

Abstract
Radioadaptive response (RAR) describes phenomena where small conditioning doses of ionizing radiation (IR) reduce detrimental effects of subsequent higher IR doses. Current radiation protection regulations do not include RAR because of the large variability in expression among individuals and uncertainties of the mechanism. However, RAR should be regarded as an indispensable factor for estimation and control of individual IR sensitivity. In this article, RAR studies relevant to individual cancer risk are reviewed. Using various stains of mice, carcinogenic RAR has been demonstrated. Consistently much in vivo evidence for RAR with end points of DNA and chromosome damage is reported. Most in vivo RAR studies revealed efficient induction of RAR by chronic or repeated low-dose priming irradiation. Chronic IR-induced RAR was observed also in human individuals after environmental, occupational, and nuclear accident radiation exposure. These observations may be associated with an intrinsically distinct feature of in vivo experimental systems that mainly consist of nonproliferating mature cells. Alternatively, induction of RAR by gap junction-mediated bystander effects suggests that multicellular systems comprising densely communicating cells may be capable of responding to long-lasting low-dose-rate priming irradiation. Regulation by endocrine factors is also a plausible mechanism for RAR at an individual level. Emerging evidence suggests that glucocorticoids, known as stress hormones, participate in in vivo RAR induction following long-term low-dose-rate exposure to IR.
Keywords: Radioadaptive response, low-dose-rate radiation, intercellular signal transduction, protective bystander effect, endocrine regulation
Introduction
It is widely accepted that ionizing radiation (IR) at high doses is detrimental to the exposed organism. However, biological effects of low-dose or low-dose-rate IR remain elusive. Radioadaptive response (RAR) is a term describing phenomena where a small conditioning dose of IR (called the ‘priming dose’) reduces the biological effects of subsequent higher doses of IR (called the ‘challenge dose’). Since its discovery by Olivieri et al.1 in 1984, RAR has been confirmed using a variety of experimental systems ranging from yeasts to animal models. The end points are such effects as IR-induced DNA damage, chromosomal aberrations, cell transformation, cell death, and mutation in in vitro experiments and prenatal death, malformation, hematopoietic death, and carcinogenesis in in vivo experiments.2 Keen interest has been shown in RAR with the end points of carcinogenesis and the related genomic damage because IR-induced cancer is a major concern in the risk assessment of low-dose or low-dose-rate IR.
There is considerable interindividual variation in the expression of RAR. In a study analyzing RAR in human lymphocytes from numerous individuals, it was reported that RAR with the end points of chromatid or chromosome damage was observed in 50–78% of cases, and its extent (magnitude of reduction of challenge dose effects after priming dose exposure) ranged from 11% to 32%.3 A genetic constitution is thought to be the major source of the interindividual variability because the interindividual difference is not considerable in monozygotic twins, whereas dizygotic twins show greater variability.4 Due to the large variability between individuals as well as uncertainties of the mechanism, the International Commission on Radiation Protection concluded that RAR should not be included in the estimation of the potential risk to human population for low-level IR exposure.5 However, RAR could potentially modulate the IR sensitivity of some fraction of individuals so remarkably that it should be regarded as an indispensable factor in “tailor-made radiation protection,” which is a prospective radiation protection system based on estimation and control of the IR sensitivity of individuals.
Progress in RAR research in the last three decades has been well reviewed in several articles.2,3,6–9 Here, we focus on the studies of RAR investigated at the individual level with a particular interest in a possible link to cancer risk. RAR studies in which challenge irradiation was carried out in vitro are included in the scope of this review if RAR was induced in vivo by irradiating individuals with priming doses. However, RAR studies irrelevant to cancer risks are not included. A significant mechanistic role of intercellular signaling such as bystander effects and endocrine signal transduction through hormones in RAR is discussed with a particular emphasis on its implication in risk modulation by low-dose-rate IR.
Evidence of in vivo RARs
Animal data
Animal models showing in vivo RAR have been reported from several groups as summarized in Table 1. Carcinogenic RAR was first demonstrated by Bhattacharjee,10 who observed that the yield of thymic lymphoma of Swiss mice induced by 2 Gy of γ rays was remarkably decreased when the mice were preirradiated with a priming dose of 10 mGy day−1 for 5 or 10 consecutive days. Ina et al.11 reported that induction of thymic lymphomas by four fractionated doses of 1.8 Gy each (7.2 Gy in total) in C57BL/6 mice was suppressed consistently by preirradiation with 75 mGy of X-rays given 6 h before each 1.8 Gy irradiation. They also showed that induction of thymic lymphomas was more effectively suppressed by continuous whole-body irradiation with γ rays at 1.2 mGy h−1 for 450 days starting 35 days before the challenge dose of irradiation. It is interesting that chronic exposure or fractionated low-dose IR has efficiently suppressed carcinogenesis induced by the challenge dose. Modulation of cancer development through in vivo RAR has been observed in a variety of mouse strains. Mitchel et al.12 reported that the latent period for development of acute myeloid leukemia induced by a challenge dose of 1 Gy in CBA/Harwell mice was significantly increased when the mice were preirradiated with a 100-mGy priming dose 24 h prior to the challenge dose. Mitchel et al.27 also reported that a single exposure of either 10 or 100 mGy alone reduced spontaneous cancer development in p53 heterozygous mice. Kakinuma et al.13 reported that four deliveries (1 week−1) of a fractionated dose of 200 mGy (800 mGy in total) suppressed N-ethyl-N-nitrosourea-induced thymic lymphoma in B6C3F1 mice, suggesting a mechanism for RAR against chemical carcinogenesis.
Table 1.
