

Abstract
Objectives
To provide a review and summary of recent advances in the diagnosis and management of disorder(s) of sexual differentiation (DSD), an area that has developed over recent years with implications for the management of children with DSD; and to assess the refinements in the surgical techniques used for genital reconstruction.
Methods
Recent publications (in the previous 10 years) were identified using PubMed, as were relevant previous studies, using following keywords; ‘diagnosis and management’, ‘ambiguous genitalia’, ‘intersex’, ‘disorders of sexual differentiation’, ‘genitogram’, ‘endocrine assessment’, ‘gender assignment’, ‘genitoplasty’, and ‘urogenital sinus’. The findings were reviewed.
Results
Arbitrary criteria have been developed to select patients likely to have DSD. Unnecessary tests, especially those that require anaesthesia or are associated with radiation exposure, should be limited to situations where a specific question needs to be answered. Laparoscopy is an important diagnostic tool in selected patients. The routine use of multidisciplinary diagnostic and expert surgical teams has become standard. Full disclosure of different therapeutic approaches and their timing is recommended.
Conclusions
Diagnostic tests should be tailored according to the available information. Parents and/or patients should be made aware of the paucity of well-designed studies, as these conditions are rare. Unnecessary irreversible surgery should be postponed until a multidisciplinary experienced team, with the parents’ and or patients’ approval, can make a well-judged decision.
Abbreviations: DSD, disorder(s) of sexual differentiation; CAH, congenital adrenal hyperplasia; MIS, Müllerian-inhibiting substance; PAIS, partial androgen insensitivity; GD, gonadal dysgenesis; CAIS, complete androgen–insensitivity syndrome; UGS, urogenital sinus; TUM, total UGS mobilisation; ASTRA, anterior sagittal transrectal approach
Keywords: Diagnosis, Management, Ambiguous genitalia, Intersex, Genitogram, Endocrine assessment, Gender assignment, Genitoplasty, Urogenital sinus
Diagnosis
There are four general concepts and goals in the diagnosis of disorders of sexual differentiation (DSD): (1) The urgent detection of underlying endocrinopathies; (2) Gender assignment must be avoided before an expert evaluation in newborns, which usually requires a few weeks; (3) The evaluation and long-term management must be undertaken at a centre with an experienced multidisciplinary team; and (4) Open communication with patients and families is essential, participation in decision-making is to be encouraged, and ample time and opportunity should be made for continued discussion.
Presentation and clinical evaluation
The presentation of DSD can be at birth or later in life (Table 1); the clinical evaluation is shown in Table 2. The stage of female virilisation can be assessed using the Prader scale [1]. The Quigley scale is used for assessing the development of external genitalia in 46,XY children with DSD [2]. Recently, Rink et al. [3] developed the ‘PVE’ classification for genital ambiguity, where P represents stretched phallic length and width, V is the distance from the bladder neck to the vagina and the distance from the vagina to the perineal meatus, and E is the Prader number. This classification system was found to aid in the surgical planning and the analysis of surgical outcomes.
Table 1.
The presentation of DSD.
| Period | Signs/symptoms |
|---|---|
| Neonatal | |
| 1. Overt genital ambiguity (e.g. cloacal exstrophy) | |
| 2. Apparent female genitalia: | |
| Enlarged clitoris > 9 mm | |
| Posterior labial fusion: Anogenital ratio, anus to posterior fourchette/anus and the base of the clitoris is > 0.5 | |
| Inguinal/labial mass | |
| 3. Apparent male genitalia: | |
| Bilateral impalpable testes | |
| Mild hypospadias with undescended testes (palpable or not) | |
| Perineal hypospadias with bifid scrotum | |
| Micropenis < 1.9 cm | |
| 4. A family history of DSD (CAIS) | |
| 5. Genital and karyotype discordance | |
| Older individuals | |
| 1. Unrecognised genital ambiguity | |
| 2. Inguinal hernia in a female | |
| 3. Delayed/incomplete puberty | |
| Virilisation in a female | |
| Primary amenorrhoea | |
| Male breast development | |
| Gross haematuria in a male | |
Table 2.
