. 1) and striatum (Supplemental Fig. 2), a table listing all proteins significantly altered in a sample (Supplemental Table 1) and a table listing the complete set of measurements obtained by SWATH mass spectrometry (Supplemental Table 2). Supplemental material can be obtained online.
References
- 1.Parker WD, Jr, Parks JK, Swerdlow RH. Complex I deficiency in Parkinson's disease frontal cortex. Brain Research. 2008;1189:215–218. doi: 10.1016/j.brainres.2007.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 3.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nature clinical practice Neurology. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 5.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genetics. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 6.Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nature Genetics. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 7.Miller GW. Paraquat: the red herring of Parkinson's disease research. Toxicological sciences : an official journal of the Society of Toxicology. 2007;100:1–2. doi: 10.1093/toxsci/kfm223. [DOI] [PubMed] [Google Scholar]
- 8.Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nature Neuroscience. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 10.Somayajulu-Nitu M, Sandhu JK, Cohen J, Sikorska M, Sridhar TS, Matei A, Borowy-Borowski H, Pandey S. Paraquat induces oxidative stress, neuronal loss in substantia nigra region and parkinsonism in adult rats: neuroprotection and amelioration of symptoms by water-soluble formulation of coenzyme Q10. BMC Neuroscience. 2009;10:88. doi: 10.1186/1471-2202-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crabtree DM, Zhang J. Genetically engineered mouse models of Parkinson's disease. Brain research bulletin. 2012;88:13–32. doi: 10.1016/j.brainresbull.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dave KD, De Silva S, Sheth N, Ramboz S, Beck MJ, Quang C, Benkovic SA, Ahmad S, Sunkin S, Walker D, et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson's disease. Neurobiol Dis. 2014 doi: 10.1016/j.nbd.2014.06.009. in press. [DOI] [PubMed] [Google Scholar]
- 13.Gandhi S, Muqit MM, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, et al. PINK1 protein in normal human brain and Parkinson's disease. Brain : a journal of neurology. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- 14.Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends in neurosciences. 2014;37:315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo M. Drosophila as a model to study mitochondrial dysfunction in Parkinson's disease. Cold Spring Harbor perspectives in medicine. 2012:2. doi: 10.1101/cshperspect.a009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. The Journal of clinical investigation. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paxinos G, Watson C. The Rat Brain in Stereotactic Coordinates. 6 edn. Academic Press; 2007. [Google Scholar]
- 19.Hsu SM, Raine L, Fanger H. The use of antiavidin antibody and avidin-biotin-peroxidase complex in immunoperoxidase technics. American journal of clinical pathology. 1981;75:816–821. doi: 10.1093/ajcp/75.6.816. [DOI] [PubMed] [Google Scholar]
- 20.Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. Journal of microscopy. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- 21.Elozory DT, Kramer KA, Chaudhuri B, Bonam OP, Goldgof DB, Hall LO, Mouton PR. Automatic section thickness determination using an absolute gradient focus function. Journal of microscopy. 2012;248:245–259. doi: 10.1111/j.1365-2818.2012.03669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. The Anatomical record. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 23.Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, Barnes CC, Pierce K. Neuron number and size in prefrontal cortex of children with autism. JAMA : the journal of the American Medical Association. 2011;306:2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- 24.Mouton PR, Kelley-Bell B, Tweedie D, Spangler EL, Perez E, Carlson OD, Short RG, deCabo R, Chang J, Ingram DK, et al. The effects of age and lipopolysaccharide (LPS)-mediated peripheral inflammation on numbers of central catecholaminergic neurons. Neurobiology of aging. 2012;33:423. doi: 10.1016/j.neurobiolaging.2010.09.025. e427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann N Y Acad Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- 26.Ratiney H, Coenradie Y, Cavassila S, van Ormondt D, Graveron-Demilly D. Time-domain quantitation of 1H short echo-time signals: background accommodation. Magma. 2004;16:284–296. doi: 10.1007/s10334-004-0037-9. [DOI] [PubMed] [Google Scholar]
- 27.Ratiney H, Sdika M, Coenradie Y, Cavassila S, van Ormondt D, Graveron-Demilly D. Time-domain semi-parametric estimation based on a metabolite basis set. NMR in biomedicine. 2005;18:1–13. doi: 10.1002/nbm.895. [DOI] [PubMed] [Google Scholar]