In vivo RAR studies using animal models with the carcinogenic or related end points.
| Animal model | Priming dose | Challenge dose | Time interval | End point remarks | Reference | ||
|---|---|---|---|---|---|---|---|
| Radiation | Dose | Radiation | Dose | ||||
| Swiss mouse | γ Rays | 10 mGy day−1 × 5–10 days | γ Rays | 2 Gy | 24 h | Thymic lymphoma | 10 |
| C57BL/6 mouse | X-Rays | 75 mGy given before each 1.8 Gy dose | X-Rays | 1.8 Gy week−1 × 4 weeks | 6 h | Thymic lymphomas | 11 |
| C57BL/6 mouse | γ Rays | Continuous 1.2 mGy h−1 × 450 days | X-Rays | 1.8 Gy week−1 × 4 weeks | – | Thymic lymphomas | 11 |
| CBA/Harwell mouse | γ Rays | 100 mGy (500 mGy h−1) | γ Rays | 1 Gy | 24 h | Latent period for acute myeloid leukemia | 12 |
| B6C3F1 mouse | X-Rays | 200 mGy week−1 × 4 weeks | ENU | 50–200 ppm | 3 days | Thymic lymphomas | 13 |
| ICR mouse | 900 MHz radiofrequency | 120 W cm−2 × 4 h day−1 × 1–14 days | γ Rays | 3 Gy | 4 h | DNA damage (comet tail length) in peripheral leukocytes | 15 |
| ICR mouse | 900 MHz radiofrequency | 120 W cm−2 × 4 h day−1 × 7 days | γ Rays | 3 Gy | 4 h | MN induction in immature erythrocytes | 41 |
| C57BL/6 transgenic mouse | NNK | 2 mg day−1 × 4 days in the middle course of challenge irradiation | γ Rays | 1.5 mGy h−1 × 31 days | – | Chromosomal large deletion (>1 kb) in lung | 16 |
| rabbit | γ Rays | 0.3–1.8 Gy (5.6 mGy h−1) | X-Rays | 1.5 Gy | 6–38 days | Chromosomal aberrations in peripheral blood lymphocytes | 17 |
| Kunming mouse | X-Rays | 10 mGy (3.0 Gy h−1) | X-Rays | 0.75 Gy | 2.5–3 h | Chromatid aberrations in bone marrow cells and spermatocytes | 18 |
| C57BL/6 mouse | X-Rays | 2–100 mGy (3.0 Gy h−1) | X-Rays | 0.65 Gy | 2.5–3 h | Chromatid aberrations in bone marrow cells | 18 |
| Af mouse | X-Rays | 200 mGy (3.4 Gy h−1) | X-Rays | 1.5 Gy | 4 h | Chromosomal aberrations in bone marrow cells | 19 |
| C57BL/6 transgenic mouse | X-Rays | 150–375 mGy (over 3 days) | X-Rays | 2.5 Gy | 3 weeks | lacZ mutation in brain | 20 |
| C57BL/6 transgenic mouse | X-Rays | Acute 0.001–10 mGy | X-Rays | 1 Gy | 4 h | Chromosome inversions at lacZ locus in prostate | 21 |
| C57BL/6 mouse | γ Rays | 500 mGy (1.2 mGy h−1) | X-Rays | 0.4 Gy | 23 days | DNA damage in spleen analyzed by comet assay | 22 |
| C57BL/6 mouse | X-Rays | Acute 500 mGy | X-Rays | 7.5 Gy | 2 weeks | MN induction in polychromatic and normochromatic erythrocytes | 23 |
| C57BL/6 mouse | X Rays | Acute 500 mGy | Heavy particles | 5.5–5.75 Gy | 2 weeks | MN induction in polychromatic and normochromatic erythrocytes | 23 |
| C57BL/6 mouse | X Rays from CT scanner | 2 × 20 mGy week−1 × 10 weeks | γ Rays | 1–2 Gy | 5 days | γH2AX in lymphocyte-rich population of bone marrow cells | 24 |
| BALB/c mouse fetus | Chernobyl soils | 10–13 mSv day−1 for 10 days during organogenesis | γ Rays | 2.4 Sv | After born and weaned | MN induction in polychromatic erythrocytes | 25 |
| SHK white mongrel mouse | BH and protons | 160 mGy (4.3 mGy day−1) | X-Rays | 1.5 Gy | One to two generations | MN induction in bone marrow cells of F1 and F2 offsprings | 33 |
| Leopard frog | β Rays | Approximately 1 mGy year-1 | γ Rays | 4 Gy | – | Chromosome breaks in liver cells | 9 |
| Leopard frog | γ Rays | (Chronic) 1–100 mGy | γ Rays | 4 Gy | – | Chromosome breaks in liver cells | 9 |
RAR: radioadaptive response; ENU: N-ethyl-N-nitrosourea; NNK: 4-(methylnitrosamino))-1-(3-pyridyl)-1-butanone; BH: bendazol hydrochloride; MN: micronulei.
Induction of genomic damage is thought to be a critical step in IR carcinogenesis. Consistent with the above-mentioned carcinogenic RAR, much data for in vivo RAR with the end point of genomic damage, such as chromosomal aberrations, micronuclei induction, DNA double-strand breaks (DSBs), and gene mutations in lymphocytes or other somatic cells have been reported (Table 1).17–23 For example, Otsuka et al.22 analyzed DNA damage in spleen of C57BL/6N mice by a comet assay and revealed that DNA damage induced by a 0.4-Gy challenge dose was significantly reduced in the mice that had been preirradiated at 1.2 mGy h−1 for 23 days (500 mGy in total) compared with that in the sham-irradiated mice. The authors further revealed a correlation between reduced DNA damage and induction of antioxidative enzymes in the RAR condition. Induction of RAR by extremely low doses of X-rays was demonstrated in transgenic mice. By conducting a chromosomal inversion assay in the pKZ1 mouse, which contains the β-galactosidase gene in inverse orientation with respect to the β-actin promoter, Day et al.21 showed that 0.001–10 mGy followed 4 h later by a 1-Gy challenge dose caused reduction in inversions in prostates compared with those in mice irradiated with 1 Gy alone. Recently, in vivo RAR was explored using IR sources of high public concern such as nuclear medicine diagnostic devices and environmental radionuclides released from nuclear accidents. Phan et al.24 irradiated C57BL/6 mice with X-rays from a computed tomography (CT) scanner followed by irradiation of bone marrow cells withdrawn from the mice with a 1–2-Gy challenge dose. They found an approximately 10% decrease in γH2AX fluorescence level in bone marrow cells from the repeatedly CT-scanned mice (irradiation with 20 mGy twice a week for 10 consecutive weeks) compared with that in sham CT-scanned mice. The authors pointed out a requirement of repeated CT scans to confer resistance to the challenge dose because no RAR could be observed in mice receiving only a single CT scan. Howell et al.25 used an exposure plate comprised of soils collected from contaminated areas in Chernobyl, mainly containing cesium-137 and strontium-90. Pregnant BALB/c mice were irradiated on the exposure plate at 10–13 mSv d−1 for 10 days during organogenesis in the mouse fetus. The progeny mice born from the irradiated or sham-irradiated pregnant mice were exposed to the challenge dose of 2.4 Sv of γ rays after weaning. As a result, decreased micronuclei induction was observed in polychromatic erythrocytes from mice preirradiated with IR from Chernobyl soils.
