

Abstract
Hepatic encephalopathy (HE) is a major neurological complication of severe liver disease that presents in acute and chronic forms. While elevated brain ammonia level is known to be a major etiological factor in this disorder, recent studies have shown a significant role of neuroinflammation in the pathogenesis of both acute and chronic HE. This review summarizes the involvement of ammonia in the activation of microglia, as well as the means by which ammonia triggers inflammatory responses in these cells. Additionally, the role of ammonia in stimulating inflammatory events in brain endothelial cells (ECs), likely through the activation of the toll-like receptor-4 and the associated production of cytokines, as well as the stimulation of various inflammatory factors in ECs and in astrocytes, are discussed. This review also summarizes the inflammatory mechanisms by which activation of ECs and microglia impact on astrocytes leading to their dysfunction, ultimately contributing to astrocyte swelling/brain edema in acute HE. The role of microglial activation and its contribution to the progression of neurobehavioral abnormalities in chronic HE are also briefly presented. We posit that a better understanding of the inflammatory events associated with acute and chronic HE will uncover novel therapeutic targets useful in the treatment of patients afflicted with HE.
Keywords: ammonia, neuroinflammation, hepatic encephalopathy
Abbreviations: HE, hepatic encephalopathy; AHE, acute hepatic encephalopathy; ALF, acute liver failure; LPS, lipopolysaccharide; ONS, oxidative/nitrative stress; NF-κB, nuclear factor-kappaB; FHF, fulminant hepatic failure; TNF-α, tumor necrosis factor-alpha; IL, interleukin; HO, hemoxygenase; iNOS, inducible nitric oxide synthase; BBB, blood–brain barrier; BDL, bile duct ligation; cNOS, constitutive nitric oxide synthase; NOX, NADPH oxidase; RONS, reactive oxygen and nitrogen species; MAPK, mitogen-activated protein kinases; PLA2, phospholipase-A2; ECs, endothelial cells; COX2, cyclooxygenase-2; Tg, transgenic; WT, wild type; TLR, Toll-like receptor
Hepatic encephalopathy (HE) is the major neurological complication of severe liver disease. It presents in two forms, chronic HE and acute HE. Chronic HE (portal-systemic encephalopathy) usually occurs in patients with alcoholic liver cirrhosis and is characterized by impaired neurological function, including changes in personality, altered mood, diminished intellectual capacity, and abnormal muscle tone and tremor.1 Acute HE (AHE, acute liver failure, ALF; fulminant hepatic failure) generally occurs following massive liver necrosis due to viral hepatitis (hepatitis B and C), hepatic neoplasms, vascular causes, or exposure to acetaminophen and other hepatotoxins. AHE is associated with the abrupt onset of delirium, seizures, and coma.
The principal pathological change in chronic HE is characterized by Alzheimer type II astrocytosis,2,3 which is characterized by astrocytes possessing enlarged, pale nuclei, often occurring in pairs, with the nuclei frequently displaying prominent nucleoli. Cerebral edema and associated increase in intracranial pressure leading to brain herniation are the characteristic features of AHE which occurs in upto 80% of patients with AHE4–6 and represents the most frequent cause of death in these patients (70% mortality).4,7 While the basis for the edema in AHE is poorly understood, astroglial swelling (cytotoxic edema) dominates the pathology in experimental animals,8–11 as well as in humans.12 Of interest, no significant or consistent morphologic changes have been identified in neurons or other neural cells. Such findings prompted the suggestion that HE fundamentally represents a primary “astrogliopathy”.13
While the precise involvement of astrocytes in HE is incompletely understood, it is known that the enzyme responsible for ammonia metabolism in brain, glutamine synthetase, is exclusively found in astrocytes.14 Such metabolism in the setting of elevated ammonia levels elicits a number of untoward events in astrocytes which exerts negative consequences on other neural cell. For review, see Norenberg13; Norenberg et al, 1992.15
While the molecular basis for the neurological disorder in acute and chronic liver failure remains incompletely understood, elevated blood and brain ammonia levels have been strongly implicated in its pathogenesis. Factors that lead to increased levels of blood or brain ammonia have been shown to worsen HE, whereas reducing blood ammonia levels alleviate HE.16 Additionally, clinical, pathological, and biochemical changes observed in HE are reproduced by increasing blood or brain ammonia in experimental animals,2 while exposure of cultured astrocytes to ammonium salts reproduces the morphological and biochemical findings.17–20
Recent studies have shown a significant role of inflammation in the mechanism of HE, in particular, the involvement of cytokines and lipopolysaccharide (LPS; endotoxin) in the pathogenesis of acute and chronic HE.21,22 The focus of this review is to emphasize the involvement of ammonia in stimulating an inflammatory response in brain endothelial cells, microglia and astrocytes, in both acute and chronic HE.
Mechanisms of ammonia neurotoxicity
Impaired bioenergetics, electrophysiological defects, changes in intracellular pH, altered glutamateric and GABAergic neurotransmission, excitotoxicity, involvement of the peripheral benzodiazepine receptor,23 oxidative/nitrative stress (ONS) and induction of the mitochondrial permeability transition, have all been identified as major mechanistic events triggered by ammonia.24,25 Additionally, activation of intracellular signaling systems, including c-fos,26 mitogen-activated protein kinases,26 protein kinase G,27 Src kinase family,28 ciliary neurotrophic factor,26 and the transcription factors p53,29 SP-126 and nuclear factor-kappaB (NF-κB)30–32 have all been shown to be involved in the mechanism of ammonia neurotoxicity, particularly in neuroinflammation.
