

Abstract
Acute liver failure (ALF) is a rare, potentially fatal complication of severe hepatic illness resulting from various causes. In a clinical setting, severe hepatic injury is usually recognised by the appearance of jaundice, encephalopathy and coagulopathy. The central and most important clinical event in ALF is occurrence of hepatic encephalopathy (HE) and cerebral edema which is responsible for most of the fatalities in this serious clinical syndrome. The pathogenesis of encephalopathy and cerebral edema in ALF is unique and multifactorial. Ammonia plays a central role in the pathogenesis. The role of newer ammonia lowering agents is still evolving. Liver transplant is the only effective therapy that has been identified to be of promise in those with poor prognostic factors, whereas in the others, aggressive intensive medical management has been documented to salvage a substantial proportion of patients. A small fraction of patients undergo liver transplant and the remaining are usually treated with medical therapy. Therefore, identification of the complications and causes of death in such patients, and use of appropriate prognostic models to identify those who need liver transplant and those who can be managed with medical treatment is a vital component of therapeutic strategy. In this review, we discuss the various pathogenetic mechanisms and treatment options available.
Keywords: ammonia, cerebral edema, LOPA
Abbreviations: AASLD, American Association For the Study of Liver; ALF, Acute Liver Failure; ALFED, Acute Liver Failure Early Dynamic Model; BBB, Blood Brain Barrier; BCAA, Branched Chain Amino acid; CBF, Cerebral Blood Flow; CPP, Cerebral Perfusion Pressure; CVVHD, Continuous Veno-Venous Hemodialysis; FFP, Fresh Frozen Plasma; GM-CSF, Granulocyte Macrophage Colony Stimulating Factor; HE, Hepatic Encephalopathy; ICU, Intensive Care Unit; IEI, Icterus Encephalopathy Interval; INR, International Normalized Ratio; IL-1β, Interleukin-1 beta; IL6, Interlekin 6; LOLA, l-Ornithine L Aspartate; LOPA, l-Ornithine Phenyl Acetate; MAP, Mean Arterial Pressure; NAC, N-Acetyl Cysteine; NO, Nitric Oxide; OLT, Orthotopic Liver Transplantation; PCWP, Pulmonary Capillary Wedge Pressure; PEEP, Positive End Expiratory Pressure; PT, Prothrombin Time; SIMV, Synchronous Intermittent mandatory Ventilation; SIRS, Systemic Inflammatory Response Syndrome; SPEAR, Selective Parenteral and Enteral Antibiotic Regimen; TNF-α, Tumor Necrosis Factor alfa; UCD, Urea Cycle Disorder; USALF, United States Acute liver Failure Study Group
Acute liver failure (ALF) can be a fatal complication of acute hepatic injury and occurs unpredictably. It is a rare clinical entity marked by the sudden loss of hepatic function and a severe life-threatening course in a person with no prior history of liver disease. ALF represents a syndrome rather than a specific disease, having multiple causes that vary in course and outcome. ALF is difficult to identify in its early stages, resulting in frequent delays in initiation of treatment. The causes of ALF include viral hepatitis, drug induced and toxin-induced liver damage, metabolic errors, ischemia, and miscellaneous rare causes.
ALF is defined by three criteria: (1) rapid development of hepatocellular dysfunction (jaundice, coagulopathy), (2) encephalopathy, and (3) absence of a prior history of liver disease.1 However, the interval between onset of acute hepatic injury (jaundice) and onset of liver failure (encephalopathy with or without coagulopathy) in such patients (icterus-encephalopathy interval; IEI) has been described to be between 4 weeks (Indian definition)2–4 to 24 weeks (AASLD–ALF study group).5 Further, due to the diverse natural course, ALF has been sub-classified as hyper-acute (IEI ≤ 7 day), acute (IEI ≤ 4 weeks) and sub-acute ALF (IEI ≥ 5 week to ≤12 weeks) by British authors.6 Despite these differences in definitions, the central and most important clinical event in ALF is occurrence of hepatic encephalopathy (HE) and cerebral edema which is responsible for most of the fatalities in this serious clinical syndrome. The pathogenesis of encephalopathy and cerebral edema in ALF is unique and multifactorial, and evaluation of these pathogenetic processes provides insight into its effective treatment strategy. Additionally, infection and coagulopathy have been identified to ensue rapidly in these patients - which also determines outcome with resultant management challenge.7
Complications and causes of death in acute liver failure
The major complications in ALF usually associated with death include cerebral edema, seizures, infections, bleeding due to coagulopathy and renal failure. These events infrequently get further aggravated by electrolyte and acid base imbalance and hypoglycaemia. The complications of ALF vary by region and by etiology.
Cerebral edema has been documented to be the most common cause of mortality. In India 58% of ALF patients have cerebral edema at the time of hospitalization. The mortality rate of patients with cerebral edema has been reported as 82%, compared to 44% among patients without cerebral edema.2 Older studies from the UK reported that overt features of cerebral edema increase in frequency with increasing grades of HE. However, with advent of intracranial pressure estimation, it is now clear that most patients with ALF at the time of hospitalization have some degree of cerebral edema and need careful monitoring.8 Recent data suggest that cerebral edema is less frequent now than in former years, but this may reflect earlier admission to hospital and better intensive care unit care.9
Infection is a common complication in ALF that has been documented across the globe.10 An incidence of infection has been reported as high as 90% has been reported in the initial series from UK; the causative organisms being bacteria in 80% of cases and fungal infections in 32%.10 The predominant organisms are Gram-positive bacteria and the most common site of infection is respiratory tract. In more recent reports, the predominant organisms reported are Gram-negative.10,11
Renal failure has been described in 40%–80% of patients in western series, and is associated with a poor prognosis. It occurs more frequently in acetaminophen induced ALF (70%) and less frequently in ALF due to other causes (30%) in western series, suggesting that there may be a toxic effect of acetaminophen on renal tubules.10 Renal failure is reported in 10% of patients from India.12
Gastrointestinal bleeding is reported in 7–20%.2,8 However gastrointestinal bleed has rarely been implicated as the cause of death and usually occurs as a terminal event associated with other complications.
These complications may occur at presentation, or may develop subsequently. They may occur in isolation or in combinations. Management of each complication is important as it influences the ultimate outcome. Encephalopathy and cerebral edema are common presenting features over which other complications like infections, renal failure and gastrointestinal bleed may supervene, perpetuating the brain edema and consequent death. Therefore it is important to understand the pathophysiologic drivers of encephalopathy. Intervention at the level of these pathogenetic mechanisms, along with specific therapy of complications, forms the mainstay of medical management of ALF.