It should be noted that the majority of in vivo RAR studies revealed efficient suppression of IR-induced carcinogenesis or genomic damage by chronic or repeated low-dose priming irradiation. This is a distinguishing feature of in vivo RAR observations compared with those of in vitro studies, although there are a few reports that describe induction of RAR in cultured cells by priming doses delivered at marginally low-dose-rates such as 300 mGy h−1.28 The difference may be attributable to the technical difficulties inherent in in vitro experiments to irradiate cultured cells or tissues with chronic or fractionated low-dose IR. Alternatively, it may be related to an intrinsically distinct feature of in vivo experimental systems. Two aspects can be considered. The first is that the in vivo experimental systems consist of a majority of nonproliferating mature cells, which are more likely capable of spending a long time to accumulate enough priming stimuli to induce RAR. Besides, whereas the repair of IR-induced DNA damage is thought to play an important role in RAR,29–32 different DSB repair pathways are chosen depending on the cell cycle.26 It may be speculated that the DSB repair pathway in nonproliferating cells is differently induced by low-dose-rate priming irradiation. The second aspect to consider is that the cells in in vivo experimental systems are aligned in a three-dimensional structure and are associated with neighboring cells through a distinct intercellular communication different from that of cells in two-dimensional in vitro experimental systems. A highly organized multicellular structure composed of densely communicating cells could respond to low-dose-rate priming irradiation even if only a minor fraction of the structure is irradiated at any moment. The cells in in vivo experimental systems are also likely to be under the control of systemic endocrine regulation. Intercellular dense communication within multicellular systems, and/or long-term hormonal regulation, is the plausible mechanism for in vivo RAR after chronic or sporadic low-dose priming irradiation.
There is one in vivo study that described transgenerational transmission of RAR-induced radioresistance. Sorokina et al.33 observed that the combined exposure of SHK white mongrel male mice to the immunomodulator bendazol hydrochloride and 160 mGy of chronic protons reduced micronuclei induction in bone marrow cells of F1 and F2 offsprings irradiated with the challenge dose (1.5 Gy) of X-rays. Although genetic effects of IR have not been observed in humans, ample amounts of data have proved such effects in mice.34–38 A distinct mechanism for transmission of IR effects to offspring is thought to underlie the difference between humans and rodents. The results of Sorokina et al. suggest a transgenerational transmission of RAR signals in mice, which should provide an insight into the mechanisms behind the frequently observable genetic effects in this species. More studies into the mechanism of transgenerational transmission of RAR signals are required.
Human data
RAR was first discovered by analyzing X-ray-induced chromatid aberrations in cultured lymphocytes obtained from the peripheral blood of healthy human adults.1 Subsequently, a number of in vitro studies were conducted to investigate the characteristics and mechanisms of RAR in human lymphocytes.2 In accord with these in vitro studies, data have been obtained for human in vivo RAR after long-term low-dose-rate exposure to environmental, occupational, and accidental radiation as previously summarized by Tapio and Jacob.3 For instance, Ghiassi-nejad et al.39 analyzed chromosomal aberrations in lymphocytes collected from residents in a high background radiation area (HBRA) and normal background radiation area in Ramsar, Iran. Residents in the HBRA had been exposed to up to 260 mGy year−1 primarily due to high concentrations of radium-226 on the ground. When the lymphocytes collected from residents in the HBRA were exposed to 1.5 Gy of γ rays, a significantly lower frequency of chromosomal aberration was observed compared with that in 1.5 Gy-irradiated lymphocytes collected from the normal background radiation area residents. This result suggests that long-term exposure of human individuals to low-dose-rate IR induces a steady radioresistance. A potential criticism against the interpretation of the radioresistance in HBRA residents as a result of RAR is that radioresistant individuals may have been selected during stable inhabitation over multiple generations. However, sensitivity to the low-dose-rate IR from natural sources is unlikely subjected to natural selection because the effects would appear much later than reproductive age. A more sound analysis is required into the possible correlation of the radioresistance in HBRA residents with any advantageous genetic changes.
As another example, Barquinero et al.40 studied RAR in lymphocytes after in vivo exposure to medical IR. The chromosomal aberration induced by challenge irradiation with 2 Gy of X-rays in lymphocytes collected from hospital workers who had been exposed to IR of up to 28 mSv year−1 was significantly lower than that in lymphocytes taken from nonradiation workers. A common shortcoming of these and other human RAR studies is that the number of donors who provided the blood sample was limited. As a result, significance of the data was often limited by poor statistical power due to large interindividual variations in both basal IR sensitivity and RAR inducibility. However, the influence of the interindividual variation in basal IR sensitivity was thought to be eliminated by measuring IR sensitivity of lymphocytes taken from identical individuals both before and after exposure to priming low-dose-rate IR. Based on this strategy, the micronuclei induction by 3.5 Gy of γ rays was tested in short-term radiation workers who had been exposed to 3 mSv on average in about 5 weeks by Thierens et al.14 Blood samples were collected twice before and after the radiation work, and it was found that for the majority of the workers, micronuclei induction by 3.5 Gy of γ rays was lower in lymphocytes collected after radiation work.