Recently, the stimulation of ion transporters, including the Na+-K+-Cl− cotransporter-133,34 and the nonselective cation (NCCa-ATP) channel,35 as well as an increase in the level of the astrocytic plasma membrane protein aquaporin-4,36 have all been shown to be involved in the mechanism of ammonia neurotoxicity, particularly in the development of the cytotoxic brain edema associated with acute HE. While the signaling systems that are stimulated in acute HE are well established, evidence for the involvement of signaling factors in chronic HE remains sparse.
Inflammation in hepatic encephalopathy
A growing body of evidence suggests that inflammation plays an important role in the development of HE. The concept that inflammation is involved in HE was first proposed by Gans et al, 1971,37 based on findings that peripheral infections and inflammation were associated with fulminant hepatic failure (FHF). This aspect was further elaborated upon by Wilkinson et al, 1974; Liehr et al, 1976; Wyke et al, 1982.38–40 Izumi et al 199541 documented increased levels of IL-6 in plasma in both FHF and in chronic HE. Subsequently, Wigmore et al, 199842 and Wright et al 200743 reported increased levels of serum tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6) and their arterio-venous differences, consistent with a brain cytokine efflux in patients with acute HE, supporting the presence of a central nervous system inflammatory component (neuroinflammation) in HE when associated with peripheral infections. Noteworthy, blood-derived cytokines, likely arising from necrotic liver and/or from sepsis, are well-known to freely cross blood–brain barrier under normal conditions.44
-
a.
Role of inflammation in the brain edema associated with acute HE
As noted above, a major complication of acute HE is the development of cytotoxic brain edema. The involvement of neuroinflammation in acute HE was established by Chung et al, 2001,45 who showed a protective effect of the anti-inflammatory agent, indomethacin (a potent inhibitor of cyclooxygenase-2), against the development of brain edema in rats with a portacaval anastomosis that were infused with ammonia45; indomethacin also normalized the intracranial pressure in patients associated with acute HE.46 Subsequently, Jiang et al, 200947 documented that a member of the tetracycline class of antibiotic, minocycline, also attenuated the encephalopathy grade and prevented brain edema formation in experimental ALF. It should noted, however, that minocycline itself induces liver damage,48,49 which precludes its possible therapeutic use for the treatment of acute HE.
It was further shown that AHE results in a lesser degree of brain edema in transgenic mice deficient in TNF-α, IL-1β and IL-6 receptors, as compared to wild type mice,50 and that etanercept, a TNF-α neutralizing molecule, prevented the onset of coma stages of HE as well as liver injury induced by AOM (Chastre et al, 2012).51 Induction of endotoxemia with LPS in rats was subsequently shown to aggravate the brain edema and encephalopathy associated with ALF.43 Collectively, these findings suggest an important role of “central” inflammation in the development of the brain edema in ALF. For general reviews on neuroinflammation, see references.21,52
Consistent with the role of inflammation in the brain edema associated with AHE, we previously reported that treatment of cultured astrocytes with inflammatory cytokines (TNF-α, IL-1β, IL-6 and IFN-γ) caused astrocyte swelling.53 Moreover, the swelling caused by these cytokines was markedly potentiated when astrocytes were pre-treated with ammonia for 24 h, and then exposed to cytokines for an additional 24 h. Additionally, astrocyte cultures exposed to a combination of cytokines (TNF-α, IL-1β, IL-6 and IFN-γ) were recently shown to activate NF-κB and that such effect was potentiated by ammonia,53 while blocking the activation of NF-κB prevented the astrocyte swelling.53 Since cytokines are well-known inducers of ONS and activation of NF-κB,54 it appears that in addition to ammonia, cytokines also contribute to the ONS and to the exacerbation of the astrocyte swelling acute HE.
Similarly, exposure of cultured astrocytes to a combination of ammonia and recombinant IL-1β resulted in significant additive increases in the expression of both hemoxygenase-1 (HO-1) and inducible nitric oxide synthase (iNOS).55 These findings are consistent with the worsening effects of ammonia and inflammation on the CNS complications of ALF.55 Together, these studies strongly suggest a synergistic interaction between ammonia, reactive oxygen/nitrogen species and inflammatory cytokines in the astrocyte swelling/brain edema associated AHE.53
-
b.
Microglial activation and its contribution to the development of astrocyte swelling and brain edema in ALF
While the precise pathophysiological mechanisms responsible for the development of astrocyte swelling/brain edema in AHE have not been fully elucidated, it was recently proposed that microglial activation and the subsequent synthesis and release of pro-inflammatory cytokines are involved.38,43,47,56 Microglia are well-known to induce inflammatory responses in brain,57 as they represent the primary resident immune cells of the CNS. Microglia can be activated by a wide number of factors, including glutamate, pro-inflammatory cytokines (derived as a consequence of liver necrosis and/or sepsis), LPS, as well as by extracellular potassium.57 Activated microglia, in turn, can release the pro-inflammatory cytokines IL-1α, IL-1β and TNF-α, all of which play a crucial role in neurodegeneration.57
Upregulation of CD11b/c immunoreactivity, consistent with microglial activation, was observed in brains of rats with AHE at the time of coma.47 Additionally, increased IL-1β, TNF-α, and IL-6 mRNAs were observed in brains of rats with AHE, and the magnitude of expression of these pro-inflammatory cytokines correlated well with the progression of the encephalopathy and the onset of brain edema.47 Microglial activation was also identified in experimental models of hyperammonemia,58 as well as in brain tissue obtained at autopsy from patients with ALF.59
Increased OX-42 and OX-6 immunoreactivities in microglia (suggestive of their activation), was shown to be accompanied by increased expression of interleukins (IL-1β, IL-6) and TNF-α in the cortex of rats with AHE.47 Minocycline treatment prevented both the microglial activation, as well as the upregulation of IL-1β, IL-6 and TNF-α, which was associated with a concomitant attenuation of the encephalopathy and brain edema. These findings are consistent with the involvement of microglial activation in the development of the brain edema in AHE.47
We recently identified the synthesis and release of the cytokines IL-6 and TNF-α when primary cultures of microglia were exposed to ammonia (1 mM for 72 h or 5 mM for 24 h).60 Additionally, exposure of microglia to ammonia resulted in ONS60 and in the activation of the transcription factor NF-κB. Since inflammation is known to influence cell volume in other conditions,61 we recently examined the potential role of microglia in the mechanism of ammonia-induced astrocyte swelling. Accordingly, conditioned media (CM) derived from ammonia-treated (5 mM, NH4Cl) cultured microglia, when added to cultured astrocytes, resulted in significant cell swelling (32%).60 Such swelling was synergistically increased when astrocytes were additionally exposed to ammonia. We also found that CM from ammonia-treated microglia containing Tempol (a superoxide scavenger), or uric acid (a peroxynitrite scavenger), when added to astrocytes, resulted in a marked reduction in cell swelling as compared to the effect of CM from ammonia-only treated microglia.60
In unpublished observations, we found the activation of the toll-like receptor-4 (TLR4; an inflammatory response factor), in ammonia-treated cultured microglia. Additionally, astrocytes exposed to CM from TLR4 gene-silenced microglia that were then treated with ammonia, showed a lesser degree of astrocyte swelling as compared to similarly treated controls, strongly suggesting a contribution of microglial TLR4 to the development of the cytotoxic brain edema associated with AHE.