Pathogenetic drivers
The underlying pathogenesis of encephalopathy, consequent cerebral edema and raised intracranial hypertension (ICT) in ALF is complex and has been extensively studied. Ammonia has been implicated as the major neurotoxin in ALF.13 In addition, Systemic Inflammatory Response Syndrome (SIRS) and loss of autoregulation of cerebral blood flow have been implicated as other important pathogenetic events in accentuating encephalopathy and cerebral edema in ALF.14
Role of Ammonia
In animal models as well as in patients with ALF, marked swelling of astrocytes has been documented. In animal models of ALF, expression of various astrocytic proteins has been reported.13 Astrocytes are specialized neuroglial cells, initially considered as passive supporters of the neuronal framework, which are now recognized to play crucial roles in brain metabolism, maintaining the blood brain barrier, modulating synaptic transmission, and neural inflammation. Changes in astrocytes are documented to be due to hyperammonemia in ALF. Astrocyte swelling due to increased brain water and alteration in the functional property of the astrocyte causes encephalopathy and cerebral edema resulting in intracranial hypertension, which often results in brain herniation, the most important mechanism of death in ALF.15 The source of circulating ammonia is primarily derived from glutamine metabolism in the intestinal epithelium.7 Intestinal epithelium undergoes rapid turnover and uses glutamine as a source of energy. Intestinal epithelium contains glutaminase as well as glutamine synthase, and glutaminase converts glutamine to glutamate and ammonia. Some amount of ammonia is also generated by the urease activity of gut flora and renal production (kidney also contains glutaminase and glutamine synthase).9 The circulating ammonia to some extent is excreted by the kidney, used by the muscles to re-synthesise glutamine (muscle also has glutamine synthase and glutaminase), but predominantly is converted to urea (Kreb's urea cycle present in periportal hepatocytes) as well as glutamine (by glutamine synthase present in perivenous hepatocytes) in the liver. Brain also contains glutamine synthase as well as glutaminase. Therefore, brain can synthesise glutamine from ammonia as well as metabolise glutamine to glutamate and ammonia.16 Thus, ammonia is predominantly metabolized in the liver and is converted to urea and glutamine. The enzymes for a complete urea cycle are exclusively localized in the liver. Urea is water soluble and is excreted in urine which is the major pathway for ammonia disposal. Thus, the ability of liver to metabolise ammonia is grossly compromised in ALF resulting in hyperammonemic state, which exerts deleterious effects contributing to HE.16
The neurotoxic effect of ammonia in ALF
|
The arterial ammonia level measured within 24 h of patient presentation has also been demonstrated to correlate with patient outcome in ALF, unlike the case in cirrhosis.4,17 A study conducted by us documented that non-survivors had significantly higher median ammonia levels than survivors (174.7 v 105.0 mmol/L; P, 0.001) in patients with ALF.4 An arterial ammonia level of >124 mmol/L was found to predict mortality with 78.6% sensitivity and 76.3% specificity, and had 77.5% diagnostic accuracy. Patients with higher ammonia levels also developed more complications, including deeper encephalopathy, cerebral edema, need for ventilation, and seizures. Logistic regression analysis showed that pH, presence of cerebral edema, and arterial ammonia at admission were independent predictors of mortality (odds ratios 6.6, 12.6, and 10.9, respectively).4 However, ALF is a dynamic process where variables determining prognosis at admission change over time, and thus the clinical course varies accordingly. The ALF Early dynamic model (ALFED) is based on whether the levels of predictive variables remain persistently high or increase over 3 days above the discriminatory cut-off values. The model has four variables: arterial ammonia ≥123 μmols/l, serum bilirubin ≥15 mg/dl, international normalized ratio ≥5 and hepatic encephalopathy >Grade II. These values if present on day 3 of presentation are independent predictors of mortality. The scores have been depicted in Table 1; total scores vary from 0 to 6.
Table 1.
ALFED Model.18
| Variables over 3 days | Score |
|---|---|
| Hepatic Encephalopathy Persistent or progressed to Grade >2 |
2 |
| INR Persistent or increased to ≥5 |
1 |
| Arterial ammonia Persistent or increased to ≥123 mmol/L |
2 |
| Serum bilirubin Persistent or increased to ≥15 mg/dl |
1 |
Total score calculated by adding individual scores of all 4 variables.
The performance of the ALFED model was superior to the King's College Hospital criteria and the Model for End stage Liver Disease score, even when their 3-day serial values were taken into consideration. An ALFED score of ≥4 had a high positive predictive value (85%) and negative predictive value (87%) in the validation cohort.18 The most widely used prognostic model is the King's college criteria (Table 2). However, in Indian situation, its relevance is questionable. In India paracetamol poisoning associated ALF is practically non-existent,2–4 and therefore the rognostic criteria for paracetamol poisoning is not practiced. The King's prognostic criteria for non-paracetamol poisoning include 5 parameters (Table 2), of which 3 parameters are not prevalent in Indian situation. Most Indian patients with ALF are 20–30 years old at presentation, almost all patients have an I-E interval of <7 days and Non A, Non B virus (its exact nature is not known) and drug idiosyncrasy as etiology of ALF are absent among Indian patients. Therefore the ALFED model may be more applicable in countries like India and probably in other developing countries in Asia and Africa where two third of world's population lives.
Table 2.
ALF Prognostic Model (King's College Criteria).
| Non Acetaminophen ALF | Acetaminophen ALF |
|---|---|
| INR >6.5 (or PT >100 s) or Any 3 of the following:
|
Arterial pH <7.3 or INR >6.5 (or PT >100 s) and Serum creatinine >3.4 mg%; and stage 3 or 4 HE or Arterial lactate >3.5 mmol/L at 4 h or Arterial lactate level >3 mmol/L at 12 h (after fluid resuscitation) |
Infections and Systemic Inflammatory Response Syndrome (SIRS) in Acute Liver Failure
Liver is an important component of the immune system. In ALF, rapid immune paralysis is well known and overt sepsis has been reported in 55%–90% of patients.19 A strong association between occurrence of infection and course of encephalopathy has been clearly documented.3,20 In a prospective evaluation of 96 acetaminophen induced ALF cases with early encephalopathy, occurrence of infection was associated with progression of encephalopathy to deeper grades in about 80% of the individuals.21 In this study, ALF patients without evidence of overt infection (n = 168), who progressed from early to advanced encephalopathy had a number of components of SIRS.21 Fifty percent of those with 2–3 component of SIRS progressed to advanced encephalopathy in contrast to 25% of the patients without any components of SIRS who had progression of encephalopathy. The components of SIRS are highlighted in Table 3.22
Table 3.
SIRS Criteria (Two or More of the Following).
|
Astrocytes form an integral component of the blood–brain barrier (BBB). Ammonia gets accumulated in astrocytes, theoretically altering the permeability of the BBB. It thus ‘primes’ the brain to participate in the systemic inflammation stimulated predominantly by sepsis. Endotoxins and cytokines, the major mediators of the systemic inflammatory response, are unable to cross the BBB. They interact with receptors in the endothelial cells and astrocyte processes on the BBB and can disrupt the capillary tight junctions. They stimulate multiple downstream pathways associated with NO signalling, COX and cytokines. In the late stages of acute liver failure, pro-inflammatory cytokines are observed to efflux from the brain, indicating brain production of TNF-α, IL-6 and IL-1β during uncontrollable intracranial hypertension.23 This inflammatory response is considered to mediate vasodilatation and oxidative stress, both adding to the cytotoxic effects of ammonia.24,25 Even peripheral cytokines subsequent to sepsis in patients with liver failure can cross the BBB. Cytokines have been documented to increase intracranial hypertension in pig models of ALF.23 Liver failure increases an individual's susceptibility to sepsis.
Loss of Cerebral Auto Regulation
Usually cerebral blood flow (CBF) is independent of change in systemic blood pressure and blood flow to various regions of brain varies depending upon the metabolic need of neuron (glucose and oxygen). This delicate balance of regional CBF and the metabolic requirement is lost in patient and animal models of ALF.26,27 This may cause increase in CBF, cerebral congestion which aggravates cerebral edema. CBF correlates very closely with the brain water and ICP in experimental ammonia induced brain edema.28 Normally, the brain autoregulation can maintain a normal CBF at arterial pressures from 65 to 140 mm Hg. This loss of autoregulation of CBF in ALF signifies that even mildly elevated cerebral perfusion pressure (CPP) will increase CBF and hydrostatic pressure across the BBB, while episodes of arterial hypotension may stop capillary blood flow, leading to cerebral hypoxia. This phenomenon has been called ‘dissociated cerebral vasoparalysis’.29 CBF is variable in ALF, and may be excessive at some time point and inadequate at others. CBF is reduced in most patients with grade 3 or 4 encephalopathy, even with maintained mean arterial pressure.30 However, increased CBF precedes high ICP and cerebral herniation.31 This loss of CBF autoregulation is rare in patients with cirrhosis or sepsis. Even though the exact mechanism of loss of autoregulation of CBF is poorly understood, it is a definite event in ALF perpetuating the process of cerebral edema. This loss of autoregulation is restored within 1 day of liver transplant and within 4 days of spontaneous recovery.32 Even hypothermia restored cerebral autoregulation in a series of 14 patients with ALF.33
Aggravating Factors of Encephalopathy in Acute Liver Failure
In the rat model, hyponatremia along with ammonia have been documented to induce brain edema,34 and hypernatremia in patients with ALF seems to protect against brain edema.35 About 25% of patients with ALF in a report from India had hyponatremia, which was prominent in patients with severe encephalopathy.4 However pathogenesis of hyponatremia in ALF is unclear. Use of sedatives in patients with ALF presenting with agitation causing advanced encephalopathy is a common clinical experience. Alkaline pH is also known to drive ammonia in to the astrocyte in patients with hepatic encephalopathy. These factors are reversible and therefore important to be assessed in such patients.