Mechanisms for in vivo RAR
Protective bystander effects
It is widely believed that the initial event for RAR is the generation of DSB,35 although there are some exceptions that suggest RAR is triggered by nongenotoxic agents such as radiofrequency fields.15,41 Initiated by a few DSB per cell, signal transduction cascades involving de novo protein synthesis are elicited42 and are thought to result in activation of effector factors that play direct roles either in enhancement of DNA repair, induction of molecular chaperon, synchronization of the cell cycle, or induction of antioxidants.43 The signaling factor p53 is crucial in various experimental systems for RAR.2 In vitro studies revealed that RAR is a transient but quasi-sustainable response in which radioresistance is typically elevated during a limited time period of about 20 h following a time interval of about 4 h after priming irradiation.44 Shimizu et al.45 and Sasaki et al.44 proposed a model of a molecular mechanism for RAR emphasizing a pivotal role of signaling factors as shown in Figure 1. Here, in response to a small number of DSB produced by a priming dose in the cell nucleus, a long-lasting signal transduction circuit circulating between cell nucleus and cell membrane is postulated, which would be maintained by p38 mitogen-activated protein kinases, phospholipase C, and protein kinase C (PKc). The RAR signal is thought to be transferred to neighboring cells through growth factors, reactive oxygen species, and/or nitric oxide (NO), which bring about protective bystander effects.
Figure 1.
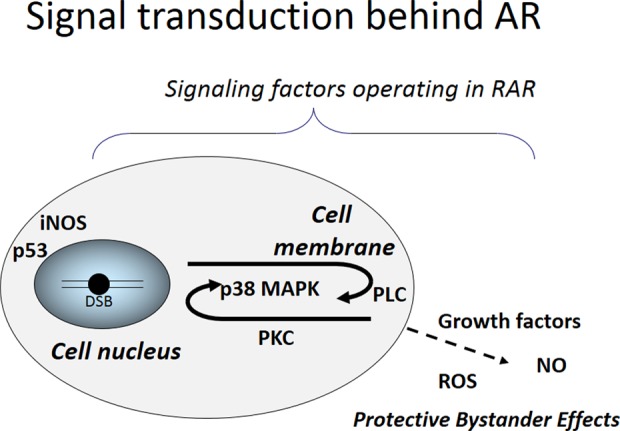
Signal transduction functioning in RAR. RAR: radioadaptive response; DSB: double-strand break; iNOS: inducible nitric oxide synthase; PKC: protein kinase C; PLC: phospholipase C; NO: nitric oxide; ROS: reactive oxygen species; MAPK: mitogen-activated protein kinase. Modified from Nenoi et al.72 with permission of Radiation Biology Research Communications.
The IR-induced bystander response was originally characterized by the cellular effects expressed in unirradiated cells located in some vicinity to an irradiated cell or cells.46 It was initially described in 1992 by Nagasawa and Little,47 who observed an elevated frequency (20–40%) of sister chromatid exchanges in Chinese hamster ovary cells in the condition where only 0.1-1% of cell nuclei were actually traversed by an α–particle track. The bystander effect is mediated by two modes of signal transduction from irradiated cells to unirradiated bystander cells; one is transmission of signaling molecules through a gap junction assembly spanning plasma membranes of adjacent two cells, and the other is interaction of factors secreted from irradiated cells with their specific receptors in bystander cells. It was recently reported that RAR is induced via the bystander mechanism (referred to as protective bystander effects). By measuring DSB in primary normal human fibroblast MRC-5 cells irradiated with 1 Gy of X-rays, Ojima et al.48 observed that the mean number of DSB per cell significantly decreased when nondividing confluent cells were preirradiated with 3–5 mGy of X-rays 4 h prior to the challenge irradiation. The authors further found that the effect of the preirradiation was diminished when the cells were incubated with lindane, an inhibitor of gap junction assembly, for 2 h before the priming irradiation. The result clearly indicated that the RAR was induced depending on signaling molecules transmitted through the gap junction. By investigating RAR with the end point of chromosomal aberrations in human H1299 lung cancer cells, Takahashi et al.49 observed that the RAR was blocked by aminoguanidine, an inducible NO synthase inhibitor, or 2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy PTIO), an NO radical scavenger. The authors further observed that an RAR-like response was induced by treatment of cells with isosorbide dinitrate, an NO-generating agent, alone. On the basis of these observations, they concluded that RAR was induced via NO radicals as intercellular signaling mediators. In addition, by incubating mouse embryonic fibroblasts in the media transferred from the replica culture irradiated with 0.1–1 Gy of X-rays, Klammer et al.50 found that the activity of DNA-PKc-dependent nonhomologous end joining was slightly but significantly elevated in unirradiated cells with similar kinetics compared with directly irradiated cells. Although the mechanisms are yet to be determined, involvement of enhanced DSB repair activity in protective bystander effects could be suggested.
RAR is a cellular response whereby radioresistance is elevated only during a limited time period after priming irradiation. However, it may be expected that radioresistance is sustainably induced by IR at an extremely low-dose rate (a few DSB or less in 1 year) considering the role of bystander effects in RAR. This can be depicted by a simplified and hypothetical model of in vivo RAR illustrated in Figure 2. The model is based on three assumptions, which were derived from in vitro studies: (1) RAR is initiated by a DSB, and the resistance to subsequent DSB is induced in cells during a limited time period of 4–24 h after generation of initial DSB as shown in Figure 2(a); (2) during the time period in which resistance to DSB is induced, the RAR signal is also transmitted to neighboring cells (up to Nth cells) in all directions from the cell in which the initial DSB was generated (Figure 2(b)); and (3) the cells can repeatedly become radioresistant as long as they receive the RAR signal. On the basis of these assumptions, every cell in the three-dimensional structure can receive an RAR signal from its neighboring (2N + 1)3 cells as shown by the gray cube in Figure 2(b). When the dose rate and number of DSB produced by 1 Gy are represented by R (in gray per hour) and λ (per gray), respectively, the probability that radioresistance is induced in each cell, P, can be described as:
Figure 2.