While the above studies strongly suggest that activated microglia contribute to the cytotoxic edema in acute HE, Rangroo-Thrane et al62 reported that while microglia are indeed activated in the azoxymethane-induced animal model of ALF, such activation did not contribute to the cytotoxic brain edema. Instead, these authors noted that the azoxymethane-induced vasogenic edema was associated with a breakdown of the blood–brain barrier (BBB), as well as the activation of microglia. It should be noted, however, that a generalized breakdown of the BBB (as occurs in vasogenic edema) is not a feature of ALF in humans. Further, the liver toxin used in this study, azoxymethane, was recently shown to induce a breakdown of the BBB in vivo.63 Subsequently, a direct toxic effect on brain endothelial cells by azoxymethane was observed in an in vitro model of the BBB, resulting in a severe breakdown of the BBB.64 These findings suggest that the use of azoxymethane-induced hepatotoxicity as an experimental model of ALF is not suitable for the study of acute HE.
Rangroo-Thrane et al, 201262 further commented that microglial activation did not occur in ammonia infused mice, despite the mice developing pathologically increased plasma ammonia levels and severe neurological dysfunction. While considerable evidence documents that microglial activation contributes to the pathogenesis of AHE (as noted above), the reason for the absence of microglial activation in their study is not clear. However, these investigators used a relatively high concentration of ammonia (7.5 mmol/kg NH4Cl, i.p.) which is not pathophysiologically relevant as this concentration of ammonia induces acute deterioration of neurological function, development of myoclonic seizures and death within 5–10 min after ammonia injection,65–67 thereby precluding the subsequent identification of microglial activation. The basis for such deterioration following treatment with high levels of ammonia is not known, but it suggests the possibility of some generalized negative systemic event(s) (e.g., in heart or lung) occurring as a consequence of the hyperammonemia.
Microglial activation was also reported in animal models of chronic HE due to bile duct ligation (BDL)68,69 or to portacaval shunts.70,71 However, a recent report did not observe microglial activation in the BDL model of chronic HE.72 While these studies68,72 employed an identical animal model of chronic HE, identical time point of examination (4 weeks), as well as used the same microglial marker (Iba1), the reason for such discrepancies is unclear.
Neurobehavioral abnormalities are the major clinical consequences of chronic HE.56 While significant evidence indicates the involvement of inflammation in the astrocyte swelling/brain edema in acute HE, relatively few studies have examined the potential role of inflammation in the neurobehavioral defects associated with chronic HE. Recently, however, Rodrigo et al, 201056 reported that chronic/moderate hyperammonemia or BDL resulted in microglial activation, increased inducible nitric oxide synthase activity, as well as increased brain levels of interleukin-1β, and prostaglandin E2, and that these changes were associated with impaired cognitive and motor functions. These authors further demonstrated that ibuprofen, a non-steroidal anti-inflammatory agent, reduced the microglial activation and restored cognitive and motor functions in hyperammonemic and in rats with BDL. Additionally, rats that underwent portal vein ligation were shown to exhibit impaired locomotor activity, as well as increased inflammatory events, including the presence of protein tyrosine nitration, RNA oxidation, and IL-6 mRNA increase in brain.73 These authors also reported that prevention of protein tyrosine nitration and RNA oxidation with indomethacin, a non-specific cyclooxygenase-2 inhibitor, prevented brain protein tyrosine nitration, RNA oxidation, as well as disturbances in locomotor activity associated with chronic HE. Taken together, these reports suggest that inflammation also contributes to the neurobehavioral deficits observed in chonic HE.
Astrocytes in the inflammatory response in hepatic encephalopathy
As noted above, swelling of astrocytes (cytotoxic brain edema) is the most prominent neuropathological abnormality in AHE. Additionally, a direct effect of ammonia on the activation of inflammatory factors in cultured astrocytes has been identified. Activation of NF-κB, and the tumor suppressor gene p53 were shown in ammonia-treated cultured astrocytes.29–31 Additional studies in support of an astrocytic inflammatory response in vitro, is derived from recent reports demonstrating that inflammatory factors such as constitutive nitric oxide synthase (cNOS) and NADPH oxidase (NOX) and the consequent generation of reactive oxygen and nitrogen species (RONS), phospholipase-2 (PLA2) and mitogen-activated protein kinases (MAPKs), were all activated in cultured astrocytes by ammonia.74,75 Moreover, inhibition of these factors was shown to reduce the ammonia-induced astrocyte swelling in culture.74,75 Altogether, these findings strongly suggest that ammonia is capable of inducing an inflammatory response in astrocytes, independent of any influence from other neural cells or from any extracellular stimuli, and that such effects may contribute to the cytotoxic brain edema in AHE.