Hyponatremia, alkalosis, SIRS have been well identified by now to increase susceptibility of astrocytes to increased arterial ammonia levels. Therefore control and prevention of these factors, along with ammonia lowering therapy, today remains the most rational approach to manage patients with hepatic encephalopathy. Figure 1 summarizes the essential mechanism of encephalopathy and cerebral edema in ALF and the possible therapeutic interventions directed towards these pathogenetic changes.
Figure 1.
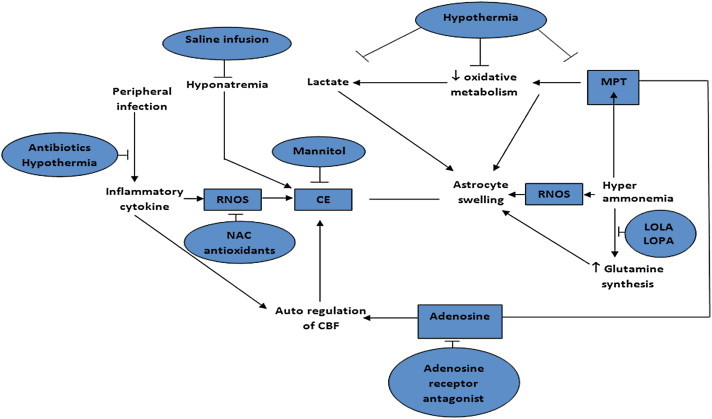
Mechanism of encephalopathy in ALF and therapeutic intervention directed to these pathogentic mechanisms.
Management of encephalopathy in acute liver failure
Current specific management of encephalopathy in ALF is based on measures that reduce or eliminate ammonia, prevent sepsis and; if sepsis has set in, treat it aggressively. While these rationales remains the mainstay of therapeutic approach, treatment in an intensive care unit with availability of organ support systems to prevent death due to organ failure also continues to be the other important aspect in the management of hepatic encephalopathy in ALF. The essential steps of management of hepatic encephalopathy are depicted in Table 4.
Table 4.
Principles in the Management of Hepatic Encephalopathy.
|
However certain details of specific support in encephalopathy in ALF need to be discussed in greater detail and can be categorized as 1) medical therapy directed to control encephalopathy and cerebral edema 2) ammonia lowering strategies 3) other forms of therapy directed to control and prevent complications like hemodynamic instability, renal failure, coagulopathy, hypoglycaemia and respiratory arrest. 4) liver transplant and bioartificial liver support systems.
Medical Therapy Directed to Control Encephalopathy and Cerebral Edema
Neurologic Support
All patients should be nursed in a 20°–30° head up tilt to improve venous drainage. Patient turning and other tactile stimulation should be minimized. Respiratory suctioning should be limited to less than 15 s duration at a time. One to two ml of lidocaine instilled in the endotracheal tube and ventilation with 100% oxygen prior to suctioning may be helpful in preventing surges of raised ICP. Control of agitation is important, as valsalva maneuver from psychomotor agitation may lead to elevation of the ICP. A diligent search for treatable causes of agitation like distended bladder, and bedsores should be made (Table 5). Small intravenous doses of short acting benzodiazepines should be reserved as a last resort for serious psychomotor agitation.
Table 5.
Factors that Increase Intracranial Pressure.
| Valsalva maneuver | Arterial hypertension |
| Positive end expiratory pressure (PEEP) | Vomiting, shivering |
| Head and body turning or moving | Psychomotor agitation |
| Neck veins compression | Noxious stimuli |
| Respiratory suctioning | Seizures |
| Fever | Vasodilatory agents |
| Horizontal decubitus | Isometric muscle exercise |
| Severe hypoxemia | Trendelenburg position |
| Any degree of hypercapnia | Coughing, sneezing |
Intracranial Pressure Monitoring
Traditional signs of elevated ICP are unreliable in ALF. Computed tomography of the head is not a reliable way to estimate ICP in ALF. Currently many centres install an extradural ICP monitoring device once the patient has developed grade 3 or 4 encephalopathy. Prior endotracheal intubation is necessary under propofol anesthesia. Aggressive correction of coagulopathy and thrombocytopenia must be undertaken with a goal of achieving a platelet count of >50,000/mm3 and an INR <1.7 or PT within 2–3 s of normal. Correction of the PT usually requires bolus administration of 2 units of fresh frozen plasma (FFP) followed by a continuous FFP infusion at a rate of 75–150 ml/h. The transducer to measure the ICP is placed by a neurosurgeon in the ICU under local anesthesia. The standard site is the right frontal area chosen to avoid the (right handed) dominant lobe, sagittal sinus, and motor cortex.
Epidural transducers are used most commonly and carry the lowest complication rate (3.8%). Subdural bolts and parenchymal monitors carry complication rates (mainly bleeding) of 20% and 22% respectively.36 Colonization of ICP devices increases significantly after about 5 days of insertion, and the risk of superficial wound infection and meningitis rises.
In adults, the average ICP ranges from 0 to 10 mmHg. The maximal permissible upper limit is 20 mmHg. The overall goal is to maintain an ICP of less than 20 mmHg and CPP (MAP—ICP) above 50 mmHg at all times. A slow progressive increase in ICP to >25 mmHg, or pressure waves rising to 30–50 mmHg lasting for 5–20 min, reflect falling cerebral compliance. Prolonged (>2 h) elevation of ICP to >40 mmHg or a reduction in CPP < 50 mmHg are considered a contraindication for liver transplantation.
Reverse Jugular Venous Monitoring
Jugular bulb venous saturation is frequently used in neurosurgical intensive care, and involves insertion of a fine catheter in a retrograde fashion until its tip lies in the jugular bulb. Blood returning from the cerebral circulation can then be sampled. A jugular venous saturation of <55% represents an ischemic brain, while a saturation of >85% represents a hyperemic brain.37 Measures to improve oxygen supply are indicated in the former situation, and measures to reduce cerebral blood flow in the latter situation.
Osmotherapy
Mannitol lowers the ICP by reducing the brain water and changing the rheological characteristics of blood. When the ICP rises to over 20–25 mmHg for more than 5 min, an intravenous bolus of mannitol should be given (0.5–1 g/kg, 20% solution, over 5 min). Repeated boluses may be administered as long as the serum osmolality is <320 mOsm/L. The response to a mannitol bolus may be expected 15–60 min post-injection. In about 20% of patients a paradoxical increase in ICP occurs after mannitol infusion.38 High doses can result in acute renal failure and damage to the BBB. Mannitol works best in mild to moderate intracranial hypertension and is less effective when the ICP is greater than 60 mmHg.39 In oliguric/anuric patients, mannitol should only be given in combination with continuous veno venous or arteriovenous hemodiafiltration.
Thiopentone
Based on data suggesting that barbiturates could be of value in controlling the intracranial hypertension of head injury, intravenous thiopental was assessed in 13 patients with fulminant hepatic failure complicated by unresponsive intracranial hypertension. The ICP was reduced in all cases, and in eight cases thiopentone infusion achieved stable normal intracranial and cerebral perfusion pressure. Five patients made a complete recovery.40 The recommended dose of pentobarbital is a loading dose of 3–5 mg/kg (maximum 500 mg) over 15 min, followed by a continuous infusion of 0.5–2.0 mg/h. Barbiturate therapy must be used with simultaneous continuous ICP and arterial blood pressure monitoring.