Clinical evaluations of DSD.
| Evaluation | Finding |
|---|---|
| History | |
| 1. The maternal history of androgen exposure | |
| 2. A family history of neonatal death might indicate CAH | |
| 3. History of consanguinity, autosomal recessive disorder | |
| 4. Family history of females who are childless or have amenorrhoea (CAIS) | |
| Physical examination | |
| 1. The gonads | |
| a. If the gonads are palpable CAH can be excluded. | |
| b. Ovotestes can be suspected by asymmetry of tissue texture of the poles of the gonad | |
| c. Impalpable gonads raise the possibility of a virilised female with CAH | |
| 2. Rugated scrotum with increased pigmentation raises the possibility of CAH | |
| 3. Normal phallic length in a newborn, measured when stretched from the penopubic junction to the tip of the glans, should be ⩾ 2.5 cm and the normal diameter ⩾ 0.9 cm | |
| 4. Normal clitoral width 2–6 mm; clitoral length > 9 mm is unusual | |
| 5 Anogenital ratio: if >0.5 indicates virilisation | |
Laboratory evaluation
First-line testing in newborns includes karyotyping with X- and Y-specific probe detection (even when a prenatal karyotype is available). The results of karyotyping can take 2–3 days. Fluorescence in situ hybridisation can be used to identify X and Y chromosomes within a few hours. The laboratory evaluation should also include an assay for serum electrolytes, to exclude a salt-wasting form of congenital adrenal hyperplasia (CAH), a measurement of 17-hydroxyprogesterone (usually after 48 h to avoid interference with maternal progesterone), testosterone, gonadotrophins, and Müllerian-inhibiting substance (MIS). Urine analysis is also required to exclude proteinuria associated with Denys Drash syndrome.
Radiology
The radiological assessment can include ultrasonography, MRI or CT, and a retrograde genitogram (Fig. 1) to assess the Müllerian structures and the kidneys. In a recent study the role of the genitogram during the preoperative evaluation in females with CAH has been evaluated. Genitography did not reveal the urogenital sinus (UGS) anatomy completely in 25% of the patients [4]. However, a well-conducted genitogram shows important anatomical details that can be used in operative planning and eliminates the need for an endoscopic examination in a separate session.
Figure 1.

A genitogram in a patient with Prader 4 CAH, showing the anatomy of the UGS. Note that distance number 5 indicates the length of the confluence of the urethra and the vagina to the perineum, and distance number 4 is the urethral length.
Because of radiation exposure and the need for sedation the use of CT and MRI should be reserved for patients with suspect or inconclusive findings on ultrasonography.
Endoscopy, laparoscopy and gonadal biopsy
Preoperative endoscopy and laparoscopy (See Video 1) can provide useful anatomical details that can help in the diagnosis and surgical planning [4]. The presence of a cervix and a uterus exclude 46,XY DSD, except for persistent Müllerian duct syndrome and dysgenetic gonads. Preoperative endoscopy is considered an integral part of genitoplasty, as it facilitates the insertion of a Fogarty balloon catheter. A biopsy as well as laparoscopy is not required when the diagnosis is clearly established biochemically or by gene studies, as the histology can be confidently predicted. It is only required when an ovotestis or dysgenetic gonad is suspected, to determine the definitive diagnosis [5].
Gender assignment
This should be based on the judgement of a multidisciplinary team including paediatric subspecialists in endocrinology, urology, psychology, genetics, neonatology, social work, nursing, and medical ethics [6,7]. The assignment depends on many factors, including the genes involved. Despite the significant progress made over recent years in understanding the genetic basis of human sexual development, a specific molecular diagnosis is identified in about 20% of cases of DSD. Most virilised 46,XX infants will have CAH. By contrast, only half of 46,XY children with DSD will be given a definitive diagnosis [8,9]. Nevertheless, the molecular diagnosis alone cannot dictate the gender assignment. There are several other factors that cannot be ignored, including genital appearance, prenatal androgen exposure, surgical options, the need for lifelong hormone therapy, fertility potential, family wishes, and social circumstances.
Disorders that are preferably raised as males are:
-
(1)
XY karyotype, relative virilisation, partial androgen insensitivity (PAIS), 5α-reductase or 17β-hydroxysteroid dehydrogenase deficiency.
-
a.
At puberty, two-thirds of those reared as females virilise and live as males [10].
-
b.
Fertility is possible in 5α-reductase deficiency [10].
-
c.
Dissatisfaction is reported by 25% of patients with PAIS regardless of sex of rearing [11].
-
(2)
Micropenis and 46,XY cloacal exstrophy [12].
-
(3)
46,XX CAH in older patients who have normal male external genitalia but with a delayed diagnosis. By contrast, evidence supports the current recommendation to raise markedly virilised 46,XX newborns with CAH as female [13].
Disorders that are preferably raised as females are:
-
(1)
46,XY individuals with PAIS or genital gonadal dysgenesis (GD) with poorly virilised genitalia [11].