Cerebral endothelial cells (ECs)
ECs are the first resident brain cells exposed to blood-borne ‘‘noxious agents’’ (i.e., ammonia, CKs, LPS). Since ECs are also capable of activating a number of inflammatory factors, including inducible nitric oxide synthase (iNOS), NADPH oxidase (NOX), phospholipase A2 (PLA2), cyclooxygenase-2 (COX2), as well as NF-κB,76 it is possible that these inflammatory factors may generate “cell swelling mediators”, including arachidonic acid, RONS, prostaglandins and CKs,76 that may ultimately result in astrocyte swelling/brain edema in acute HE. In support of this view, we recently reported that cultured ECs treated with ammonia showed evidence of ONS77–80 and the activation of NF-κB.77,79
As noted above, we also found increased TLR4 protein expression and its associated signaling cascades, the myeloid differentiation primary response gene 88 (MyD88), and the TIR-domain-containing adapter-inducing interferon-β (TRIF) protein expression in ammonia-treated cultured rat brain ECs.79 Additionally, exposure of cultured ECs to ammonia stimulated the release of cytokines and free radicals, all of which are known to enhance inflammatory responses in other conditions. Further, receptors for CKs, including IL-1β and TNF-α, have been identified in brain ECs in experimental models of sepsis,81 and that activation of these receptors further stimulated the synthesis and release of additional CKs leading to an exacerbation of the brain edema in acute HE.82
To examine whether ammonia, CKs and LPS stimulate ECs to release inflammatory mediators that may contribute to the astrocyte swelling and brain edema in AHE, primary cultures of rat brain endothelial cells were treated with ammonia, CKs and LPS and the conditioned medium derived from these treatments were added to cultured astrocytes and cell volume was determined. We found that CM from ammonia-treated ECs when added to astrocytes caused significant cell swelling, and such swelling was potentiated when astrocytes were exposed to CM from ECs treated with a combination of ammonia, LPS and CKs.77,79 We also found an additive effect when astrocytes were exposed to ammonia, along with CM from ammonia-treated ECs. CM derived from ECs treated with ammonia, along with antioxidants or the NF-κB inhibitor BAY 11-7082, when added to astrocytes, resulted in a significant reduction in cell swelling.77,79 Additionally, astrocytes exposed to CM from TLR4 gene-silenced ECs that were treated with ammonia, cytokines or LPS, alone or in combination, showed less cell swelling as compared to similarly treated controls.77,79 Altogether, these findings suggest that ECs are indeed capable of activating a plethora of inflammatory factors that play a critical role in the development of the brain edema associated with AHE.
We additionally found increased TLR4 protein expression in brain ECs after treatment of rats with the liver toxin thioacetamide (TAA), and that the administration of TAA to transgenic mice lacking TLR4 exhibited a lesser amount of brain edema, as compared to wild type mice.79 These studies strongly suggest that ECs significantly contribute to the astrocyte swelling/brain edema in acute HE, in part, as a consequence of increased TLR4 protein expression triggered by blood-borne noxious agents (e.g., LPS, cytokines). While a major role of brain endothelial cells has been shown in the pathogenesis of AHE, whether endothelial cells also play a significant role in the pathogenesis of chronic HE, remains to be established.
Role of nuclear factor-kappaB in vivo
As noted above, the activation of NF-κB represents a major inflammatory response in brain. We recently examined whether transgenic (Tg) mice that have a functional inactivation of astrocytic NF-κB (NF-κB null mice) are resistant to ALF-associated brain edema. We found that wild type (WT) mice when treated with TAA showed a significant increase in brain water content, along with prominent astrocyte swelling and associated increase in iNOS immunoreactivity.32 By contrast, NF-κB null mice treated with TAA did not exhibit brain edema, histological changes, nor displayed an increase in iNOS immunoreactivity.32 Additionally, astrocyte cultures derived from NF-κB null mice showed no cell swelling nor morphological abnormalities when exposed to ammonia. By contrast, ammonia significantly increased cell swelling in cultured astrocytes from WT mice and displayed cytological abnormalities.32 In aggregate, these findings indicate that the activation of NF-κB represents an important inflammatory factor in the development of astrocyte swelling/brain edema in the setting of AHE.
Summary
Inflammation, in conjunction with ammonia, plays an important role in the pathogenesis of HE. The application of ammonia to cultured astrocytes stimulates a number of inflammatory signaling systems (NF-κB, PLA2, NOS, NOX, COX2) that leads to astrocyte swelling/brain edema in acute hepatic encephalopathy. Additionally, ammonia activates microglia and stimulates brain endothelial cells; some of these events also occur in experimental animals and in humans with HE. The consequences of this inflammatory response (synthesis and release of inflammatory factors from activated microglia and endothelial cells), along with hyperammonemia, leads to severe astrocyte swelling/brain edema in acute hepatic encephalopathy. The presence of both central and peripheral inflammation has also been shown to contribute to the neurobehavioral abnormalities observed in chronic HE. A schematic diagram illustrating mechanisms by which inflammation contributes to the astrocyte dysfunction in HE is presented in Figure 1. Studies focusing on the identification of molecular mechanisms and cell–cell interactions during inflammation and hyperammonia may provide useful therapeutic targets for the treatment of both acute and chronic hepatic encephalopathy.