A hypothetical model for in vivo RAR depending on intercellular communication via gap junction. (a) It is postulated that the resistance to DNA DSBs is induced in cells during a limited time period of 4–24 h after generation of the initial DSB (indicated by a horizontally long oval), and (b) it is also postulated that the RAR signal is transmitted to neighboring cells (up to the Nth cells) in all directions from the cell in which the initial DSB was generated. It is also postulated that the cells can become radioresistant repeatedly as long as they receive the RAR signal. (c) Based on these assumptions, cells in the three-dimensional structure can receive multiple RAR signals from neighboring cells. RAR: radioadaptive response; DSB: double-strand break. Modified from Nenoi et al.73 with permission of National Institute of Radiological Sciences.
where M = (2N + 1)3.
The yield of DSB induced by IR in a cell was found to be linearly dependent on dose with a rate of approximately 30 Gy−1 (λ = 30 Gy−1).51,52 The propagation distance has been reported to vary in the range of 0.1–3 mm, roughly corresponding to >3 cells.53 However, no data are available to date on the transmission of protective bystander signals. If we postulate that the protective bystander RAR signal could be transmitted up to a third of the neighboring cells (N = 3) and a dose rate of 3 mSv in 5 weeks (R = 3.4 × 10−6 Gy h−1) is used as an example, we obtain P = 0.50.
This calculation suggests that resistance to DSB is induced in 50% of cells as long as irradiation is continued, and therefore, the induced radioresistance would likely be experimentally detectable. Thus, it is suggested that the cancer incidence after low-dose-rate IR could be reduced by an RAR mediated by the protective bystander effect. However, it should be noted that the outcome of RAR by low-dose-rate IR would be a persistent elevation of radioresistance, which would be indistinguishable from the general dose rate effects.
Potential association of endocrine factors
In vivo RAR can be regarded as a type of homeostatic control, where constancy in the internal environment of the body is maintained by various sensing, feedback, and control mechanisms. Because the endocrine response is a key mechanism for homeostatic control, regulation through endocrine factors is also a plausible mechanism for RAR at the individual level. The release of glucocorticoids (cortisol in humans, rabbits, and squirrels or corticosterone in mice and rats) from the adrenal cortex is a typical response of vertebrates to stressors such as intrinsic ROS and extrinsic assaults including infectious agents, toxic substances, and temperature extremes.54,55 The major role of glucocorticoid is to protect individuals against the excessive actions of immune and inflammatory responses. However, it was shown that glucocorticoids play a role in mitigating the harmful effects of a variety of stressors56 and moderate hyperadrenocorticism (increased secretion of glucocorticoids) induced by low levels of stressors making individuals resistant to these stressors. By examining blood samples collected from Chernobyl workers who carried out cleanup operations at the destroyed reactor from 1986 to 1988 (approximately 120 mSv of exposure on average over 1–3 months), Souchkevitch and Lyasko57 revealed a statistically significant increase in cortisol level compared with the controls. Similarly, Boonstra et al.58 reported a significantly higher level of corticosterone in meadow voles irradiated with low-dose-rate γ rays (22.6 μGy h−1 over 2.5 years) than those in the control or higher dose-rate (3840 μGy h−1 over 1.5 years)-irradiated groups. These results suggest a potential role of glucocorticoids in RAR induced in vivo after a long-term low-dose-rate exposure to IR.
In a normal condition, glucocorticoid receptor (GR) remains in the cytoplasm in large macromolecular complexes bound to chaperones such as HSP90. Upon ligand binding, GR dissociates from the complex and translocates into the cell nucleus where it activates or represses transcription of various genes depending on physiological conditions.59–61 Vares et al.62 searched for potential recognition sequences for transcription factors in the upstream region of the genes whose expression was modulated in the liver of C57BL/6J mice after long-term (400 days) irradiation at low dose rates (2.3–910 μGy h−1). As a result, the potential recognition sequence for GR was found with a significantly higher frequency than that in unmodulated genes consistent with the observations that the glucocorticoid level was modulated after a low-dose-rate irradiation. This result further supports the idea that glucocorticoids may be involved in in vivo RAR after low-dose-rate IR. However, the level of the dose response of GR and glucocorticoids after exposure to IR seems complicated, as Liu63 has shown a decreased GR level in splenic T cells after a low-dose (30–75 mGy) X-irradiation. It is also elusive whether activated GR suppresses IR carcinogenesis. Whereas p53 is a key factor in the suppression of carcinogenesis after exposure to IR, GR is known to interact with p53 in both a complementary and antagonistic fashion depending on physiological and pathological conditions.64 The physical interaction of GR and p53 in the presence of ligand causes their cytoplasmic sequestration and degradation through the proteasome pathway by recruiting the E3 ubiquitin ligase Hdm2, resulting in inhibition of each other’s transactivation properties.65–67 Consistently, it was reported that a short-term in vitro exposure of BALB/c 3T3 cells to physiological concentrations of cortisol consistently resulted in increased DNA damage and cellular transformation.68 In addition, using a p53 heterozygous mouse model with an elevated corticosterone level due to chronic restraint stress, Feng et al.69 demonstrated that the carcinogenic effect of IR was enhanced through reduced p53 activity. These observations suggest an enhancement of the carcinogenic effects of IR by glucocorticoids. In contrast, it was demonstrated that activated GR caused translocation of p53 to the cell nucleus, leading to enhanced transcription of p53-target genes.70 GR was also found to stimulate p21 gene transcription through the steroid response element in the promoter in rat hepatoma cells.71 More studies are required to clarify how glucocorticoids modulate cancer susceptibility depending on physiological and pathological conditions.