-
(2)
46,XX individuals with CAH with milder degrees of virilisation [13].
-
(3)
All patients with 46,XY CAIS with no ambiguous genitalia who were assigned as female in infancy. All such patients later identify themselves as females [14].
Surgical management
Feminising genitoplasty: The genitalia are reconstructed at 2–6 months old; reconstruction in early infancy is relatively easier due to the advantageous effects of maternal oestrogen on tissue. Moreover, the potential complications related to the continuity between the urinary tract and peritoneum via the Fallopian tubes are circumvented. However, minor surgical revisions might be needed at the time of puberty [15]. These refined surgical procedures are mainly used for vaginal stenosis, as vaginal dilatation is not advisable before puberty. The efficacy of early (<12 months old) vs. late surgery (in adolescence and adulthood) has not been evaluated in controlled clinical trials. Preoperative preparation should include antibiotics and a ‘steroid-stress’ dose [16]. A rectal enema can be also used. Reconstruction includes three components, i.e. clitoroplasty, labioplasty and vaginoplasty. Postnatal steroid therapy seems to be associated with an improvement in the external appearance of genitalia in patients with less severe clitoromegaly [17]. Thus clitoroplasty may only be used in severe cases (Prader III–V) and be done at the same time as the common UGS repair. Clitoroplasty should respect the innervation and vascularity, to preserve not only the appearance but also the function (Fig. 2).
Figure 2.

Clitoroplasty: Note the preservation of the dorsal neurovascular bundle and the sensitive mucosal collar around the glans of the clitoris.
The technique of vaginoplasty depends on the location of the confluence in relation to the bladder neck, which is a more critical factor than the length of the common channel [18]. The ‘cutback’ vaginoplasty is appropriate only for simple cases. The perineal omega-shaped skin-flap vaginoplasty is applicable to a low confluence and as an adjunct to other forms of vaginoplasty [19]. Total UGS mobilisation (TUM) (Fig. 3) is particularly useful in those with a low and intermediate confluence [16,18].
Figure 3.
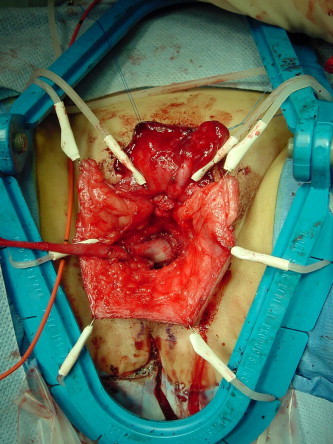
TUM: Note the confluence between the vagina and the urethra, as indicated by the Fogarty catheter balloon, was brought down to the level of the perineum.
Partial UGS mobilisation with a limited anterior dissection to the inferior border of pubis has been reported to be equivalent to TUM, but with less risk of urinary incontinence [20]. The ‘pull-through’ vaginoplasty is used for a very high confluence [16].
Recently, Pippi Salle et al. [21] reported the surgical outcome of the anterior sagittal transrectal approach (ASTRA) in the management of patients with a high UGS. ASTRA was found to be useful in providing optimal exposure, facilitating vaginal dissection and separation from the urethra, and allowing reconstruction of the bladder neck musculature, with minimal morbidity. Complete vaginal replacement can be achieved by several techniques, including replacement by intestine [22], skin, or more recently, oral mucosa free grafts [23]. Complete vaginal replacement is usually required in patients with vaginal agenesis. With the current use of vaginal dilators, few women with CAIS need surgery to lengthen the vagina [24]. If vaginoplasty is the only indicated operation, delaying it until puberty can minimise the complications [22].
Despite the great advances in medical and surgical care for this rare disease, women with CAH still have major sexual and reproductive functional deficits [25]. Gastaud et al. [25] evaluated the long-term outcome of 35 women with CAH (Prader stages I–V) who had been treated from birth to adolescence in the same clinics. None of the women (when aged 18–43 years) expressed doubts about their gender assignment. However, homosexual inclinations were noted in 20% and more than a third of the patients had never had heterosexual intercourse with vaginal penetration. Moreover, most (81%) of those who had vaginal penetration experienced pain. Only 17% (six of 35) had children.
Masculinising genital surgery is surgically more challenging than feminising genitoplasty [11]. Different innovative surgical approaches have been described for repairing severe hypospadias with chordee correction. Preoperative testosterone can be given to facilitate surgery. In some cases an adult-sized testicular implant can be inserted after sufficient pubertal scrotal development [26]. The prophylactic removal of asymptomatic discordant structures, such as a utriculus or Müllerian remnants, is indicated only if they become symptomatic. The surgical outcome in under-masculinised males is variable and depends on the severity of the hypospadias and the adequacy of phallic size.