Figure 1.
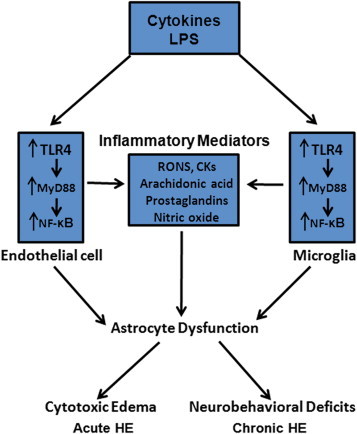
Mechanisms of neuroinflammation in hepatic encephalopathy. Inflammatory cytokines and lipopolysaccahride (LPS, endotoxin) activate endothelial cells and microglia that subsequently leads to the stimulation of the Toll-like receptor-4 (TLR4). TLR4 then activates down-stream factors, including NF-κB and myeloid protein88 (MyD88)-dependent pathways, which subsequently leads to the production of inflammatory mediators, including reactive oxygen and nitrogen species (RONS), arachidonic acid, and prostaglandins. These inflammatory mediators ultimately impact on astrocytes, leading to their dysfunction resulting in brain edema in acute HE, and likely contribute to the neurobehavioral deficits in chronic HE.
Conflicts of interest
All authors have none to declare.
Acknowledgments
This work was supported by a Merit Review from the Department of Veterans Affairs and by a National Institutes of Health grant DK063311.
References
- 1.Lockwood A.H., Yap E.W., Wong W.H. Cerebral ammonia metabolism in patients with severe liver disease and minimal hepatic encephalopathy. J Cereb Blood Flow Metab. 1991;11:337–341. doi: 10.1038/jcbfm.1991.67. [DOI] [PubMed] [Google Scholar]
- 2.Norenberg M.D. The astrocyte in liver disease. In: Fedoroff S., Hertz L., editors. Vol. 2. Academic Press; New York: 1981. pp. 303–352. (Advances in Cellular Neurobiology). [Google Scholar]
- 3.Martin H., Voss K., Hufnagl P., Wack R., Wassilew G. Morphometric and densitometric investigations of protoplasmic astrocytes and neurons in human hepatic encephalopathy. Exp Pathol. 1987;32:241–250. doi: 10.1016/s0232-1513(87)80035-x. [DOI] [PubMed] [Google Scholar]
- 4.Lee W.M. Acute liver failure. Semin Respir Crit Care Med. 2012;33:36–45. doi: 10.1055/s-0032-1301733. [DOI] [PubMed] [Google Scholar]
- 5.Jalan R., Olde Damink S.W., Hayes P.C., Deutz N.E., Lee A. Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol. 2004;41:613–620. doi: 10.1016/j.jhep.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Larsen F.S., Hansen B.A., Blei A.T. Intensive care management of patients with acute liver failure with emphasis on systemic hemodynamic instability and cerebral edema: a critical appraisal of pathophysiology. Can J Gastroenterol. 2000;14(suppl D):105D–111D. doi: 10.1155/2000/493629. [DOI] [PubMed] [Google Scholar]
- 7.Shawcross D.L., Wendon J.A. The neurological manifestations of acute liver failure. Neurochem Int. 2012;60:662–671. doi: 10.1016/j.neuint.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Norenberg M.D. A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest. 1977;36:618–627. [PubMed] [Google Scholar]
- 9.Voorhies T.M., Ehrlich M.E., Duffy T.E., Petito C.K., Plum F. Acute hyperammonemia in the young primate. Physiologic and neuropathological correlates. Pediatr Res. 1983;17:970–975. doi: 10.1203/00006450-198312000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Traber P.G., Dal Canto M.C., Ganger D., Blei A.T. Electron microscopic evaluation of brain edema in rabbits with galactosamine-induced fulminant hepatic failure: ultrastructure and integrity of the blood–brain barrier. Hepatology. 1987;7:1272–1277. doi: 10.1002/hep.1840070616. [DOI] [PubMed] [Google Scholar]
- 11.Swain M.S., Blei A.T., Butterworth R.F., Kraig R.P. Intracellular pH rises and astrocytes swell after portacaval anastomosis in rats. Am J Physiol Regul Integr Comp Physiol. 1991;261:R1491–R1496. doi: 10.1152/ajpregu.1991.261.6.R1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kato M., Hughes R.D., Keays R.T., Williams R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology. 1992;15:1060–1066. doi: 10.1002/hep.1840150615. [DOI] [PubMed] [Google Scholar]
- 13.Norenberg M.D. The role of astrocytes in hepatic encephalopathy. Neurochem Pathol. 1987;6:13–33. doi: 10.1007/BF02833599. [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Hernandez A., Bell K.P., Norenberg M.D. Glutamine synthetase: glial localization in brain. Science. 1977;195:1356–1358. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- 15.Norenberg M.D., Neary J.T., Bender A.S., Dombro R.S. Hepatic encephalopathy: a disorder in glial-neuronal communication. Prog Brain Res. 1992;94:261–269. doi: 10.1016/s0079-6123(08)61756-2. [DOI] [PubMed] [Google Scholar]
- 16.Conn H., Lieberthal M. Williams & Wilkins Co; Baltimore: 1978. The Hepatic Coma Syndrome and Lactulose. [Google Scholar]
- 17.Gregorios J.B., Mozes L.W., Norenberg L.O.B., Norenberg M.D. Morphologic effects of ammonia on primary astrocyte cultures. I: light microscopic studies. J Neuropathol Exp Neurol. 1985;44:397–403. doi: 10.1097/00005072-198507000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Gregorios J.B., Mozes L.W., Norenberg M.D. Morphologic effects of ammonia on primary astrocyte cultures. II: electron microscopic studies. J Neuropathol Exp Neurol. 1985;44:404–414. doi: 10.1097/00005072-198507000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Blei A.T., Olafsson S., Therrien G., Butterworth R.F. Ammonia-induced brain edema and intracranial hypertension in rats after portacaval anastomosis. Hepatology. 1994;19:1437–1444. [PubMed] [Google Scholar]