Conclusion
Carcinogenic RAR has been demonstrated in various strains of mice, and the majority of in vivo RAR studies revealed efficient induction of RAR by chronic or repeated low-dose priming irradiation. In vivo RAR in humans after long-term low-dose-rate exposure to environmental, occupational, and nuclear accident IR was also reported. The recent finding of protective bystander effects via gap junction-mediated intercellular signal transduction suggests that exposure of only a small fraction of cells to IR could induce RAR at the individual level, providing insights into the mechanism for in vivo RAR after very low-dose-rate exposure. In addition, by viewing RAR as a type of homeostatic control, regulation through endocrine factors is thought to be a plausible mechanism for RAR at the individual level. Emerging evidence suggests that glucocorticoids, known as stress hormones, are involved in in vivo RAR after long-term low-dose-rate exposure to IR. In vivo RAR induced by exposure to low-dose-rate IR should be reflected in the reduced effects of the low-dose-rate IR itself, and this effect should be indistinguishable from the general dose rate effects. Thus, in vivo RAR induced by low-dose-rate IR does not seem to have a drastic effect on the conventional radiation protection system. However, to establish a scientific basis for estimation and control of the IR sensitivity of individuals as an essential factor for prospective tailor-made radiation protection, more in vivo studies as well as studies of the underlying mechanism are necessary, with a particular focus on genetic components associated with interindividual differences of RAR.
Footnotes
Conflict of interest: The authors declared no conflicts of interest.
Funding: This work was partially supported by JSPS KAKENHI 25340041.
References
- 1. Olivieri G, Bodycote J, Wolff S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science 1984; 223: 594–597. [DOI] [PubMed] [Google Scholar]
- 2. Vares G, Wang B, Tanaka K, et al. Radiation-induced adaptive response with reference to evidence and significance: a review. Indian J Rad Res 2006; 3: 16–34. [Google Scholar]
- 3. Tapio S, Jacob V. Radioadaptive response revisited. Radiat Environ Biophys 2006; 46: 1–12. [DOI] [PubMed] [Google Scholar]
- 4. Kalina I, Némethová G. Variability of the adaptive response to low dose radiation in peripheral blood lymphocytes of twins and unrelated donors. Folia Biol (Praha) 1997; 43: 91–95. [PubMed] [Google Scholar]
- 5. International Commission on Radiation Protection (ICRP). Low-dose extrapolation of radiation-related cancer risk. Publication 99, Annals of the ICRP 2005; 35(4): 1–142. [DOI] [PubMed] [Google Scholar]
- 6. Matsumoto H, Hamada N, Takahashi A, et al. Vanguards of paradigm shift in radiation biology: radiation-induced adaptive and bystander responses. J Rad Res 2007; 48: 97–106. [DOI] [PubMed] [Google Scholar]
- 7. Matsumoto H, Tomita M, Otsuka K, et al. A new paradigm in radioadaptive response developing from microbeam research. J Radiat Res 2009; 50(Suppl A): A67–A79. [DOI] [PubMed] [Google Scholar]
- 8. Mitchel RE. Low doses of radiation are protective in vitro and in vivo: evolutionary origins. Dose Response 2006; 4: 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mitchel RE. Low doses of radiation reduce risk in vivo. Dose Response 2006; 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhattacharjee D. Role of radioadaptation on radiation-induced thymic lymphoma in mice. Mut Res 1996; 358: 231–235. [DOI] [PubMed] [Google Scholar]
- 11. Ina Y, Tanooka H, Yamada T, et al. Suppression of thymic lymphoma induction by life-long low-dose-rate irradiation accompanied by immune activation in C57BL/6 mice. Radiat Res 2005; 163: 153–158. [DOI] [PubMed] [Google Scholar]
- 12. Mitchel RE, Jackson JS, McCann RA, et al. The adaptive response modifies latency for radiation-induced myeloid leukemia in CBA/H mice. Radiat Res 1999; 152: 273–279. [PubMed] [Google Scholar]
- 13. Kakinuma S, Yamauchi K, Amasaki Y, et al. Low-dose radiation attenuates chemical mutagenesis in vivo. J Radiat Res 2009; 50: 401–405. [DOI] [PubMed] [Google Scholar]
- 14. Thierens H, Vral A, Barbé M, et al. Chromosomal radiosensitivity study of temporary nuclear workers and the support of the adaptive response induced by occupational exposure. Int J Radiat Biol 2002; 78: 1117–1126. [DOI] [PubMed] [Google Scholar]
- 15. Jiang B, et al. Adaptive response in mice exposed to 900 MHz radiofrequency fields: primary DNA damage. PLoS One 2012; 7: e32040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ikeda M, Masumura K, Sakamoto Y, et al. Combined genotoxic effects of radiation and a tobacco-specific nitrosamine in the lung of gpt delta transgenic mice. Mutat Res 2007; 626: 15–25. [DOI] [PubMed] [Google Scholar]
- 17. Liu SZ, Cai L, Sun SQ. Induction of a cytogenetic adaptive response by exposure of rabbits to very low dose-rate gamma radiation. Int J Radiat Biol 1992; 62: 187–190. [DOI] [PubMed] [Google Scholar]
- 18. Cai L, Liu SZ. Induction of cytogenetic adaptive response of somatic and germ cells in vivo and in vitro by low-dose X-irradiation. Int J Radiat Biol 1990; 58: 187–194. [DOI] [PubMed] [Google Scholar]
- 19. Mosse L, Kostrova I, Subbot S, et al. Melanin decreases clastogenic effects of ionizing radiation in human and mouse somatic cells and modifies the radioadaptive response. Radiat Environ Biophys 2000; 39: 47–52. [DOI] [PubMed] [Google Scholar]
