

Abstract
Background:
Infantile Systemic Hyalinosis (ISH) is a very rare autosomal recessive disorder characterized by connective tissue involvement as hyaline deposition in skin, gastrointestinal tract, muscles, glands and other organs.
Cases Presentation:
We report eight Iranian children (4 male and 4 female) with ISH referred to our hospital from 1996 to 2013. The illness had been diagnosed by clinical manifestations and disease progression. Six of them died and two are alive but very sick.
Conclusion:
ISH is a very rare disorder with poor prognosis. Seventy five percent of our 8 patients died before 2 years old due to severe diarrhea, malabsorption and/or infection.
Keywords: Infantile Systemic Hyalinosis, Joint Contractures, Skin Thickness, Blond Hair, Hyaline Deposits
Introduction
Infantile Systemic Hyalinosis (ISH) is an autosomal recessive disorder characterized by extensive involvement of connective tissue due to deposition of amorphous hyaline [1] .
This fatal disorder usually presents at first months of life, especially first six months with widespread involvement of joints, skin, gastrointestinal tract and other parts. Joints usually are the first involved areas with swelling, pain and limited range of motions. Patients become disabled. Bones start to become osteoporotic and prone to multiple bone fractures. Skin is another involved part which becomes thickened and covered by purplish papules especially gathered on face, neck and ears. The scattered fibrous nodules on chest wall, hypertrophic gum and prominent nodules around the anus are the other manifestations of involved skin [2] .
Because of hyaline deposition in intestinal walls these patients have protein loosing entropathy and intermittent diarrhea that lead to short stature, failure to thrive, severe malnutrition, infection and septicemia and death [3] .
Cases Presentation
A retrospective case series based on medical records of 8 referred patients from August 1996 till September 2013 to Imam Khomeini Hospital, Department of Pediatric Rheumatology, Tehran have been surveyed.
A total of 8 patients, 4 (50%) males and 4 (50%) females between 1 and 8 (mean 6.3) months, were diagnosed to have ISH according to their clinical symptoms and pathological findings that showed hyaline depositions in skin biopsies.
Six of these patients died between the age of 24 and 28 months. Two of them died because of respiratory failure and pneumonia, two others died with diarrhea and one of unknown cause.
Only one patient had positive familial history of an involved sister, both (12.5%) died before the age of 18 months with septicemia. All patients had good prenatal progress and birth weights over 2200g.
In seven (87.5%) patients, first manifestations of disease were joint stiffness and limitation of movement that progressed to fixed flexion contractures. Joint stiffness began at age 1 to 7 months with a mean age of 2.5 months and only in one case it was present from birth on. Diarrhea as one of the main complaints began in average at the age of 3 months and only in one case first manifestation of disease was noticed at 40 days of life.
Skin changes occurred after involvement of joints in almost all cases with skin thickening, hyper pigmentated plaques, purplish papules especially around ears and neck (Fig. 1). Gingival hypertrophy was seen in all cases (Fig. 2). Clinical presentations of ISH in our patients were shown in table 1.
Fig. 1:
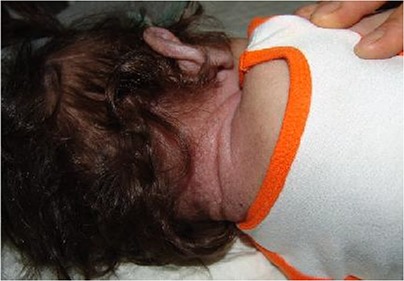
Thickened and hyperpigmented skin around neck in Infantile Systemic Hyalinosis
Fig. 2:
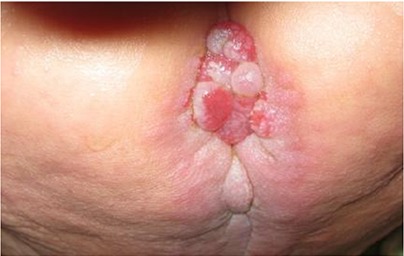
Perianal nodules in 14-month old patient with Infantile Systemic Hyalinosis
Table 1:
Clinical presentations and frequency in 8 patients with ISH
| Features | n (%) |
|---|---|
| Joint contracture | 8 (100) |
| Skin thickness & nodules | 8 (100) |
| Diarrhea | 8 (100) |
| Preanal nodules | 4 (50) |
| Gingival hyperplasia | 8 (100) |
| Failure to thrive | 8 (100) |
| Blond hair | 2 (25) |
| Osteoporosis | 7 (87.5) |
In four (50%) patients, perianal nodules were seen (Fig. 3). Internal otosclerosis existed in two (25%) cases. Two (25%) cases had blond hairs without similarity to their parents. All patients had normal mental and cognitive functions. One patient (12.5%) had consanguineous parents. All cases had osteopenic bones on their first radiography. Height and weight growth indices were decreased in all patients.
Fig. 3:

Joint contractures, hypertrophic gum and blond hair in a patient with Infantile Systemic Hyalinosis
Six of patients had terminated follow-up visits and took conservative treatments with duration of 6 to 18 months. In the meantime severe diarrhea sustained and joint contractions progressed, two others never came back for examination and only once answered our phone calls. Six of patients (75%) died and two are alive but very sick.
Discussion
ISH was first described in 1978 by Nezelof and his colleagues in a 1-month Portuguese boy with painful limitation of joints movement, gingival and skin hypertrophy [4] . Since then, until 2000, about 18 cases have been reported worldwide. In 2008, following 19 reported cases from Saudi Arabia and 3 from Iran, the number of affected patients increased [2,5,6] .