- 20.Clemmesen J.O., Larsen F.S., Kondrup J., Hansen B.A., Ott P. Cerebral herniation in acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29:648–653. doi: 10.1002/hep.510290309. [DOI] [PubMed] [Google Scholar]
- 21.Coltart I., Tranah T.H., Shawcross D.L. Inflammation and hepatic encephalopathy. Arch Biochem Biophys. 2013 Aug 15;536(2):189–196. doi: 10.1016/j.abb.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Odeh M. Pathogenesis of hepatic encephalopathy: the tumour necrosis factor-alpha theory. Eur J Clin Invest. 2007;37:291–304. doi: 10.1111/j.1365-2362.2007.01778.x. [DOI] [PubMed] [Google Scholar]
- 23.Norenberg M.D., Rama Rao K.V., Jayakumar A.R. Signaling factors in the mechanism of ammonia neurotoxicity. Metab Brain Dis. 2009;24:103–117. doi: 10.1007/s11011-008-9113-6. [DOI] [PubMed] [Google Scholar]
- 24.Norenberg M.D., Jayakumar A.R., Rama Rao K.V., Panickar K.S. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab Brain Dis. 2007;22:219–234. doi: 10.1007/s11011-007-9062-5. [DOI] [PubMed] [Google Scholar]
- 25.Rama Rao K.V., Jayakumar A.R., Norenberg M.D. Brain edema in acute liver failure: mechanisms and concepts. Metab Brain Dis. 2014 doi: 10.1007/s11011-014-9502-y. In press. [DOI] [PubMed] [Google Scholar]
- 26.Bodega G., Suárez I., Almonacid L. Effect of ammonia on ciliary neurotrophic factor mRNA and protein expression and its upstream signalling pathway in cultured rat astroglial cells: possible implication of c-fos, Sp1 and p38MAPK. Neuropathol Appl Neurobiol. 2007;33:420–430. doi: 10.1111/j.1365-2990.2007.00831.x. [DOI] [PubMed] [Google Scholar]
- 27.Konopacka A., Konopacki F.A., Albrecht J. Protein kinase G is involved in ammonia-induced swelling of astrocytes. J Neurochem. 2009;109(suppl 1):246–251. doi: 10.1111/j.1471-4159.2009.05802.x. [DOI] [PubMed] [Google Scholar]
- 28.Aspinall R.J., Weis S.M., Barnes L. A Src family kinase inhibitor improves survival in experimental acute liver failure associated with elevated cerebral and circulating vascular endothelial growth factor levels. Liver Int. 2011;31:1222–1230. doi: 10.1111/j.1478-3231.2011.02554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panickar K.S., Jayakumar A.R., Rao K.V., Norenberg M.D. Ammonia-induced activation of p53 in cultured astrocytes: role in cell swelling and glutamate uptake. Neurochem Int. 2009;55:98–105. doi: 10.1016/j.neuint.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 30.Schliess F., Görg B., Fischer R. Ammonia induces MK-801-sensitive nitration and phosphorylation of protein tyrosine residues in rat astrocytes. FASEB J. 2002;16:739–741. doi: 10.1096/fj.01-0862fje. [DOI] [PubMed] [Google Scholar]
- 31.Sinke A.P., Jayakumar A.R., Panickar K.S., Moriyama M., Reddy P.V., Norenberg M.D. NFkappaB in the mechanism of ammonia-induced astrocyte swelling in culture. J Neurochem. 2008;106:2302–2311. doi: 10.1111/j.1471-4159.2008.05549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jayakumar A.R., Bethea J.R., Tong X.Y., Gomez J., Norenberg M.D. NF-κB in the mechanism of brain edema in acute liver failure: studies in transgenic mice. Neurobiol Dis. 2011;41:498–507. doi: 10.1016/j.nbd.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jayakumar A.R., Liu M., Moriyama M. Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J Biol Chem. 2008;283:33874–33882. doi: 10.1074/jbc.M804016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jayakumar A.R., Valdes V., Norenberg M.D. The Na-K-Cl cotransporter in the brain edema of acute liver failure. J Hepatol. 2011;54:272–278. doi: 10.1016/j.jhep.2010.06.041. [DOI] [PubMed] [Google Scholar]
- 35.Jayakumar A.R., Valdes V., Tong X.Y., Shamaladevi N., Gonzalez W., Norenberg M.D. Sulfonylurea receptor 1 contributes to the astrocyte swelling and brain edema in acute liver failure. Transl Stroke Res. 2014;5:28–37. doi: 10.1007/s12975-014-0328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rama Rao K.V., Jayakumar A.R., Tong X., Curtis K.M., Norenberg M.D. Brain aquaporin-4 in experimental acute liver failure. J Neuropathol Exp Neurol. 2010;69:869–879. doi: 10.1097/NEN.0b013e3181ebe581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gans H., Mori K., Lindsey E. Septicemia as a manifestation of acute liver failure. Surg Gynecol Obstet. 1971;132:783–790. [PubMed] [Google Scholar]
- 38.Wilkinson S.P., Arroyo V., Gazzard B.G., Moodie H., Williams R. Relation of renal impairment and haemorrhagic diathesis to endotoxaemia in fulminant hepatic failure. Lancet. 1974;1:521–524. doi: 10.1016/s0140-6736(74)92711-1. [DOI] [PubMed] [Google Scholar]
- 39.Liehr H., Grün M., Brunswig D. Endotoxaemia in acute hepatic failure. Acta Hepatogastroenterol (Stuttg) 1976;23:235–240. [PubMed] [Google Scholar]