Hypothermia
Moderate hypothermia (32 °C–35 °C) using cooling blankets leads to a reduction in CBF, cerebral metabolism, ammonia uptake by the brain, and glutamine synthesis, and reduces intracranial pressure in patients with ALF.41 Data from studies in patients undergoing liver transplantation for ALF suggests that an increase in intracranial pressure can be prevented during the dissection and reperfusion phases of the operation if the patients are maintained hypothermic during surgery.42
However hypothermia as a therapy has not shown any improvement in survival among patients with ALF but has been used as a therapeutic bridge to transplantation.43 Hypothermia in the above mentioned studies has been used for not more than 8 h, and prolonged hypothermia is well known to cause cardiac arrhythmia and coagulopathy. Therefore routine use of hypothermia for short duration may be of help particularly in patients with ALF.
Prophylactic Phenytoin
It has been suggested that subclinical seizure activity in patients with deep encephalopathy on ventilation may remain unrecognized and may lead to exacerbation of cerebral hypoxia and edema. Hence it was recommended that all patients with grade III or IV encephalopathy should be monitored for subclinical seizure activity and treated with prophylactic phenytoin. However, in a randomized, controlled clinical trial of 42 patients, it was found that prophylactic use of phenytoin did not prevent cerebral edema, seizures or improve survival.44
Hyperventilation
Patients with ALF often hyperventilate spontaneously, and attempts to control this are of no use. Hyperventilation reduces CBF, and may be useful in the subgroup of patients with cerebral hyperemia as reflected by jugular bulb venous saturation of more than 75%. Prophylactic hyperventilation does not reduce the frequency of intracranial hypertension in ALF, but a moderate reduction in pCO2 to 25–30 mmHg is helpful in decreasing the ICP once cerebral edema has begun to develop.45 Excessive hyperventilation may lead to cerebral vasoconstriction.
Ammonia Lowering Strategies
Benefits of lactulose have been seriously questioned. In a recent meta-analysis, only few high quality studies have shown any beneficial effect with lactulose and its popularity stems from a long clinical experience rather than strong objective evidence.46 Antibiotics have their inherent adverse effects, especially on prolonged use. Concepts on protein restriction are being revised.47 Newer agents to reduce ammonia like phenyl acetate and probiotics, having capacity to consume ammonia, are being introduced. Novel approaches like l-carnitine, flumazenil in deep coma even when not precipitated by benzodiazepines is being vigorously investigated.
l-Ornithine l-Aspartate (LOLA)
LOLA is a stable salt of the amino acids ornithine and aspartic acid. These two amino acids get converted to glutamate in the muscles and hepatocytes. Glutamate is the substrate on which the enzyme glutamine synthase (present in the muscle as well as liver), acts and combines it with ammonia to make glutamine and thereby reduces blood ammonia levels.48 In liver failure, the enzyme glutamine synthase in the muscle is up-regulated, and therefore it is hypothesized that LOLA may reduce arterial ammonia levels. It has been shown that LOLA reduces arterial ammonia levels accompanied by improvement in mental status in patients with hepatic encephalopathy associated with chronic liver disease.49 In rat model of acute liver failure (ALF), LOLA has been shown to prevent brain edema and hepatic encephalopathy which was accompanied by up regulation of muscle glutamine synthase activity and plasma glutamine levels were also raised indicating that LOLA in ALF may be beneficial.50 A randomized placebo controlled double blind trial of LOLA in patients with ALF was recently published.51 However LOLA therapy was not associated with decrease in ammonia levels or improvement in encephalopathy or survival (Figure 2).
Figure 2.
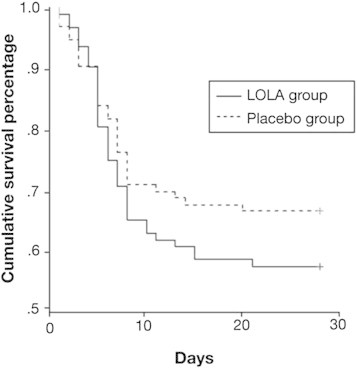
Kaplan–Meier curves showing the cumulative survival percentage of ALF patients randomized to placebo or LOLA infusion (P = 0.22 by the log rank test at 1 df).
In the above mentioned study ammonia levels were measured for 6 days among patients receiving LOLA as well as placebo, however the rate of decline of ammonia was also similar between the two arms (Figure 3). Peculiarly, patients receiving LOLA had more frequent seizures.
Figure 3.
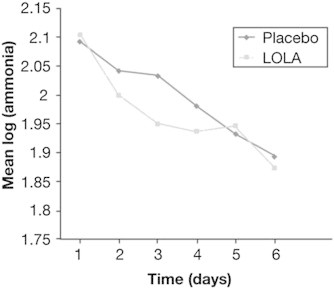
Log transformed mean ammonia levels among patients randomized to placebo or LOLA during the initial 6 days.
Very high glutamine levels in the systemic circulation are found in ALF.52,53 LOLA could theoretically further increase ammonia detoxification by the skeletal muscle by increased glutamine synthesis. This glutamine is recirculated back to the intestine and kidney where it is broken down to ammonia and glutamate by glutaminase, thus LOLA is ineffective in reducing the ammonia levels.
l-Ornithine Phenyl Acetate (LOPA)
Brusilow in 1979 suggested use of alternative pathway for ammonia disposal in Urea Cycle Disorders (UCD).54 The drugs proposed were phenyl acetate and sodium benzoate. Phenyl acetate combines with glutamine to form phenylacetylglutamine which is water soluble and is excreted in urine. One molecule of phenyl acetate removes two molecule of Glutamine and this became the mainstay of management in UCD in the ensuing time.55
Recently Davies et al from the university college of London (UCL) documented that in bile duct ligated rat model of cirrhosis, LOPA could reduce arterial ammonia, associated with decrease in cerebral glutamine/myoinositol ratio, brain water content and increase in arterial glutamine concentration along with increase in urinary phenylacetylglutamine levels.56 Ytrebo et al from the same UCL group in a devascularised pig model of ALF documented that LOPA prevented rise of plasma and brain ammonia concentration and prevented rise in intracranial pressure.57 This latter study also documented increase in urinary phenylacetylglutamine excretion along with increase in muscle glutamine synthetase activity. Ornithine has been used in these two studies to induce glutamate formation in the muscle. However whether these agents will be effective in humans needs evaluation. Further, a phenyl acetate blood level exceeding >6 mmol/L is associated with toxicity and in presence of renal failure its use will be limited.
Sodium benzoate
Sodium Benzoate is usually used as a food preservative. It combines with glycine to form hippurate which is water soluble and gets excreted in urine easily and thereby may decrease arterial ammonia levels. However, therapeutic trials in humans show conflicting results.58,59 It has never been used in patients with acute liver failure. However our own experience does not support its ammonia lowering effect in patients with ALF.
Rifaximin
Rifaximin is an oral antibiotic with a broad spectrum of activity against enteric bacteria. It is minimally absorbed from the gut (<0.4%). Safety profile and tolerability is comparable to placebo. No dose adjustment is required in patients with renal insufficiency. Current data suggests that rifaximin is as effective as established therapies like lactulose or neomycin in treatment of HE and has a better safety profile and tolerability in patients with chronic liver disease.51,60–63 Its' possible benefit in lowering ammonia in patients with ALF has not yet been explored.
Probiotics
Probiotics promote growth of non-urease producing bacteria in the colon thereby reducing generation and absorption of ammonia by suitably altering the gut flora. Probiotics have been shown to reduce ammonia and improve cognitive function in patients with minimal HE.64 There is no data on its role in ALF.
Miscellaneous interventions to lower ammonia and improve encephalopathy
In a recently published trial by USALF study group, 21 days transplant free survival was documented to be significantly better in the NAC treated than the placebo treated patients, however overall survival between the treated and control group was similar.65 In a controlled trial of 44 patients it was found that the use of dexamethasone could not prevent the development of cerebral edema or improve survival.66 In a recent prospective randomized trial involving 30 patients with ALF, it was found that use of hypertonic (30%) sodium chloride infusion to maintain serum sodium levels between 145 and 155 mmol/L led to a significant decrease in ICP.35
Therapies Directed Towards Management of Complications
Prevention and Treatment of Infection
Aseptic nursing techniques should be followed. Intravenous catheters should be changed every 72 h and removed catheter tips cultured. Urinary catheters should be removed from anuric patients and as soon as possible from recovering patients. The vast majority of the bacterial infections occur within 72 h of admission. Hence prophylactic broad spectrum parenteral antibiotics should be started at admission.