- 28.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Molecular & cellular proteomics : MCP. 2012;11 doi: 10.1074/mcp.O111.016717. O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stauch KL, Purnell PR, Fox HS. Quantitative Proteomics of Synaptic and Nonsynaptic Mitochondria: Insights for Synaptic Mitochondrial Vulnerability. Journal of Proteome Research. 2014 doi: 10.1021/pr500295n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villeneuve L, Tiede LM, Morsey B, Fox HS. Quantitative proteomics reveals oxygen-dependent changes in neuronal mitochondria affecting function and sensitivity to rotenone. Journal of Proteome Research. 2013;12:4599–4606. doi: 10.1021/pr400758d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scopes RK. Measurement of protein by spectrophotometry at 205 nm. Analytical biochemistry. 1974;59:277–282. doi: 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- 32.Kayala MA, Baldi P. Cyber-T web server: differential analysis of high-throughput data. Nucleic acids research. 2012;40:W553–W559. doi: 10.1093/nar/gks420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- 34.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al. TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 35.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Advances in enzymology and related subjects of biochemistry. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 38.Chang DT, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol. 2006;80:241–268. doi: 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. The Biochemical journal. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson's disease. Science translational medicine. 2012;4 doi: 10.1126/scitranslmed.3003985. 141ra190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen PE, Geballe MT, Stansfeld PJ, Johnston AR, Yuan H, Jacob AL, Snyder JP, Traynelis SF, Wyllie DJ. Structural features of the glutamate binding site in recombinant NR1/NR2A N-methyl-D-aspartate receptors determined by site-directed mutagenesis and molecular modeling. Molecular pharmacology. 2005;67:1470–1484. doi: 10.1124/mol.104.008185. [DOI] [PubMed] [Google Scholar]
- 42.Huxtable RJ. Physiological actions of taurine. Physiological reviews. 1992;72:101–163. doi: 10.1152/physrev.1992.72.1.101. [DOI] [PubMed] [Google Scholar]
- 43.Kumari N, Prentice H, Wu JY. Taurine and its neuroprotective role. Advances in experimental medicine and biology. 2013;775:19–27. doi: 10.1007/978-1-4614-6130-2_2. [DOI] [PubMed] [Google Scholar]
- 44.Dominy J, Jr, Thinschmidt JS, Peris J, Dawson R, Jr, Papke RL. Taurine-induced long-lasting potentiation in the rat hippocampus shows a partial dissociation from total hippocampal taurine content and independence from activation of known taurine transporters. Journal of Neurochemistry. 2004;89:1195–1205. doi: 10.1111/j.1471-4159.2004.02410.x. [DOI] [PubMed] [Google Scholar]
- 45.Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochemical research. 2002;27:21–26. doi: 10.1023/a:1014890219513. [DOI] [PubMed] [Google Scholar]
- 46.Leon R, Wu H, Jin Y, Wei J, Buddhala C, Prentice H, Wu JY. Protective function of taurine in glutamate-induced apoptosis in cultured neurons. Journal of Neuroscience Research. 2009;87:1185–1194. doi: 10.1002/jnr.21926. [DOI] [PubMed] [Google Scholar]
- 47.Hansen SH, Andersen ML, Birkedal H, Cornett C, Wibrand F. The important role of taurine in oxidative metabolism. Advances in experimental medicine and biology. 2006;583:129–135. doi: 10.1007/978-0-387-33504-9_13. [DOI] [PubMed] [Google Scholar]
- 48.Schaffer SW, Azuma J, Mozaffari M. Role of antioxidant activity of taurine in diabetes. Canadian journal of physiology and pharmacology. 2009;87:91–99. doi: 10.1139/Y08-110. [DOI] [PubMed] [Google Scholar]
- 49.Hansen SH, Andersen ML, Cornett C, Gradinaru R, Grunnet N. A role for taurine in mitochondrial function. Journal of biomedical science. 2010;17(Suppl 1):S23. doi: 10.1186/1423-0127-17-S1-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelborghs S, Marescau B, De Deyn PP. Amino acids and biogenic amines in cerebrospinal fluid of patients with Parkinson's disease. Neurochemical research. 2003;28:1145–1150. doi: 10.1023/a:1024255208563. [DOI] [PubMed] [Google Scholar]
- 51.Ghisla S, Thorpe C. Acyl-CoA dehydrogenases. A mechanistic overview. European journal of biochemistry / FEBS. 2004;271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 52.Gualano B, de Salles Painelli V, Roschel H, Lugaresi R, Dorea E, Artioli GG, Lima FR, da Silva ME, Cunha MR, Seguro AC, et al. Creatine supplementation does not impair kidney function in type 2 diabetic patients: a randomized, double-blind, placebo-controlled, clinical trial. European journal of applied physiology. 2011;111:749–756. doi: 10.1007/s00421-010-1676-3. [DOI] [PubMed] [Google Scholar]
