

Abstract
Objectives
To provide a summary of the recent major advances in the field of molecular genetics and understanding of psychosexual development, as these developments have resulted in changes in terminology and classification of disorders of sexual differentiation (DSD)/intersex; and to provide a quick and simplified review of the basic information.
Methods
Recent publications (over the last 10 years) were identified by a PubMed search, as were relevant previous studies, using the keywords; ‘sex chromosomes’, ‘psychosexual development’, ‘classifications’, ‘disorders of sexual differentiation’, ‘Chicago consensus’, ‘gonadal malignancy’, ‘intersex’ and ‘ambiguous genitalia’.
Results
The newly proposed terminology and classification has eliminated some confusion for both patient and family, as well as among health professionals. The new advances have facilitated the categorisation of gonadal malignancy in patients with DSD into high-, intermediate- and low-risk groups.
Conclusions
The major changes in terminology and classification of DSD should be considered as the first steps on a long road of research effort. The current available data remain far from sufficient. More molecular genetics studies will allow a better understanding of the causes of each condition of DSD.
Abbreviations: CIS, carcinoma in situ; DSD, disorder(s) of sexual differentiation; SRY, sex-determining region on the Y chromosome; SF-1, steroidogenic factor 1; WT-1, Wilms’ tumour-1 gene; MIS, Müllerian-inhibiting substance; MGD, mixed gonadal dysgenesis; CAH, congenital adrenal hyperplasia; hCG, human chorionic gonadotrophin; CAIS, complete androgen insensitivity syndrome; PMDS, persistent Müllerian duct syndrome
Keywords: Sex chromosomes, Psychosexual development, Classification, Chicago Consensus, Gonadal malignancy, Intersex
Normal sex development
Before the seventh week of gestation both XX and XY foetuses have an identical reproductive anatomy. At 7 weeks those foetuses with a Y chromosome begin to develop testes, while those with no Y chromosome start to develop ovaries that also require an active genetic pathway. Thereafter, gonadal differentiation and function determine the final phenotype.
Many genes are involved in normal sexual differentiation. The sex-determining region on the Y chromosome (SRY) initiates the molecular events of testis formation [1–3]. The SOX9 gene is required for Sertoli-cell differentiation; haplo-insufficiency of SOX9 results in sex reversal in XY individuals, and SOX9 duplication is the only known autosomal cause of XX sex reversal (an XX karyotype with male phenotype). Steroidogenic factor 1 (SF-1) plays a critical role in steroidogenesis, fertility and male sexual differentiation. SF-1 mutations cause cryptorchidism, micropenis and XY sex reversal. DAX-1 on the X chromosome is up-regulated in the ovary and functions as an anti-testis factor. DAX-1 duplication can repress SRY and cause a disorder of sexual differentiation (DSD) with a female phenotype in an individual with 46,XY chromosomes. The Wilms tumour (WT-1) gene is involved in both gonadal and renal development. Three distinct phenotypes are seen with WT-1 mutations: (i) the Wilms tumour, aniridia, genitourinary anomalies, and mental retardation syndrome, caused by continuous deletion of the WT-1 and PAX-6 genes. (ii) Denys–Drash syndrome (a triad of progressive renal disease, 46,XY karyotype with undervirilisation, and Wilms tumour. Affected individuals usually have ambiguous genitalia or normal female external genitalia, and streak gonads [4]. Nephrotic syndrome presents within the first 2 years of life and progresses rapidly to end-stage renal failure within a few years. iii) Frasier syndrome (46,XY DSD, gonadal dysgenesis, and renal failure), in which there is an altered ratio of the two splice isoforms of the WT-1 protein [5]. Affected individuals have normal female external genitalia but fail to develop secondary sexual characteristics [4]. Patients are at risk of gonadoblastoma developing in the dysgenetic gonads. Glomerulonephropathy gradually progresses to renal failure in the second or third decade of life.
Internal genitalia
The Wolffian and Müllerian ducts exist in both sexes. In males, testicular Sertoli cells begin secreting Müllerian-inhibiting substance (MIS) in the seventh week, which acts only locally (paracrine) to induce ipsilateral Müllerian duct regression [6]. Shortly afterwards, Leydig cells begin producing testosterone through stimulation via placental human chorionic gonadotrophin (hCG). Testosterone acts both locally (paracrine) and systemically (endocrine) to stabilise the Wolffian duct, and promotes the development of the epididymis, the vas deferens and the seminal vesicle. In females the lack of these hormones leads to Wolffian duct regression and permits Müllerian duct maturation into tubes, uterus, cervix and upper vagina.