Rolando et al stratified patients at the time of admission based on the presence or absence of clinically evident infection. Those who were infected received either cefuroxime or selective parenteral and enteral antibiotics (SPEAR regimen). The SPEAR regime consisted of oral colistin, tobramycin and amphotericin B with a 5-day course of an intravenous cephalosporin. Those who were not infected were randomized to SPEAR or no antibiotics. The outcome was similar among those randomized to cefuroxime and SPEAR in the infected group. In contrast SPEAR significantly decreased the infection rate compared to no treatment in the initially 'non infected' group.67 Addition of oral non-absorbable antibiotics does not confer any advantage over parenterally administered antibiotics. In the above studies, many patients continued to have positive cultures 1 and 2 days after antibiotics were started. Hence, surveillance cultures should be continued even after starting antibiotics.
Any unexplained decrease in blood pressure, drop in urinary output, worsening encephalopathy, and development of severe acidosis or disseminated intravascular coagulation should be considered as signs of sepsis. Fungal infection should be strongly considered in the setting of unresponsive fever, leukocytosis, and deterioration of neurologic status after initial improvement and presence of renal failure. GM-CSF has been reported to improves neutrophil function both in vitro and in vivo among ALF patients.68
Hemodynamic Support
ALF patients have a high cardiac output, low systemic vascular resistance, and relative arterial hypotension. An adequate cardiovascular filling pressure (PCWP: 8–14 mmHg) must be assured. The goal is to maintain the mean arterial pressure (MAP) above 60 mmHg. Vasopressor agents are indicated if the MAP is <60 mmHg despite adequate intravascular volume. Oxygen consumption may decrease with the use of vasopressor agents despite the increased arterial pressure, as a result of reduction in oxygen delivery and extraction rates. Reduced oxygen extraction may be prevented with concurrent use of prostacyclin. Arterial hypotension in spite of an adequate intravascular volume should be considered a sign of bacterial or fungal sepsis and treated accordingly.
Arterial hypotension in ALF may lead to a critical reduction in CPP, even with a mildly increased ICP. The coexistence of arterial hypotension and intracranial hypertension requires extremely careful management.
Coagulopathy
The coagulopathy of ALF may be clinically silent or manifested by bleeding from mucosal membranes. Infusion of FFP is indicated only for active bleeding or before invasive procedures. The risk of bleeding from stress ulceration of gastric mucosa is reduced by the prophylactic use of sucralfate.
Mechanical Ventilation of Patients with Liver Failure
The lungs are relatively spared in ALF and standard modes of mechanical ventilation can be used. Most patients will tolerate ventilation with minimal sedation. The risks of paralysis may outweigh the benefits in the majority of patients, but may have a role in selected patients with refractory intracranial hypertension. Barbiturates and midazolam produce a dose dependent reduction in metabolic rate, CBF and cerebral blood volume. A traumatic intubation may induce a sudden rise in the ICP and provoke herniation. Hence tracheal intubation must be gentle. Standard ventilation modes, most commonly synchronized intermittent mandatory ventilation (SIMV) are used. The usual initial ventilator settings are: respiratory rate of 14–18 breaths/minute; tidal volume of 10–12 times the body weight in kilograms; inspiratory period of 25%–33%; pause time of 10% of total cycle; and PEEP of 2–4 mmHg. High levels of PEEP (more than 10–15 mm Hg), lead to worsening or facilitating the development of cerebral edema, and a decrease in hepatic arterial blood flow. Propofol inhibits sympathetic vasoconstrictor activity and has negative inotropic effect. Among the muscle relaxants, pancuronium, vecuronium, and rocuronium exhibit a prolonged elimination in the presence of liver failure. Atracurium is degraded by the non enzymatic Hoffman elimination and hydrolysis by non specific plasma esterases. Neither hepatic nor renal disease leads to accumulation of atracurium.
Microcirculation
NAC has been found to improve tissue oxygen extraction. This effect is potentiated when epoprostenol and NAC are infused together. NAC may promote dilatation of vasoconstricted microcirculatory units by replenishing tissue sulfhydryl groups, which are necessary to maintain adequate activity of endothelium-derived relaxing factor. NAC infusion was shown to facilitate hemodynamic stability in association with a mean 46% and 29% increase in global tissue oxygen consumption after 30 min in patients with acetaminophen and other causes of ALF respectively.69 Prostacyclin infusion also improved oxygen delivery and consumption. More recently, another group found a more variable systemic hemodynamic response to NAC, with clear responders and non-responders. Overall, a small (6%), non sustained improvement in tissue oxygen consumption was found.70
Renal Support
Hemodialysis in ALF patients requires standard heparinization in spite of the coagulopathy, due to coexisting antithrombin III deficiency. Only a lactate free bicarbonate buffer as the dialysis fluid and biocompatible dialysis membranes like polysulphone or polyacrylnitrate should be used. Continuous forms of renal support therapy like CVVHD are preferable to conventional hemodialysis or intermittent hemofiltration, which may reduce CPP by increasing cerebral edema through rapid osmolar shifts or reducing the MAP. Dialysis reduces ammonia levels along with blood urea and glutamine.
Nutritional and Metabolic Support
Almost half of ALF patients develop hypoglycaemia. Development of hypoglycaemia can be sudden and may confound the interpretation of mental changes. Glucose requirements in these patients are highly variable and require close monitoring. Blood sugar should be monitored at 2–3 h intervals. Whenever the blood glucose level is lower than 60 mg/dl, an intravenous bolus of 50–100 ml of 50% dextrose should be administered. The amount of water administered as a solvent for dextrose should be minimized by providing solutions concentrated to 25%–50%.
Recent evidence suggests that the glucose transport across the blood brain barrier is increased because of upregulation of glucose carriers in ALF patients. Hyperglycaemia may contribute to raised ICP because increased glucose influx leads to cerebral lactic acid accumulation.71 Hence it may be prudent to keep blood glucose within the normal limits.
ALF is a hypercatabolic state. Energy requirements in ALF are increased by as much as 60% and are further elevated by the presence of complicating infection. Whole body protein catabolism may be increased up to four times the normal rate. Massive amino acid losses in the urine occur. A combination of parenteral dextrose and lipid emulsions, and at least 40 gm protein/day should be administered initially. It has been shown that lipid emulsions may be used safely in patients with ALF.72 There is no reason to restrict proteins in ALF. Branched chain amino acids (BCAA) offer no additional advantage, with the exception of the patients requiring frequent dialysis, in whom large BCAA losses may occur. Hypokalemia, hypomagnesemia, hypophosphatemia and hypocalcemia are common and must be corrected. Either the enteral or parenteral route may be used for feeding, but the former route has obvious advantages.
Liver Transplantation and Bio-artificial Liver Support
Liver Transplantation
Overall, the 1 year survival after cadaveric orthotopic liver transplantation (OLT) ranges from 50% to 75%. In experienced centres, outcome with split liver grafts is comparable with that after use of full size organs. Survival is substantially reduced among patients with sepsis and multiorgan failure before OLT. Auxiliary partial OLT, as a form of temporary liver support in patients with ALF, has been reported to have results similar to those of conventional transplantation. Withdrawal of immunosuppression, leading to graft atrophy may be possible in upto 65% patients surviving 1 year. Living donor liver transplantation has been more often used in children. The 1 year survival rate after living related OLT in a total of 35 paediatric cases in 3 series was 59%–90%.73–75 The 1 year survival in the largest series of living related OLT in adults, including 53 patients has reported a 75% survival at 1 year.76
Non Transplant Therapies for Liver Support
Therapeutic interventions that have been used to treat ALF in lieu of OLT are listed in Table 6.
Table 6.
Attempted Non-OLT Therapies for ALF.
ALF toxin removal
|
Extracorporeal Liver Support may be broadly classified as artificial which contain no biologic component (eg. MARS), or bioartificial which include viable liver cells in culture within bioreactors or involve perfusion of the patients' blood through an isolated human, or porcine whole liver. However, none of these bioartificial support system till date has been shown to improve survival among patients with ALF.