Summary
Great advances have been made over recent years in the diagnosis and management of DSD. Diagnostic tests should be tailored according to the available information. Unnecessary tests, especially those that require anaesthesia for removal or are associated with radiation exposure, should be limited to situations where a specific question needs to be answered.
Parents and or patients should be made aware of the paucity of well-designed studies, as these conditions are rare. Unnecessary irreversible surgery should be postponed until a multidisciplinary experienced team, with the parents’ and or patients’ approval, can make a well-judged decision.
Source of funding
None.
Conflict of interest
None.
Footnotes
Peer review under responsibility of Arab Association of Urology.

Appendix A. Supplementary data
A laparoscopic examination of a patient with 46,XY ovotesticular DSD. Note the right streak ovary is attached to a dysgenetic testis. On the left side there is a streak gonad and an abdominal testis. Note also the presence of uterus and Fallopian tubes on both sides?
References
- 1.Prader A. Incidence of congenital adrenogenital syndrome. Helvetica Paediatrica Acta. 1958;13:426–431. [PubMed] [Google Scholar]
- 2.Quigley C.A., De Bellis A., Marschke K.B., el-Awady M.K., Wilson E.M., French F.S. Androgen receptor defects. Historical, clinical, and molecular perspectives. Endocrine Rev. 1995;16:271–321. doi: 10.1210/edrv-16-3-271. [DOI] [PubMed] [Google Scholar]
- 3.Rink R.C., Adams M.C., Misseri R. A new classification for genital ambiguity and urogenital sinus anomalies. BJU Int. 2005;95:638–642. doi: 10.1111/j.1464-410X.2005.05354.x. [DOI] [PubMed] [Google Scholar]
- 4.Vanderbrink B.A., Rink R.C., Cain M.P., Kaefer M., Meldrum K.K., Misseri R. Does preoperative genitography in congenital adrenal hyperplasia cases affect surgical approach to feminizing genitoplasty? J Urol. 2010;184(Suppl. 4):1793–1798. doi: 10.1016/j.juro.2010.05.082. [DOI] [PubMed] [Google Scholar]
- 5.Aaronson I.A., Aaronson A.J. How should we classify intersex disorders? J Pediatr Urol. 2010;6:443–446. doi: 10.1016/j.jpurol.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Lee P.A. A perspective on the approach to the intersex child born with genital ambiguity. J Pediatr Endocrinol Metab. 2004;17:133–140. doi: 10.1515/jpem.2004.17.2.133. [DOI] [PubMed] [Google Scholar]
- 7.Palmer B.W., Wisniewski A.B., Schaeffer T.L., Mallappa A., Tryggestad J.B., Krishnan S. A model of delivering multi-disciplinary care to people with 46 XY DSD. J Pediatr Urol. 2012;8:7–16. doi: 10.1016/j.jpurol.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed S.F., Cheng A., Dovey L., Hawkins J.R., Martin H., Rowland J. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab. 2000;85:658–665. doi: 10.1210/jcem.85.2.6337. [DOI] [PubMed] [Google Scholar]
- 9.Morel Y., Rey R., Teinturier C., Nicolino M., Michel-Calemard L., Mowszowicz I. Aetiological diagnosis of male sex ambiguity: a collaborative study. Eur J Pediatr. 2002;161:49–59. doi: 10.1007/s00431-001-0854-z. [DOI] [PubMed] [Google Scholar]
- 10.Cohen-Kettenis P.T. Gender change in 46,XY persons with 5 alpha-reductase-2 deficiency and 17 beta-hydroxysteroid dehydrogenase-3 deficiency. Arch Sex Behav. 2005;34:399–410. doi: 10.1007/s10508-005-4339-4. [DOI] [PubMed] [Google Scholar]
- 11.Migeon C.J., Wisniewski A.B., Gearhart J.P., Meyer-Bahlburg H.F.L., Rock J.A., Brown T.R. Ambiguous genitalia with perineoscrotal hypospadias in 46,XY individuals: long-term medical, surgical, and psychosexual outcome. Pediatrics. 2002;110 doi: 10.1542/peds.110.3.e31. e31-e31. [DOI] [PubMed] [Google Scholar]
- 12.Meyer-Bahlburg H.F.L. Gender identity outcome in female-raised 46,XY persons with penile agenesis, cloacal exstrophy of the bladder, or penile ablation. Arch Sex Behav. 2005;34:423–438. doi: 10.1007/s10508-005-4342-9. [DOI] [PubMed] [Google Scholar]