- 40.Wyke R.J., Canalese J.C., Gimson A.E., Williams R. Bacteraemia in patients with fulminant hepatic failure. Liver. 1982;2:45–52. doi: 10.1111/j.1600-0676.1982.tb00177.x. [DOI] [PubMed] [Google Scholar]
- 41.Izumi S., Hughes R.D., Langley P.G., Pernambuco J.R., Williams R. Acute phase response after liver transplantation for fulminant hepatic failure and cirrhosis. Transpl Int. 1995;8:340–345. [PubMed] [Google Scholar]
- 42.Wigmore S.J., Walsh T.S., Lee A., Ross J.A. Pro-inflammatory cytokine release and mediation of the acute phase protein response in fulminant hepatic failure. Intensive Care Med. 1998;24:224–229. doi: 10.1007/s001340050554. [DOI] [PubMed] [Google Scholar]
- 43.Wright G., Shawcross D., Olde Damink S.W., Jalan R. Brain cytokine flux in acute liver failure and its relationship with intracranial hypertension. Metab Brain Dis. 2007;22:375–388. doi: 10.1007/s11011-007-9071-4. [DOI] [PubMed] [Google Scholar]
- 44.Banks W.A., Kastin A.J., Broadwell R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation. 1995;2:241–248. doi: 10.1159/000097202. [DOI] [PubMed] [Google Scholar]
- 45.Chung C., Gottstein J., Blei A.T. Indomethacin prevents the development of experimental ammonia-induced brain edema in rats after portacaval anastomosis. Hepatology. 2001;34:249–254. doi: 10.1053/jhep.2001.26383. [DOI] [PubMed] [Google Scholar]
- 46.Clemmesen J.O., Hansen B.A., Larsen F.S. Indomethacin normalizes intracranial pressure in acute liver failure: a twenty-three-year-old woman treated with indomethacin. Hepatology. 1997;26:1423–1425. doi: 10.1002/hep.510260608. [DOI] [PubMed] [Google Scholar]
- 47.Jiang W., Desjardins P., Butterworth R.F. Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: protective effect of hypothermia. J Cereb Blood Flow Metab. 2009;29:944–952. doi: 10.1038/jcbfm.2009.18. [DOI] [PubMed] [Google Scholar]
- 48.Böcker R., Estler C.J., Ludewig-Sandig D. Evaluation of the hepatotoxic potential of minocycline. Antimicrob Agents Chemother. 1991;35:1434. doi: 10.1128/aac.35.7.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malcolm A., Heap T.R., Eckstein R.P., Lunzer M.R. Minocycline-induced liver injury. Am J Gastroenterol. 1996;91:1641–1653. [PubMed] [Google Scholar]
- 50.Bemeur C., Qu H., Desjardins P., Butterworth R.F. IL-1 or TNF receptor gene deletion delays onset of encephalopathy and attenuates brain edema in experimental acute liver failure. Neurochem Int. 2010;56:213–215. doi: 10.1016/j.neuint.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 51.Chastre A., Bélanger M., Beauchesne E., Nguyen B.N., Desjardins P., Butterworth R.F. Inflammatory cascades driven by tumor necrosis factor-alpha play a major role in the progression of acute liver failure and its neurological complications. PLoS One. 2012;7:e49670. doi: 10.1371/journal.pone.0049670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butterworth R.F. The liver-brain axis in liver failure: neuroinflammation and encephalopathy. Nat Rev Gastroenterol Hepatol. 2013;10:522–528. doi: 10.1038/nrgastro.2013.99. [DOI] [PubMed] [Google Scholar]
- 53.Rama Rao K.V., Jayakumar A.R., Tong X., Alvarez V.M., Norenberg M.D. Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J Neuroinflammation. 2010;7:66. doi: 10.1186/1742-2094-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frank-Cannon T.C., Alto L.T., McAlpine F.E., Tansey M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chastre A., Jiang W., Desjardins P., Butterworth R.F. Ammonia and proinflammatory cytokines modify expression of genes coding for astrocytic proteins implicated in brain edema in acute liver failure. Metab Brain Dis. 2010;25:17–21. doi: 10.1007/s11011-010-9185-y. [DOI] [PubMed] [Google Scholar]
- 56.Rodrigo R., Cauli O., Gomez-Pinedo U. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology. 2010;139:675–684. doi: 10.1053/j.gastro.2010.03.040. [DOI] [PubMed] [Google Scholar]
- 57.Garden G.A., Möller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 58.Görg B., Bidmon H.J., Häussinger D. Gene expression profiling in the cerebral cortex of patients with cirrhosis with and without hepatic encephalopathy. Hepatology. 2013;57:2436–2447. doi: 10.1002/hep.26265. [DOI] [PubMed] [Google Scholar]
- 59.Butterworth R.F. Neuroinflammation in acute liver failure: mechanisms and novel therapeutic targets. Neurochem Int. 2011;59:830–836. doi: 10.1016/j.neuint.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 60.Rao K.V., Brahmbhatt M., Norenberg M.D. Microglia contribute to ammonia-induced astrocyte swelling in culture. Metab Brain Dis. 2013;28:139–143. doi: 10.1007/s11011-012-9339-1. [DOI] [PubMed] [Google Scholar]