Molecular Adsorbent Recirculating System (MARS)
MARS (Teraklin AG, Rostock, Germany) is a dialysis treatment which uses a recirculating dialysate containing 20% albumin that is regenerated on line by dialysis against a bicarbonate buffered dialysate, followed by passage through a column with uncoated charcoal and a second column with an anion exchange resin. This allows the removal of both water soluble substances, such as urea, creatinine, and ammonia, and of albumin bound substances, such as phenol, bile acids, bilirubin, BCAA, and short chain fatty acids. MARS treatment can also remove cytokines like TNF-α and IL-6. MARS therapy has been commercially available since 1999, and 38 patients with ALF have been reported to the International MARS registry till 2002.77 Majority of these cases were drug induced. Overall 19 patients survived, of whom 6 were transplanted. Preliminary data shows that MARS treatment improves encephalopathy, and leads to a reduction in bilirubin and ammonia levels.78 However no randomized trials of MARS therapy in ALF are available. At our centre we conducted a randomised study comparing MARS with conventional management in patients with ALF (n = 20) and could not document any survival benefit of MARS over the conventional management (unpublished observation).
Treatment of underlying etiology
The management of ALF is supportive. In some specific etiologies, drugs have been shown to be effective. In patients with HBV-ALF, antivirals have been recommended. The choice of antivirals is between tenofovir and entecavir, the former being preferred in patients with renal failure due to its safety profile. The doses of antivirals need to be modified as per the creatinine clearance. In patients with antituberculosis drug induced or any other suspected drug induced ALF, the suspected drug should be immediately stopped. In HEV-ALF, presently there is no data on the use of Ribavirin, though this has been shown to be effective in managing chronic HEV in solid organ transplant patients and selected patients with HEV related ACLF.79,80 In patients with paracetamol toxicity, activated charcoal has been shown to be effective in gastric decontamination. High doses of NAC are recommended in such patients and should be started within 48 h of ingestion.5
Pregnancy and acute liver failure
There is increased risk of maternal and fetal complications in pregnant patients with ALF.81 The principles of management of abortions, preterm labor, premature rupture of membranes and still birth remain unchanged. Termination of pregnancy is not indicated. There is an increased risk of bleeding due to associated coagulopathy. Management of pregnant patients with ALF is similar to other ALF patients. Complications need to be managed on a case to case basis. A detailed management of ALF in pregnancy has been published in a previous issue of this journal.81
Conclusion
Hepatic encephalopathy in ALF is its protean manifestation. Hyperammonemia, SIRS, loss of cerebral autoregulation, hyponatremia and imbalance in acid-base homeostasis are major pathogenetic mechanism of encephalopathy in ALF. Cerebral edema and infection are the major causes of death in this disease. Specific strategy for neurological support, prevention of infection and its treatment, ammonia lowering therapies, maintenance of acid-base balance, close monitoring in an ICU setup with adequate support for failing organs remains the mainstay of medical management of encephalopathy in ALF. Patients with ominous prognostic criteria benefit from liver transplant. However liver support devices have not been very beneficial in ALF till date.
Conflicts of interest
All authors have none to declare.
References
- 1.Tandon B.N., Bernauau J., O'Grady J. Recommendations of the International Association for the Study of the Liver Subcommittee on nomenclatureof acute and subacute liver failure. J Gastroenterol Hepatol. 1999;14:403–404. doi: 10.1046/j.1440-1746.1999.01905.x. [DOI] [PubMed] [Google Scholar]
- 2.Acharya S.K., Dasarathy S., Kumer T.L. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology. 1996;23:1448–1455. doi: 10.1002/hep.510230622. [DOI] [PubMed] [Google Scholar]
- 3.Acharya S.K., Panda S.K., Saxena A., Gupta S.D. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol. 2000;15:473–479. doi: 10.1046/j.1440-1746.2000.02073.x. [DOI] [PubMed] [Google Scholar]
- 4.Bhatia V., Singh R., Acharya S.K. Predictive value of arterial ammonia for complications and outcome in acute liver failure. Gut. 2006;55:98–104. doi: 10.1136/gut.2004.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee W.M., Squires R.H., Jr., Nyberg S.L., Doo E., Hoofnagle J.H. Acute liver failure: summary of a workshop. Hepatology. 2008;47:1401–1415. doi: 10.1002/hep.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Grady J.G., Schalm S.W., Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342:273–275. doi: 10.1016/0140-6736(93)91818-7. [DOI] [PubMed] [Google Scholar]
- 7.Shawcross D., Deutz N.E.P., O Damink S.W.M., Jalan R. Hepatic encephalopathy in liver failure A multi organ perspective. In: Arroyo V., Forns X., Gracia-Pagan J.C., Rodes J., editors. Progress in the Treatment of Liver Disease. Ars Medica; Barcelona: 2003. pp. 51–60. [Google Scholar]
- 8.Blei A.T. Brain edema in acute liver failure. Crit Care Clin. 2008;24:99–114. doi: 10.1016/j.ccc.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Bernal W., Hall C., Karvellas C.J., Auzinger G., Sizer E., Wendon J. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46:1844–1852. doi: 10.1002/hep.21838. [DOI] [PubMed] [Google Scholar]
- 10.Shalimar, Acharya S.K., Lee W.M. 2013. Worldwide Differences in Acute Liver Failure; pp. 33–46.www.futuremedicine.com [Google Scholar]
- 11.Karvellas C.J., Pink F., McPhail M. Predictors of bacteraemia and mortality in patients with acute liver failure. Intensive Care Med. 2009;35:1390–1396. doi: 10.1007/s00134-009-1472-x. [DOI] [PubMed] [Google Scholar]
- 12.Kumar R., Shalimar, Bhatia V. Antituberculous therapy induced acute liver failure. magnitude, profile, prognosis and predictors of outcome. Hepatology. 2010;51:1665–1674. doi: 10.1002/hep.23534. [DOI] [PubMed] [Google Scholar]
- 13.Butterworth R. Molecular neurobiology of acute liver failure. Semin Liver Dis. 2003;23:251–258. doi: 10.1055/s-2003-42643. [DOI] [PubMed] [Google Scholar]
- 14.Cordoba J., Minguez B. Hepatic encephalopathy. Semin Liver Dis. 2008;28:70–80. doi: 10.1055/s-2008-1040322. [DOI] [PubMed] [Google Scholar]
- 15.Bjerring P.N., Eefsen M., Hansen B.A., Larsen F.S. The brain in acute liver failure. A tortuous path from hyperammonemia to cerebral edema. Metab Brain Dis. 2009;24:5–14. doi: 10.1007/s11011-008-9116-3. [DOI] [PubMed] [Google Scholar]
- 16.Strauss G.I., Knudsen G.M., Kondrup J., Moller K., Larsen F.S. Cerebral metabolism of ammonia and amino acids in patients with fulminant hepatic failure. Gastroenterology. 2001;121:1109–1119. doi: 10.1053/gast.2001.29310. [DOI] [PubMed] [Google Scholar]
- 17.Ong J.P., Aggarwal A., Krieger D. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003;114:188–193. doi: 10.1016/s0002-9343(02)01477-8. [DOI] [PubMed] [Google Scholar]