- 13.Berenbaum S., Chrousos G., Clayton P., Cutler G., Keizer-Schrama S.D., Donahoe P.K. Consensus statement on 21-hydroxylase deficiency from The European Society for Paediatric Endocrinology and The Lawson Wilkins Pediatric Endocrine Society. Hormone Res. 2002;58:188–195. doi: 10.1159/000065490. [DOI] [PubMed] [Google Scholar]
- 14.Mazur T. Gender dysphoria and gender change in androgen insensitivity or micropenis. Arch Sex Behav. 2005;34:411–421. doi: 10.1007/s10508-005-4341-x. [DOI] [PubMed] [Google Scholar]
- 15.Eroglu E., Tekant G., Gundogdu G., Emir H., Ercan O., Soylet Y. Feminizing surgical management of intersex patients. Pediatr Surg Int. 2004;20:543–547. doi: 10.1007/s00383-004-1208-5. [DOI] [PubMed] [Google Scholar]
- 16.Leslie J.A., Cain M.P., Rink R.C. Feminizing genital reconstruction in congenital adrenal hyperplasia. Indian J Urol: J Urol Soc India. 2009;25:17–26. doi: 10.4103/0970-1591.45532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulshreshtha B., Khadgawat R., Eunice M., Ammini A.C. Congenital adrenal hyperplasia. Results of medical therapy on appearance of external genitalia. J Pediatr Urol. 2010;6:555–559. doi: 10.1016/j.jpurol.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Jenak R., Ludwikowski B., Gonzalez R. Total urogenital sinus mobilization. A modified perineal approach for feminizing genitoplasty and urogenital sinus repair. J Urol. 2001;165(6 Part 2):2347–2349. doi: 10.1016/S0022-5347(05)66200-3. [DOI] [PubMed] [Google Scholar]
- 19.Freitas Filho L.G., Carnevale J., Melo C.E., Laks M., Calcagno Silva M. A posterior-based omega-shaped flap vaginoplasty in girls with congenital adrenal hyperplasia caused by 21-hydroxylase deficiency. BJU Int. 2003;91:263–267. doi: 10.1046/j.1464-410x.2003.03085.x. [DOI] [PubMed] [Google Scholar]
- 20.Rink R.C., Metcalfe P.D., Kaefer M.A., Casale A.J., Meldrum K.K., Cain M.P. Partial urogenital mobilization: a limited proximal dissection. J Pediatr Urol. 2006;2:351–356. doi: 10.1016/j.jpurol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Salle J.L., Lorenzo A.J., Jesus L.E., Leslie B.A.I., Said A., Macedo F.N. Surgical treatment of high urogenital sinuses using the anterior sagittal transrectal approach: a useful strategy to optimize exposure and outcomes. J Urol. 2012;187:1024–1031. doi: 10.1016/j.juro.2011.10.162. [DOI] [PubMed] [Google Scholar]
- 22.Burgu B., Duffy P.G., Cuckow P., Ransley P., Wilcox D.T. Long-term outcome of vaginal reconstruction: comparing techniques and timing. J Pediatr Urol. 2007;3:316–320. doi: 10.1016/j.jpurol.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Samuelson M.L., Baker L.A. Autologous buccal mucosa vulvovaginoplasty for high urogenital sinus. J Pediatr Urol. 2006;2:486–488. doi: 10.1016/j.jpurol.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Wisniewski A.B., Migeon C.J., Meyer-Bahlburg H.F.L., Gearhart J.P., Berkovitz G.D., Brown T.R. Complete androgen insensitivity syndrome. Long-term medical, surgical, and psychosexual outcome. J Clin Endocrinol Metab. 2000;85:2664–2669. doi: 10.1210/jcem.85.8.6742. [DOI] [PubMed] [Google Scholar]
- 25.Gastaud F., Bouvattier C., Duranteau L., Brauner R., Thibaud E., Kutten F. Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. J Clin Endocrinol Metabo. 2007;92:1391–1396. doi: 10.1210/jc.2006-1757. [DOI] [PubMed] [Google Scholar]
- 26.Mouriquand P.D.E., Mure P.Y. Current concepts in hypospadiology. BJU Int. 2004;93:26–34. doi: 10.1111/j.1464-410X.2004.04706.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A laparoscopic examination of a patient with 46,XY ovotesticular DSD. Note the right streak ovary is attached to a dysgenetic testis. On the left side there is a streak gonad and an abdominal testis. Note also the presence of uterus and Fallopian tubes on both sides?