External genitalia
The testosterone produced by testicular Leydig cells undergoes peripheral conversion to dihydrotestosterone, under the effect of 5α-reductase. Dihydrotestosterone induces posterior fusion of the genital folds and growth of the genital tubercle into a phallic structure. Male external genitalia are complete by 12–16 weeks. Subsequent phallic growth is a result of foetal pituitary luteinising hormone stimulation of testicular Leydig cell testosterone production [2]. By 12 weeks, the non-hormone-dependent separation of the vagina and the urethra is complete in females. Excess androgen exposure before this separation can cause labial fusion and development of a phallic urethra or urogenital sinus, but later exposure causes only clitoral enlargement and scrotalisation of the labial folds.
Psychosexual development
Psychosexual development has three main components: ‘gender identity’ which signifies the child’s self-recognition as a boy or a girl, and usually starts at 3 years of age [7]. ‘Gender role’ refers to sex-typical behaviours such as toy preferences [8] and physical aggression, and ‘sexual orientation’ refers to the direction(s) of sexual interest (heterosexual, bisexual, and homosexual) [9]. Many factors affect psychosexual development, e.g. exposure to androgens, sex chromosome genes and brain structure, as well as the society and family perspectives. Gender dysphoria indicates unhappiness with the assigned sex and it results from an inconsistency between the assignment and the inherent identity later in life [10]. Although gender dissatisfaction occurs more frequently in individuals with DSD than in the general population it cannot be easily predicted from the karyotype, prenatal androgen exposure, degree of genital virilisation, or assigned gender [7–10].
Several factors influence prenatal androgen exposure including the timing, dose and type of androgen exposure, and brain receptor availability, as well as the social environment. The prenatal period is thought to be critical for brain masculinisation [9]. Girls with congenital adrenal hyperplasia (CAH) with marked genital virilisation play more with boys’ toys, and the incidence of homosexuality later in life is higher in this group [9,11]. Animal studies showed marked but complex effects of androgens on sex differentiation of the brain and on behaviour.
Moreover, sex chromosome genes might also influence the brain structure and behaviour directly. However, studies in individuals with complete androgen-insensitivity syndrome (CAIS) do not indicate a behavioural role for Y-chromosome genes [12].
Brain structural differences between males and females have been identified across species [13,14]. However, knowing the structure of the brain is not currently beneficial to gender assignment.
What is DSD?
The Lawson Wilkins Paediatric Endocrine Society and the European Society for Paediatric Endocrinology recently reviewed the management of intersex disorders [15]. Two main factors necessitated a re-examination of the nomenclature, i.e. the advances in molecular genetics, and the increased awareness of ethical issues and patient advocacy concerns [16]. Terms such as ‘intersex’, ‘pseudohermaphroditism’, ‘hermaphroditism’, and ‘sex reversal’ are particularly controversial. These terms are perceived as potentially demeaning by patients, who can function normally as either males or females, and can be confusing to practitioners and parents alike [16].
The term DSD, as defined by a congenital discrepancy between external genitalia, gonadal and chromosomal sex, has been proposed to replace the term ‘intersex’. The changes in terminology considered the flexibility to incorporate new molecular genetic information and to accommodate the spectrum of phenotypic variation. The updated taxonomy reflects the primary disease and does not establish or dictate gender assignment.
The Chicago Consensus [15] new nomenclature is based on the primary genetic defect. It includes three broad categories: (1) Sex chromosome DSD; (2) 46,XX DSD; and (3) 46,XY DSD. An example of DSD categorisation based on the Chicago consensus is shown in Table 1.
Table 1.
An example of DSD categorisation based on the Chicago consensus.