- 18.Kumar R., Shalimar, Sharma H. Prospective derivation and validation of early dynamic model for predicting outcome in patients with acute liver failure. Gut. 2012;61:1068–1075. doi: 10.1136/gutjnl-2011-301762. [DOI] [PubMed] [Google Scholar]
- 19.Bhatia V., Singhal A., Panda S.K., Acharya S.K. A 20-year single-center experience with acute liver failure during pregnancy: is the prognosis really worse? Hepatology. 2008;48:1577–1585. doi: 10.1002/hep.22493. [DOI] [PubMed] [Google Scholar]
- 20.Rolando N., Wade J., Davalos M., Wendon J., Philpott-Howard J., Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734–739. doi: 10.1053/jhep.2000.17687. [DOI] [PubMed] [Google Scholar]
- 21.Vaquero J., Polson J., Chung C. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125:755–764. doi: 10.1016/s0016-5085(03)01051-5. [DOI] [PubMed] [Google Scholar]
- 22.Muckart D.J., Bhagwanjee S. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference definitions of the systemic inflammatory response syndrome and allied disorders in relation to critically injured patients. Crit Care Med. 1997;25:1789–1795. doi: 10.1097/00003246-199711000-00014. [DOI] [PubMed] [Google Scholar]
- 23.Wright G., Shawcross D., Olde Damink S.W., Jalan R. Brain cytokine flux in acute liver failure and its relationship with intracranial hypertension. Metab Brain Dis. 2007;22:375–388. doi: 10.1007/s11011-007-9071-4. [DOI] [PubMed] [Google Scholar]
- 24.Chung C., Gottstein J., Blei A.T. Indomethacin prevents the development of experimental ammonia-induced brain edema in rats after portacaval anastomosis. Hepatology. 2001;34:249–254. doi: 10.1053/jhep.2001.26383. [DOI] [PubMed] [Google Scholar]
- 25.Gorg B., Bidmon H.J., Keitel V. Inflammatory cytokines induce protein tyrosine nitration in rat astrocytes. Arch Biochem Biophys. 2006;449:104–114. doi: 10.1016/j.abb.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Strauss G.I., Hogh P., Moller K., Knudsen G.M., Hansen B.A., Larsen F.S. Regional cerebral blood flow during mechanical hyperventilation in patients with fulminant hepatic failure. Hepatology. 1999;30:1368–1373. doi: 10.1002/hep.510300608. [DOI] [PubMed] [Google Scholar]
- 27.Dethloff T.J., Knudsen G.M., Larsen F.S. Cerebral blood flow autoregulation in experimental liver failure. J Cereb Blood Flow Metab. 2008;28:916–926. doi: 10.1038/sj.jcbfm.9600589. [DOI] [PubMed] [Google Scholar]
- 28.Larsen F.S., Gottstein J., Blei A.T. Cerebral hyperemia and nitric oxide synthase in rats with ammonia-induced brain edema. J Hepatol. 2001;34:548–554. doi: 10.1016/s0168-8278(00)00069-6. [DOI] [PubMed] [Google Scholar]
- 29.Larsen F.S., Adel Hansen B., Pott F. Dissociated cerebral vasoparalysis in acute liver failure. A hypothesis of gradual cerebral hyperaemia. J Hepatol. 1996;25:145–151. doi: 10.1016/s0168-8278(96)80066-3. [DOI] [PubMed] [Google Scholar]
- 30.Wendon J.A., Harrison P.M., Keays R., Williams R. Cerebral blood flow and metabolism in fulminant liver failure. Hepatology. 1994;19:1407–1413. [PubMed] [Google Scholar]
- 31.Larsen F.S. Cerebral circulation in liver failure: Ohm's law in force. Semin Liver Dis. 1996;16:281–292. doi: 10.1055/s-2007-1007241. [DOI] [PubMed] [Google Scholar]
- 32.Strauss G., Hansen B.A., Kirkegaard P., Rasmussen A., Hjortrup A., Larsen F.S. Liver function, cerebral blood flow autoregulation, and hepatic encephalopathy in fulminant hepatic failure. Hepatology. 1997;25:837–839. doi: 10.1002/hep.510250409. [DOI] [PubMed] [Google Scholar]
- 33.Jalan R., Olde Damink S.W., Deutz N.E., Hayes P.C., Lee A. Restoration of cerebral blood flow autoregulation and reactivity to carbon dioxide in acute liver failure by moderate hypothermia. Hepatology. 2001;34:50–54. doi: 10.1053/jhep.2001.25386. [DOI] [PubMed] [Google Scholar]
- 34.Cordoba J., Gottstein J., Blei A.T. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol. 1998;29:589–594. doi: 10.1016/s0168-8278(98)80154-2. [DOI] [PubMed] [Google Scholar]
- 35.Murphy N., Auzinger G., Bernel W., Wendon J. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004;39:464–470. doi: 10.1002/hep.20056. [DOI] [PubMed] [Google Scholar]
- 36.Blei A.T., Olafsson S., Webster S., Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet. 1993;341:157–158. doi: 10.1016/0140-6736(93)90016-a. [DOI] [PubMed] [Google Scholar]
- 37.Bernal W., Wendon J. Acute liver failure; clinical features and management. Eur J Gastroenterol Hepatol. 1999;11:977–984. doi: 10.1097/00042737-199909000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Hanid M.A., Davies M., Mellon P.J. Clinical monitoring of intracranial pressure in fulminant hepatic failure. Gut. 1980;21:866–869. doi: 10.1136/gut.21.10.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munoz S.J. Difficult management problems in fulminant hepatic failure. Semin Liver Dis. 1993;13:395–413. doi: 10.1055/s-2007-1007368. [DOI] [PubMed] [Google Scholar]
- 40.Forbes A., Alexander G.J., O'Grady J.G. Thiopental infusion in the treatment of intracranial hypertension complicating fulminant hepatic failure. Hepatology. 1989;10:306–310. doi: 10.1002/hep.1840100309. [DOI] [PubMed] [Google Scholar]
- 41.Chatauret N., Rose C., Butterworth R.F. Mild hypothermia in the prevention of brain edema in acute liver failure: mechanisms and clinical prospects. Metab Brain Dis. 2002;17:445–451. doi: 10.1023/a:1021982523691. [DOI] [PubMed] [Google Scholar]
- 42.Jalan R., OD S.W., Deutz N.E., Lee A., Hayes P.C. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet. 1999;354:1164–1168. doi: 10.1016/s0140-6736(98)12440-6. [DOI] [PubMed] [Google Scholar]
- 43.Stravitz R.T., Larsen F.S. Therapeutic hypothermia for acute liver failure. Crit Care Med. 2009;37:S258–S264. doi: 10.1097/CCM.0b013e3181aa5fb8. [DOI] [PubMed] [Google Scholar]
- 44.Bhatia V., Batra Y., Acharya S.K. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol. 2004;41:89–96. doi: 10.1016/j.jhep.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 45.Ede R.J., Gimson A.E., Bihari D., Williams R. Controlled hyperventilation in the prevention of cerebral oedema in fulminant hepatic failure. J Hepatol. 1986;2:43–51. doi: 10.1016/s0168-8278(86)80007-1. [DOI] [PubMed] [Google Scholar]
- 46.Als-Nielsen B., Gluud L.L., Gluud C. Non-absorbable disaccharides for hepatic encephalopathy: systematic review of randomised trials. BMJ. 2004;328:1046. doi: 10.1136/bmj.38048.506134.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shawcross D., Jalan R. Dispelling myths in the treatment of hepatic encephalopathy. Lancet. 2005;365:431–433. doi: 10.1016/S0140-6736(05)17832-5. [DOI] [PubMed] [Google Scholar]
- 48.Olde Damink S.W., Deutz N.E., Dejong C.H., Soeters P.B., Jalan R. Interorgan ammonia metabolism in liver failure. Neurochem Int. 2002;41:177–188. doi: 10.1016/s0197-0186(02)00040-2. [DOI] [PubMed] [Google Scholar]
- 49.Kircheis G., Wettstein M., Dahl S., Haussinger D. Clinical efficacy of L-ornithine-L-aspartate in the management of hepatic encephalopathy. Metab Brain Dis. 2002;17:453–462. doi: 10.1023/a:1021934607762. [DOI] [PubMed] [Google Scholar]