Pathogenesis is still unknown. Histopathology of the skin and other involved organ biopsies showed deposition of amorphous eosinophilic hyaline material in the dermis and viscera [7] .
Increased type IV collagen is major responsible for joint rigidity and hyaline deposition in GI tract. Intestinal lymphangectasia is another cause for protein losing entropathy [8–10] . On the other hand, Juvenile Hyaline Fibromatosis (JHF) according to same mutations and symptoms seems to be another form of ISH [9,11] .
Both diseases have autosomal ressesive pattern of heredity. JHF presents with nodules and papules, particularly on scalp, ears, nose and hands, gingival hypertrophy and joint contractures. JHF eventually progresses to osteoporosis in late childhood. Patients live usually 2 to 3 decades [3] .
The responsible gene for both has been mapped on chromosome 4q21. A mutation in the gene encoding ANTRX2/capillary morphogenesis protein 2 (CMG2) is the main cause of these diseases. CMG2 is a trans membrane protein which is induced during capillary morphogenesis. This protein CMG2 has an extracellular region that contains a von Willebrand type A domain that shows strong binding to laminin and collagen IV, both of which are markedly induced in endothelial cell morphogenesis, If a mutation affects the laminin sites, severe form of ISH occurs [9,12] .
In the Middle East, 19 reported cases from Saudi Arabia and 8 from Iran, revealed that this region has a higher incidence rate. Perhaps marriage between families and clans are main reason [2,6,13] .
ISH is a very devastative disease and no effective treatment has been yet reported, studies are still incomplete and continued. Oral d-penicillamin improves joint movement and flexibility in only a few cases. Physiotherapy, nutritional support and pain reductions are only effective therapy that partially improves the quality of life in these patients. Accurate diagnosis of this disease and its genetic analysis will enable families to prevent unnecessary and disturbing procedures for their children and reduce the risk of its recurrence in their families [10,13,14] .
Conclusion
ISH is a very rare fatal disorder and our knowledge of this genetic disorder is very limited. Reporting 8 patients from Iran and representing their clinical manifestations and follow up results may become helpful for future studies. In our study, 75% patients died before 2 years old due to severe diarrhea, malabsorption and/or infection.
References
- 1. Shin HT, Paller A, Hoganson G, et al. Infantile systemic hyalinosis. J Am Acad Dermatol 2004; 50 (Suppl 2): S61– 4. [DOI] [PubMed] [Google Scholar]
- 2. Aghighi Y, Bahremand S, Nematollahi LR. Infantile systemic hyalinosis: report of three Iranian children and review of the literature. Clin Rheumatol 2007; 26 (1): 128– 30. [DOI] [PubMed] [Google Scholar]
- 3. Dhingra M, Amladi S, Savant S, et al. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: divergent expressions of the same genetic defect?. Indian J Dermatol Venereol Leprol 2008; 74 (4): 371– 4. [DOI] [PubMed] [Google Scholar]
- 4. Nézelof C, Letourneux-Toromanoff B, Griscelli C, et al. Painful disseminate fibromatosis (systemic hyalinosis): a new hereditary collagen dysplasia. Arch Fr Pediatr 1978; 35 (10): 1063– 74. [PubMed] [Google Scholar]
- 5. Lindvall LE, Kormeili T, Chen E, et al. Infantile systemic hyalinosis: Case report and review of the literature. J Am Acad Dermatol 2008; 58 (2): 303– 7. [DOI] [PubMed] [Google Scholar]
- 6. Al-Mayouf SM, Al Mehaidib A, Bahabri S, et al. Infantile systemic hyalinosis: a fatal disorder commonly diagnosed among Arabs. Clin Exp Rheumatol 2005; 23 (5): 717– 20. [PubMed] [Google Scholar]
- 7. Glover MT, Lake BD, Atherton DJ. Clinical, histologic, and ultrastructural findings in two cases of infantile systemic hyalinosis. Pediatr Dermatol 1992; 9 (3): 255– 8. [DOI] [PubMed] [Google Scholar]
- 8. Büyükgebiz B, Oztürk Y, Arslan N, et al. A rare cause of protein-losing enteropathy and growth retardation in infancy: infantile systemic hyalinosis Turk J Pediatr 2003; 45 (3): 258–60. [PubMed] [Google Scholar]
- 9. Dowling O, Difeo A, Ramirez MC, et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 2003; 73 (4): 957– 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Al Reheili K, Al Mehaidib A, Alsaleem K, et al. Intestinal lymphangiectasia in a patient with infantile systemic hyalinosis syndrome: a rare cause of protein-losing enteropathy. Ann Saudi Med 2012; 32 (2): 206– 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Urbina F, Sazunic I, Murray G. Infantile systemic hyalinosis or juvenile hyaline fibromatosis?. Pediatr Dermatol 2004; 21 (2): 154– 9. [DOI] [PubMed] [Google Scholar]
- 12. Hanks S, Adams S, Douglas J, et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 2003; 73 (4): 791– 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stucki U, Spycher MA, Eich G, et al. Infantile systemic hyalinosis in siblings: clinical report, biochemical and ultrastructural findings, and review of the literature. Am J Med Genet 2001; 100 (2): 122– 9. [DOI] [PubMed] [Google Scholar]
- 14. Sahn EE, Salinas CF, Sens MA, et al. Infantile systemic hyalinosis in a black infant. Pediatr Dermatol 1994; 11 (1): 52–60. [DOI] [PubMed] [Google Scholar]