- 53.NINDS NET-PD Investigators. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66:664–671. doi: 10.1212/01.wnl.0000201252.57661.e1. [DOI] [PubMed] [Google Scholar]
- 54.NINDS NET-PD Investigators. A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clinical neuropharmacology. 2008;31:141–150. doi: 10.1097/WNF.0b013e3181342f32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bender A, Koch W, Elstner M, Schombacher Y, Bender J, Moeschl M, Gekeler F, Muller-Myhsok B, Gasser T, Tatsch K, Klopstock T. Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial. Neurology. 2006;67:1262–1264. doi: 10.1212/01.wnl.0000238518.34389.12. [DOI] [PubMed] [Google Scholar]
- 56.Adhihetty PJ, Beal MF. Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. Neuromolecular medicine. 2008;10:275–290. doi: 10.1007/s12017-008-8053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rakshi JS, Uema T, Ito K, Bailey DL, Morrish PK, Ashburner J, Dagher A, Jenkins IH, Friston KJ, Brooks DJ. Frontal, midbrain and striatal dopaminergic function in early and advanced Parkinson's disease A 3D [(18)F]dopa-PET study. Brain : a journal of neurology. 1999;122(Pt 9):1637–1650. doi: 10.1093/brain/122.9.1637. [DOI] [PubMed] [Google Scholar]
- 58.Graham JW, Williams TC, Morgan M, Fernie AR, Ratcliffe RG, Sweetlove LJ. Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. The Plant cell. 2007;19:3723–3738. doi: 10.1105/tpc.107.053371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao Z, Gandhi S, Burchell VS, Plun-Favreau H, Wood NW, Abramov AY. Cell metabolism affects selective vulnerability in PINK1-associated Parkinson's disease. Journal of cell science. 2011;124:4194–4202. doi: 10.1242/jcs.088260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazzio E, Soliman KF. The role of glycolysis and gluconeogenesis in the cytoprotection of neuroblastoma cells against 1-methyl 4-phenylpyridinium ion toxicity. Neurotoxicology. 2003;24:137–147. doi: 10.1016/s0161-813x(02)00110-9. [DOI] [PubMed] [Google Scholar]
- 61.Buono P, D'Armiento FP, Terzi G, Alfieri A, Salvatore F. Differential distribution of aldolase A and C in the human central nervous system. Journal of neurocytology. 2001;30:957–965. doi: 10.1023/a:1021828421792. [DOI] [PubMed] [Google Scholar]
- 62.Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- 63.Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 64.Ma H, Cai Q, Lu W, Sheng ZH, Mochida S. KIF5B motor adaptor syntabulin maintains synaptic transmission in sympathetic neurons. J Neurosci. 2009;29:13019–13029. doi: 10.1523/JNEUROSCI.2517-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lammermann K, Brunner B, Kurz-Drexler A, Vogel F, Reichert AS, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. The Journal of Biological Chemistry. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox biology. 2014;2:82–90. doi: 10.1016/j.redox.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Jr, Schwarzschild MA. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson's disease. J Neurosci. 2001;21:RC143. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu K, Xu YH, Chen JF, Schwarzschild MA. Caffeine's neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity shows no tolerance to chronic caffeine administration in mice. Neuroscience letters. 2002;322:13–16. doi: 10.1016/s0304-3940(02)00069-1. [DOI] [PubMed] [Google Scholar]
- 69.Kalda A, Yu L, Oztas E, Chen JF. Novel neuroprotection by caffeine and adenosine A(2A) receptor antagonists in animal models of Parkinson's disease. Journal of the neurological sciences. 2006;248:9–15. doi: 10.1016/j.jns.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 70.Popat RA, Van Den Eeden SK, Tanner CM, Kamel F, Umbach DM, Marder K, Mayeux R, Ritz B, Ross GW, Petrovitch H, et al. Coffee, ADORA2A, and CYP1A2: the caffeine connection in Parkinson's disease. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2011;18:756–765. doi: 10.1111/j.1468-1331.2011.03353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Speakman JR, Talbot DA, Selman C, Snart S, McLaren JS, Redman P, Krol E, Jackson DM, Johnson MS, Brand MD. Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging cell. 2004;3:87–95. doi: 10.1111/j.1474-9728.2004.00097.x. [DOI] [PubMed] [Google Scholar]
- 72.Ivanov AS, Putvinskii AV, Antonov VF, Vladimirov Iu A. [Magnitude of the protein permeability of liposomes following photoperoxidation of lipids] Biofizika. 1977;22:621–624. [PubMed] [Google Scholar]
- 73.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Quarterly reviews of biophysics. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.