| Category of DSD |
|---|
| 1. Sex chromosome DSD |
| a. 45,X0/46,XY Mixed gonadal dysgenesis (MGD) |
| b. 45,X0/46,XY Partial gonadal dysgenesis |
| c. 46,XX/XY or 45,X0/46,XY Ovotesticular DSD |
| d. 45,X0 or 45,X0/46,XY (Turner’s syndrome) |
| e. Seminiferous tubule dysgenesis: Klinefelter’s syndrome (47,XXY) |
| 2. 46,XX (DSD) |
| a. Androgen excess: (60%–70%) |
| i. CAH (vast majority) |
| 1.21-Hydroxylase deficiency (95%) |
| 2. 11β-Hydroxylase deficiency (5%) |
| 3. 3β-Hydroxysteroid dehydrogenase deficiency |
| ii. Maternal androgens (very rare) |
| 1. Endogenous: Virilising tumours in the mother |
| 2. Exogenous (very rare) |
| b. Disorders of ovarian development |
| i. Ovotesticular DSD |
| ii. 46,XX testicular DSD (SRY translocation) |
| iii. Pure gonadal dysgenesis (e.g. SOX9 duplication) |
| c. Meyer–Rokitansky syndrome (Müllerian aplasia) |
| 3. 46,XY DSD |
| a. Disorders of testicular development |
| i. Gonadal dysgenesis (Swyer syndrome = Pure, DAX-1 Duplication. SF-1 mutation) |
| ii. Ovotesticular DSD |
| iii. Bilateral vanishing testis/testicular regression syndromes |
| b. Disorders of androgen synthesis or action: |
| i. Leydig cell agenesis, unresponsiveness |
| ii. Enzyme deficiency: |
| 1. StAR deficiency (lipoid adrenal hyperplasia) |
| 2. 3β-Hydroxysteroid dehydrogenase deficiency |
| 3. 17α-Hydroxylase deficiency |
| 4. 17,20-Lyase deficiency |
| 5. 17β-Hydroxysteroid oxidoreductase deficiency |
| 6. 5α-Reductase deficiency |
| iii. Disorders of androgen-dependent target tissue |
| 1. CAIS (testicular feminisation) |
| 2. PAIS |
| iv. Persistent Müllerian duct syndrome |
| 4. Others: Severe hypospadias and cryptorchidism, penile agenesis and cloacal exstrophy |
Potential disadvantages of the new classification include [10]: (1) The karyotype might vary among individuals with the same condition. (2) The current level of knowledge might be insufficiently complete to comprehensively classify DSD based on the underling genetic error. (3) The term ‘Different’ or ‘Discrepant’ has been suggested to replace ‘Disorder’. A comprehensive classification based on gonadal histology has been subsequently proposed [17] as follows: (i) Ovarian DSD: the gonads are composed of normal ovarian stroma with numerous follicles. (ii) Ovotesticular DSD: the gonads are essentially composed of both ovarian and testicular tissues, either in two separate gonads or within a single gonad. At the least, a well-defined ovarian follicle should be seen to diagnose the ovarian element. The testicular component comprises architecturally ordered tubules, although the intervening stroma may be more abundant than normal. (iii) Testicular DSD: The seminiferous tubules are normal, although Leydig cells might be prominent. (iv) Dysgenetic DSD: The tubules are disordered, often sparse, and the stromal tissue is abundant. These gonads have a strong propensity to undergo malignant degeneration. Aaronson’s classification does not contradict the Chicago consensus and allows a better understanding of the nomenclature based on the histology.
Gonadal malignancy
A summary of the risks of gonadal malignancy in different DSD conditions is provided in Table 2. Gonadoblastoma arises from undifferentiated germ cells and carcinoma and carcinoma in situ (CIS) arises from differentiated germ cells [15]. A scrotal testis in patients with gonadal dysgenesis is at risk of malignancy. The current recommendations are a testicular biopsy at puberty to seek signs of CIS or undifferentiated intratubular germ cell neoplasia. If positive, the option is sperm banking before treatment with local low-dose radiotherapy that is curative [18].
Table 2.
Gonadal malignancy risks in different DSD conditions.
| Risk (%) | Disorder | Treatment |
|---|---|---|
| Highest (up to 60) | ||
| MGD + Y + intra-abdominal | Gonadectomy at diagnosis (all) | |
| Fraiser | ||
| Denys–Drash + Y | ||
| PAIS + intra-abdominal | ||
| Intermediate (up to 28) | ||
| Turner + Y | Gonadectomy at diagnosis | |
| MGD + Y + scrotal | Examination every 6 months + | |
| PAIS Scrotal gonad | pubertal biopsy + | |
| 17β-hydroxylase | irradiation if CIS | |
| Lowest (5) | ||
| Ovotestis DSD | Gonadectomy at puberty Testicular tissue removal | |
| Genetically confirmed CAIS | ||
Examples of individual DSD conditions
Ovotesticular DSD (previously termed true hermaphrodites) comprises 10% of the DSD pool [19]. The gonads are asymmetrical, with an ovary on one side, usually the left and abdominal with at least one defined follicle, and a testis or ovotestis on the other side, usually the right, located anywhere from the abdomen to the scrotum. The internal ducts vary according to the associated gonads. The karyotype varies, i.e. 46,XX (60%), mosaic 46,XX/XY (30%) and 46,XY (10%). Those patients have a tendency to male phenotype with hypospadias (75%) (Figure 1). Fertility is possible, especially in females. The sex assignment varies, being preferably female in those with a 46,XX karyotype.
Figure 1.

Ovotesticular DSD: Note the tendency to a male phenotype with hypospadias.