- 61.Khanna A., Kahle K.T., Walcott B.P., Gerzanich V., Simard J.M. Disruption of ion homeostasis in the neurogliovascular unit underlies the pathogenesis of ischemic cerebral edema. Transl Stroke Res. 2014;5:3–16. doi: 10.1007/s12975-013-0307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rangroo Thrane V., Thrane A.S., Chang J., Alleluia V., Nagelhus E.A., Nedergaard M. Real-time analysis of microglial activation and motility in hepatic and hyperammonemic encephalopathy. Neuroscience. 2012;220:247–255. doi: 10.1016/j.neuroscience.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen J.H., Yamamoto S., Steers J. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J Hepatol. 2006;44:1105–1114. doi: 10.1016/j.jhep.2005.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jayakumar A.R., Ruiz-Cordero R., Tong X.Y., Norenberg M.D. Brain edema in acute liver failure: role of neurosteroids. Arch Biochem Biophys. 2013;536:171–175. doi: 10.1016/j.abb.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hermenegildo C., Marcaida G., Montoliu C., Grisolía S., Miñana M.D., Felipo V. NMDA receptor antagonists prevent acute ammonia toxicity in mice. Neurochem Res. 1996;21:1237–1244. doi: 10.1007/BF02532401. [DOI] [PubMed] [Google Scholar]
- 66.Paul V., Jayakumar A.R. Independent and combined effects of L-arginine and diazepam on ammonium chloride-induced convulsions in rats. Indian J Physiol Pharmacol. 1999;43:199–204. [PubMed] [Google Scholar]
- 67.Vanaja P., Jayakumar A.R. Evidence for an involvement of the ammonia-decreasing action of L-arginine in suppressing picrotoxin-induced convulsions in rats and its additive action with diazepam. Neurol Res. 2001;23:622–626. doi: 10.1179/016164101101198901. [DOI] [PubMed] [Google Scholar]
- 68.Chen J.R., Wang B.N., Tseng G.F., Wang Y.J., Huang Y.S., Wang T.J. Morphological changes of cortical pyramidal neurons in hepatic encephalopathy. BMC Neurosci. 2014;15:15. doi: 10.1186/1471-2202-15-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.D'Mello C., Le T., Swain M.G. Cerebral microglia recruits monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agusti A., Cauli O., Rodrigo R., Llansola M., Hernández-Rabaza V., Felipo V. p38 MAP kinase is a therapeutic target for hepatic encephalopathy in rats with portacaval shunts. Gut. 2011;60:1572–1579. doi: 10.1136/gut.2010.236083. [DOI] [PubMed] [Google Scholar]
- 71.Zemtsova I., Görg B., Keitel V., Bidmon H.J., Schrör K., Häussinger D. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology. 2011;54:204–215. doi: 10.1002/hep.24326. [DOI] [PubMed] [Google Scholar]
- 72.Wright G.A., Sharifi Y., Newman T.A. Characterisation of temporal microglia and astrocyte immune responses in bile duct-ligated rat models of cirrhosis. Liver Int. 2014 doi: 10.1111/liv.12481. In press. [DOI] [PubMed] [Google Scholar]
- 73.Brück J., Görg B., Bidmon H.J. Locomotor impairment and cerebrocortical oxidative stress in portal vein ligated rats in vivo. J Hepatol. 2011;54:251–257. doi: 10.1016/j.jhep.2010.06.035. [DOI] [PubMed] [Google Scholar]
- 74.Jayakumar A.R., Panickar K.S., Murthy ChR., Norenberg M.D. Oxidative stress and mitogen-activated protein kinase phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes. J Neurosci. 2006;26:4774–4784. doi: 10.1523/JNEUROSCI.0120-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jayakumar A.R., Rama Rao K.V., Tong X.Y., Norenberg M.D. Calcium in the mechanism of ammonia-induced astrocyte swelling. J Neurochem. 2009;109(suppl 1):252–257. doi: 10.1111/j.1471-4159.2009.05842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feuerstein G.Z., Wang X., Barone F.C. The role of cytokines in the neuropathology of stroke and neurotrauma. Neuroimmunomodulation. 1998;5:143–159. doi: 10.1159/000026331. [DOI] [PubMed] [Google Scholar]
- 77.Jayakumar A.R., Tong X.Y., Ospel J., Norenberg M.D. Role of cerebral endothelial cells in the astrocyte swelling and brain edema associated with acute hepatic encephalopathy. Neuroscience. 2012;218:305–316. doi: 10.1016/j.neuroscience.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jayakumar A.R., Norenberg M.D. Endothelial-astrocytic interactions in acute liver failure. Metab Brain Dis. 2013;28:183–186. doi: 10.1007/s11011-012-9344-4. [DOI] [PubMed] [Google Scholar]
- 79.Jayakumar A.R., Tong X.Y., Curtis K.M., Ruiz-Cordero R., Abreu M.T., Norenberg M.D. Increased toll-like receptor 4 in cerebral endothelial cells contributes to the astrocyte swelling and brain edema in acute hepatic encephalopathy. J Neurochem. 2014;128:890–903. doi: 10.1111/jnc.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Skowrońska M., Zielińska M., Wójcik-Stanaszek L. Ammonia increases paracellular permeability of rat brain endothelial cells by a mechanism encompassing oxidative/nitrosative stress and activation of matrix metalloproteinases. J Neurochem. 2012;121:125–134. doi: 10.1111/j.1471-4159.2012.07669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hashimoto M., Ishikawa Y., Yokota S., Goto F., Bando T., Sakakibara Y. Action site of circulating interleukin-1 on the rabbit brain. Brain Res. 1991;540:217–223. doi: 10.1016/0006-8993(91)90510-3. [DOI] [PubMed] [Google Scholar]
- 82.Matsumura K., Kobayashi S. Signaling the brain in inflammation: the role of endothelial cells. Front Biosci. 2004;9:2819–2826. doi: 10.2741/1439. [DOI] [PubMed] [Google Scholar]