- 20. Kind JA, Winn RN, Boerrigter ME, et al. Investigation of the radioadaptive response in brain and liver of pUR288 lacZ transgenic mice. J Toxicol Environ Health, A 2001; 63: 207–220. [DOI] [PubMed] [Google Scholar]
- 21. Day TK, Zeng G, Hooker AM, et al. Extremely low priming doses of X radiation induce an adaptive response for chromosomal inversions in pKZ1 mouse prostate. Radiat Res 2006; 166: 757–766. [DOI] [PubMed] [Google Scholar]
- 22. Otsuka K, Koana T, Tauchi H, et al. Activation of antioxidative enzymes induced by low-dose-rate whole-body gamma irradiation: adaptive response in terms of initial DNA damage. Radiat Res 2006; 166: 474–478. [DOI] [PubMed] [Google Scholar]
- 23. Wang B, Tanaka K, Ninomiya Y, et al. Relieved residual damage in the hematopoietic system of mice rescued by radiation-induced adaptive response (Yonezawa Effect). J Radiat Res 2013; 54: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Phan N, De Lisio M, Parise G, et al. Biological effects and adaptive response from single and repeated computed tomography scans in reticulocytes and bone marrow of C57BL/6 mice. Radiat Res 2012; 177: 164–175. [DOI] [PubMed] [Google Scholar]
- 25. Howell EK, Gaschak SP, Griffith KD, et al. Radioadaptive response following in utero low-dose irradiation. Radiat Res 2013; 179: 29–37. [DOI] [PubMed] [Google Scholar]
- 26. Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 2013; 49: 872–883. [DOI] [PubMed] [Google Scholar]
- 27. Mitchel RE, Jackson JS, Morrison DP, et al. Low doses of radiation increase the latency of spontaneous lymphomas and spinal osteosarcomas in cancer-prone, radiation-sensitive Trp53 heterozygous mice. Radiat Res 2003; 159: 320–327. [DOI] [PubMed] [Google Scholar]
- 28. Shadley JD, Wiencke JK. Induction of the adaptive response by X-rays is dependent on radiation intensity. Int J Radiat Biol 1989; 56: 107–118. [DOI] [PubMed] [Google Scholar]
- 29. Wiencke JK, Afzal V, Olivieri G, et al. Evidence that the [3H]thymidine-induced adaptive response of human lymphocytes to subsequent doses of X-rays involves the induction of a chromosomal repair mechanism. Mutagenesis 1986; 1: 375–380. [DOI] [PubMed] [Google Scholar]
- 30. Kleczkowska HE, Althaus FR. The role of poly(ADP-ribosyl)ation in the adaptive response. Mutat Res 1996; 358: 215–221. [DOI] [PubMed] [Google Scholar]
- 31. Varès G, Wang B, Tanaka K, et al. Mutagenic adaptive response to high-LET radiation in human lymphoblastoid cells exposed to X-rays. Mutat Res 2011; 706: 46–52. [DOI] [PubMed] [Google Scholar]
- 32. Varès G, Wang B, Tanaka K, et al. Mutagenic adaptive response to high-LET radiation in human lymphoblastoid cells exposed to low doses of heavy-ion radiation. Mutat Res 2011; 712: 49–54. [DOI] [PubMed] [Google Scholar]
- 33. Sorokina SS, Zaichkina SI, Rozanova OM, et al. Delayed effects of chronic low-dose high linear energy transfer (LET) radiation on mice in vivo. Radiat Prot Dosim 2011; 143: 305–310. [DOI] [PubMed] [Google Scholar]
- 34. Russell WL, Kelly EM. Mutation frequencies in male mice and the estimation of genetic hazards of radiation in men. Proc Nat Acad Sci U S A 1982; 79: 542–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pomerantseva MD, Goloshchapov PV, Vilkina GA, et al. Genetic effect of chronic exposure of male mice to gamma rays. Mutat Res 1984; 141: 195–200. [DOI] [PubMed] [Google Scholar]
- 36. Dubrova YE, Plumb MA. Ionising radiation and mutation induction at mouse minisatellite loci. The story of the two generations. Mutat Res 2002; 499: 143–150. [DOI] [PubMed] [Google Scholar]
- 37. Wu J, Morimyo M, Hongo E, et al. Radiation-induced germline mutations detected by a direct comparison of parents and first-generation offspring DNA sequences containing SNPs. Mutat Res 2006; 596: 1–11. [DOI] [PubMed] [Google Scholar]
- 38. Nomura T. Transgenerational effects of radiation and chemicals in mice and humans. J Radiat Res 2006; 47(Suppl B): B83–B97. [DOI] [PubMed] [Google Scholar]
- 39. Ghiassi-nejad M, Mortazavi SM, Cameron JR, et al. Very high background radiation areas of Ramsar, Iran: preliminary biological studies. Health Phys 2002; 82: 87–93. [DOI] [PubMed] [Google Scholar]
- 40. Barquinero JF, Barrios L, Caballín MR, et al. Occupational exposure to radiation induces an adaptive response in human lymphocytes. Int J Radiat Biol 1995; 67: 187–191. [DOI] [PubMed] [Google Scholar]
- 41. Jiang B, Nie J, Zhou Z, et al. Induction of adaptive response in mice exposed to 900MHz radiofrequency fields: application of micronucleus assay. Mutat Res 2013; 751: 127–129. [DOI] [PubMed] [Google Scholar]
- 42. Youngblom JH, Wiencke JK, Wolff S. Inhibition of the adaptive response of human lymphocytes to very low doses of ionizing radiation by the protein synthesis inhibitor cycloheximide. Mutat Res 1989; 227: 257–261. [DOI] [PubMed] [Google Scholar]
- 43. Coleman MA, Yin E, Peterson LE, et al. Low-dose irradiation alters the transcript profiles of human lymphoblastoid cells including genes associated with cytogenetic radioadaptive response. Radiat Res 2005; 164: 369–382. [DOI] [PubMed] [Google Scholar]
- 44. Sasaki MS, Ejima Y, Tachibanà A, et al. DNA damage response pathway in radioadaptive response. Mutat Res 2002; 504: 101–118. [DOI] [PubMed] [Google Scholar]
- 45. Shimizu T, Kato T, Jr, Tachibana A, et al. Coordinated regulation of radioadaptive response by protein kinase C and p38 mitogen-activated protein kinase. Exp Cell Res 1999; 251: 424–432. [DOI] [PubMed] [Google Scholar]