- 50.Rose C., Michalak A., Rao K.V., Quack G., Kircheis G., Butterworth R.F. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology. 1999;30:636–640. doi: 10.1002/hep.510300311. [DOI] [PubMed] [Google Scholar]
- 51.Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med;362:1071–1081. [DOI] [PubMed]
- 52.Clemmesen J.O., Kondrup J., Ott P. Splanchnic and leg exchange of amino acids and ammonia in acute liver failure. Gastroenterology. 2000;118:1131–1139. doi: 10.1016/s0016-5085(00)70366-0. [DOI] [PubMed] [Google Scholar]
- 53.Rosen H.M., Yoshimura N., Hodgman J.M., Fischer J.E. Plasma amino acid patterns in hepatic encephalopathy of differing etiology. Gastroenterology. 1977;72:483–487. [PubMed] [Google Scholar]
- 54.Brusilow S.W., Valle D.L., Batshaw M. New pathways of nitrogen excretion in inborn errors of urea synthesis. Lancet. 1979;2:452–454. doi: 10.1016/s0140-6736(79)91503-4. [DOI] [PubMed] [Google Scholar]
- 55.Larsen F.S., Wendon J. Alternative pathway therapy for hyperammonemia in liver failure. Hepatology. 2009;50:3–5. doi: 10.1002/hep.23037. [DOI] [PubMed] [Google Scholar]
- 56.Davies N.A., Wright G., Ytrebo L.M. L-ornithine and phenylacetate synergistically produce sustained reduction in ammonia and brain water in cirrhotic rats. Hepatology. 2009;50:155–164. doi: 10.1002/hep.22897. [DOI] [PubMed] [Google Scholar]
- 57.Ytrebo L.M., Kristiansen R.G., Maehre H. L-ornithine phenylacetate attenuates increased arterial and extracellular brain ammonia and prevents intracranial hypertension in pigs with acute liver failure. Hepatology. 2009;50:165–174. doi: 10.1002/hep.22917. [DOI] [PubMed] [Google Scholar]
- 58.Sushma S., Dasarathy S., Tandon R.K., Jain S., Gupta S., Bhist M.S. Sodium benzoate in the treatment of acute hepatic encephalopathy: a double-blind randomized trial. Hepatology. 1992;16:138–144. doi: 10.1002/hep.1840160123. [DOI] [PubMed] [Google Scholar]
- 59.Efrati C., Masini A., Merli M., Valeriano V., Riggio O. Effect of sodium benzoate on blood ammonia response to oral glutamine challenge in cirrhotic patients: a note of caution. Am J Gastroenterol. 2000;95:3574–3578. doi: 10.1111/j.1572-0241.2000.03295.x. [DOI] [PubMed] [Google Scholar]
- 60.Jiang Q., Jiang X.H., Zheng M.H., Jiang L.M., Chen Y.P., Wang L. Rifaximin versus nonabsorbable disaccharides in the management of hepatic encephalopathy: a meta-analysis. Eur J Gastroenterol Hepatol. 2008;20:1064–1070. doi: 10.1097/MEG.0b013e328302f470. [DOI] [PubMed] [Google Scholar]
- 61.Miglio F., Valpiani D., Rossellini S.R., Ferrieri A. Rifaximin, a non-absorbable rifamycin, for the treatment of hepatic encephalopathy. A double-blind, randomised trial. Curr Med Res Opin. 1997;13:593–601. doi: 10.1185/03007999709113333. [DOI] [PubMed] [Google Scholar]
- 62.Loguercio C., Federico A., De Girolamo V., Ferrieri A., Del Vecchio Blanco C. Cyclic treatment of chronic hepatic encephalopathy with rifaximin. Results of a double-blind clinical study. Minerva Gastroenterol Dietol. 2003;49:53–62. [PubMed] [Google Scholar]
- 63.Mas A., Rodes J., Sunyer L. Comparison of rifaximin and lactitol in the treatment of acute hepatic encephalopathy: results of a randomized, double-blind, double-dummy, controlled clinical trial. J Hepatol. 2003;38:51–58. doi: 10.1016/s0168-8278(02)00350-1. [DOI] [PubMed] [Google Scholar]
- 64.Sharma P., Sharma B.C., Puri V., Sarin S.K. An open-label randomized controlled trial of lactulose and probiotics in the treatment of minimal hepatic encephalopathy. Eur J Gastroenterol Hepatol. 2008;20:506–511. doi: 10.1097/MEG.0b013e3282f3e6f5. [DOI] [PubMed] [Google Scholar]
- 65.Lee W.M., Hynan L.S., Rossaro L. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137:856–864. doi: 10.1053/j.gastro.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Canalese J., Gimson A.E., Davis C., Mellon P.J., Davis M., Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut. 1982;23:625–629. doi: 10.1136/gut.23.7.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rolando N., Wade J.J., Stangou A. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg. 1996;2:8–13. doi: 10.1002/lt.500020103. [DOI] [PubMed] [Google Scholar]
- 68.Rolando N., Clapperton M., Wade J., Panetsos G., Mufti G., Williams R. Granulocyte colony-stimulating factor improves function of neutrophils from patients with acute liver failure. Eur J Gastroenterol Hepatol. 2000;12:1135–1140. doi: 10.1097/00042737-200012100-00011. [DOI] [PubMed] [Google Scholar]
- 69.Harrison P.M., Wendon J.A., Gimson A.E., Alexander G.J., Williams R. Improvement by acetylcysteine of hemodynamics and oxygen transport in fulminant hepatic failure. N Engl J Med. 1991;324:1852–1857. doi: 10.1056/NEJM199106273242604. [DOI] [PubMed] [Google Scholar]
- 70.Walsh T.S., Hopton P., Philips B.J., Mackenzie S.J., Lee A. The effect of N-acetylcysteine on oxygen transport and uptake in patients with fulminant hepatic failure. Hepatology. 1998;27:1332–1340. doi: 10.1002/hep.510270520. [DOI] [PubMed] [Google Scholar]
- 71.Murphy N.D., Kodakat S.K., Wendon J.A. Liver and intestinal lactate metabolism in patients with acute hepatic failure undergoing liver transplantation. Crit Care Med. 2001;29:2111–2118. doi: 10.1097/00003246-200111000-00011. [DOI] [PubMed] [Google Scholar]
- 72.Nagayama M., Okuno M., Takai T. Effect of fat emulsion in patients with liver disorders. Nutrition. 1991;7:267–270. [PubMed] [Google Scholar]
- 73.Emre S., Schwartz M.E., Shneider B. Living related liver transplantation for acute liver failure in children. Liver Transpl Surg. 1999;5:161–165. doi: 10.1002/lt.500050315. [DOI] [PubMed] [Google Scholar]
- 74.Miwa S., Hashikura Y., Mita A. Living-related liver transplantation for patients with fulminant and subfulminant hepatic failure. Hepatology. 1999;30:1521–1526. doi: 10.1002/hep.510300621. [DOI] [PubMed] [Google Scholar]
- 75.Uemoto S., Inomata Y., Sakurai T. Living donor liver transplantation for fulminant hepatic failure. Transplantation. 2000;70:152–157. [PubMed] [Google Scholar]
- 76.Ichida T., Todo S., Fujiwara K. Living related donor liver transplantation for adult fulminant hepatic failure. Hepatology. 2000;32:340A. [Google Scholar]
- 77.Steiner C., Mitzner S. Experiences with MARS liver support therapy in liver failure: analysis of 176 patients of the International MARS Registry. Liver. 2002;22(suppl 2):20–25. doi: 10.1034/j.1600-0676.2002.00003.x. [DOI] [PubMed] [Google Scholar]
- 78.Novelli G., Rossi M., Pretagostini R. MARS (Molecular Adsorbent Recirculating System): experience in 34 cases of acute liver failure. Liver. 2002;22(suppl 2):43–47. doi: 10.1034/j.1600-0676.2002.00008.x. [DOI] [PubMed] [Google Scholar]
- 79.Kamar N., Izopet J., Tripon S. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N Engl J Med. 2014;20:1111–1120. doi: 10.1056/NEJMoa1215246. [DOI] [PubMed] [Google Scholar]
- 80.Goyal R., Kumar A., Panda S.K., Paul S.B., Acharya S.K. Ribavirin therapy for hepatitis E virus-induced acute on chronic liver failure: a preliminary report. Antivir Ther. 2012;17:1091–1096. doi: 10.3851/IMP2317. [DOI] [PubMed] [Google Scholar]
- 81.Shalimar, Acharya S.K. Hepatitis E and acute liver failure in pregnancy. J Clin Exp Hepatol. 2013;3:213–224. doi: 10.1016/j.jceh.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]