- 46. Kadhim M, Salomaa S, Wright E, et al. Non-targeted effects of ionising radiation-Implications for low dose risk. Mutat Res 2013; 752: 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nagasawa H, Little JB. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Canc Res 1992; 52: 6394–6396. [PubMed] [Google Scholar]
- 48. Ojima M, Eto H, Ban N, et al. Radiation-induced bystander effects induce radioadaptive response by low-dose radiation. Radiat Prot Dosim 2011; 146: 276–279. [DOI] [PubMed] [Google Scholar]
- 49. Takahashi A, Matsumoto H, Ohnishi T. Hdm2 and nitric oxide radicals contribute to the p53-dependent radioadaptive response. Int J Rad Oncol, Biol, Phys 2008; 71: 550–558. [DOI] [PubMed] [Google Scholar]
- 50. Klammer H, Kadhim M, Iliakis G. Evidence of an adaptive response targeting DNA nonhomologous end joining and its transmission to bystander cells. Canc Res 2010; 70: 8498–8506. [DOI] [PubMed] [Google Scholar]
- 51. Löbrich M, Ikpeme S, Kiefer J. Measurement of DNA double-strand breaks in mammalian cells by pulsed-field gel electrophoresis: a new approach using rarely cutting restriction enzymes. Radiat Res 1994; 138: 186–192. [PubMed] [Google Scholar]
- 52. Löbrich M, Ikpeme S, Kiefer J. DNA double-strand break measurement in mammalian cells by pulsed-field gel electrophoresis: an approach using restriction enzymes and gene probing. Int J Radiat Biol 1994; 65: 623–630. [DOI] [PubMed] [Google Scholar]
- 53. Hamada N, Maeda M, Otsuka K, et al. Signaling pathways underpinning the manifestations of ionizing radiation-induced bystander effects. Curr Mol Pharmacol 2011; 4: 79–95. [DOI] [PubMed] [Google Scholar]
- 54. Masoro EJ. Hormesis and the antiaging action of dietary restriction. Exp Gerontol 1998; 33: 61–66. [DOI] [PubMed] [Google Scholar]
- 55. Masoro EJ. Caloric restriction and aging: an update. Exp Gerontol 2000; 35: 299–305. [DOI] [PubMed] [Google Scholar]
- 56. Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev 1984; 5: 25–44. [DOI] [PubMed] [Google Scholar]
- 57. Souchkevitch G, Lyasko L. Investigation of the impact of radiation dose on hormones, biologically active metabolites and immunoglobulins in Chernobyl accident recovery workers. Stem Cells 1997; 15(Suppl 2): 151–154. [DOI] [PubMed] [Google Scholar]
- 58. Boonstra R, Manzon RG, Mihok S, et al. Hormetic effects of gamma radiation on the stress axis of natural populations of meadow voles (Microtus pennsylvanicus). Environ Toxicol Chem 2005; 24: 334–343. [DOI] [PubMed] [Google Scholar]
- 59. Tronche F, Kellendonk C, Reichardt HM, et al. Genetic dissection of glucocorticoid receptor function in mice. Curr Opin Genet Dev 1998; 8: 532–538. [DOI] [PubMed] [Google Scholar]
- 60. Reichardt HM, Schütz G. Glucocorticoid signalling—multiple variations of a common theme. Mol Cell Endocrinol 1998; 146: 1–6. [DOI] [PubMed] [Google Scholar]
- 61. Kellendonk C, Tronche F, Reichardt HM, et al. Mutagenesis of the glucocorticoid receptor in mice. J Steroid Biochem Mol Biol 1999; 69: 253–259. [DOI] [PubMed] [Google Scholar]
- 62. Vares G, Uehara Y, Ono T, et al. Transcription factor-recognition sequences potentially involved in modulation of gene expression after exposure to low-dose-rate γ-rays in the mouse liver. J Radiat Res 2011; 52: 249–256. [DOI] [PubMed] [Google Scholar]
- 63. Liu SZ. Radiation-induced change in lymphocyte proliferation and its neuroendocrine regulation: dose-response relationship and pathophysiological implications. Nonlinearity Biol, Toxicol, Med 2004; 2: 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sengupta S, Wasylyk B. Physiological and pathological consequences of the interactions of the p53 tumor suppressor with the glucocorticoid, androgen, and estrogen receptors. Annal NY Acad Sci 2004; 1024: 54–71. [DOI] [PubMed] [Google Scholar]
- 65. Sengupta S, Vonesch JL, Waltzinger C, et al. Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. EMBO J 2000; 19: 6051–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maiyar AC, Phu PT, Huang AJ, et al. Repression of glucocorticoid receptor transactivation and DNA binding of a glucocorticoid response element within the serum/glucocorticoid-inducible protein kinase (sgk) gene promoter by the p53 tumor suppressor protein. Mol Endocrinol 1997; 11: 312–329. [DOI] [PubMed] [Google Scholar]
- 67. Sengupta S, Wasylyk B. Ligand-dependent interaction of the glucocorticoid receptor with p53 enhances their degradation by Hdm2. Genes Dev 2001; 15: 2367–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Flint MS, Baum A, Chambers WH, et al. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology 2007; 32: 470–479. [DOI] [PubMed] [Google Scholar]
- 69. Feng Z, Liu L, Zhang C, et al. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc Nat Acad Sci U S A 2012; 109: 7013–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Crochemore C, Michaelidis TM, Fischer D, et al. Enhancement of p53 activity and inhibition of neural cell proliferation by glucocorticoid receptor activation. FASEB J 2002; 16: 761–770. [DOI] [PubMed] [Google Scholar]
- 71. Cha HH, Cram EJ, Wang EC, et al. Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J Biol Chem 1998; 273: 1998–2007. [DOI] [PubMed] [Google Scholar]
- 72. Nenoi M, Vares G, Wang B. Multiple factors involved in expression of radioadaptive responses (in Japanese). Radiat Biol Res Commun 2009; 44: 294–311. [Google Scholar]
- 73. Nenoi M. Future challenges of low-dose radiation research (in Japanese). Radiol Sci 2012; 55: 35–36. [Google Scholar]