Mixed gonadal dysgenesis (MGD) is the second most common cause of ambiguous genitalia in a newborn [20]. The gonads are also asymmetrical, with a dysgenetic testis on one side composed of disordered tubules, often sparse and abundant stroma, and a streak gonad on the other side, composed of ovarian stroma but with no oocytes. The karyotype is mostly mosaic 46,XY/45X0. The term partial gonadal dysgenesis has been used to describe a variant with two dysgenetic testes in which the karyotype is mostly 46,XY.
The phenotype is usually ambiguous. Incomplete gonadal dysgenesis has also been used to describe individuals with a normal testis in the scrotum and a contralateral streak or dysgenetic gonad in which the karyotype is either mosaic 46,XY/45,X0 or 46,XY. These patients have a tendency to a male phenotype with hypospadias (Figure 2a). The internal ducts vary according to the associated gonads (Figure 2b). Fertility has not been reported in patients with MGD. The sex assignment varies, and factors to consider include prenatal androgen exposure, testicular function at and after puberty, phallic development, and gonadal location.
Figure 2.
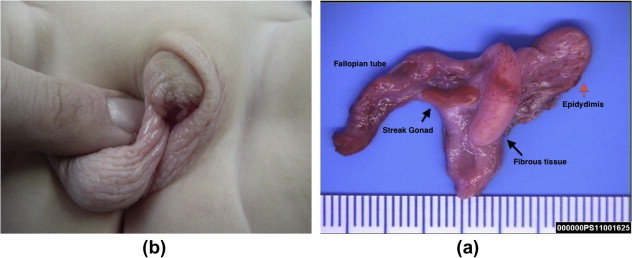
MGD (a) Note the right testis is fully descended in the scrotum; the left streak gonad is in the inguinal canal, and hypospadias and (b) the streak gonad is associated with the Fallopian tube and the epididymis.
CAH represents 60%–70% of ambiguous genitalia in a newborn [21]. The gonads are ovaries bilaterally. The Müllerian system develops normally into tubes, uterus and upper vagina, and the Wolffian system regresses. The karyotype is 46,XX. The metabolic and biochemical consequences vary. Most cases (95%) are due to an autosomal recessive deficiency of 21-hydroxylase. As a result, the adrenal gland is unable to form cortisone and the adrenal steroid hormone synthesis is diverted towards the androgen pathway, resulting in virilisation. In most cases (75%) the deficiency affects both cortisone and aldosterone production, and causes salt wasting. If the zona glumerulosa is spared, aldosterone production remains normal and only simple virilisation without salt wasting (25%) results. The second most common enzyme deficiency is 11β-hydroxylase. The characteristic feature is the accumulation of desoxycorticosterone that causes salt retention and hypertension. Virilisation of the external genitalia varies from minimal phallic enlargement to almost complete masculinisation (Figure 3). Because of the andrenocorticotropic hormone drive there is hyperpigmentation of the external genitalia and nipples. A female gender is preferred, especially in those with mild to moderate virilisation. Fertility is normally expected. A prenatal diagnosis in high-risk families is possible through the determination of an elevated 17-hydroxyprogesterone in amniotic fluid, or more recently by human leukocyte antigen genotyping of chorionic villus samples [22,23].
Figure 3.
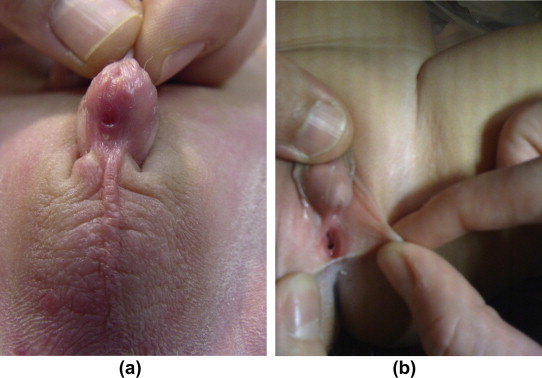
(a) A patient with CAH Prader 4 and (b) a patient with CAH Prader 2.
However, the diagnosis cannot be confirmed before the initial development of the external genitalia. Treatment with dexamethasone, which crosses the blood placental barrier, can prevent virilisation of the external genitalia, but ≈90% of foetuses would be treated unnecessarily, as the disorder is an autosomal recessive (affecting only 25% of foetuses) and male foetuses (50%) would not be affected. Medical treatment with life-long cortisone replacement after birth is mandatory. Prophylactic adrenalectomy might be indicated in patients with repeatedly escaped adrenal suppression.
In under-masculinisation of a male due to enzyme deficiencies (Table 1), the gonads are exclusively testicular tissue intra-abdominally, inguinally, or in the labia. Müllerian structures regress and Wolffian ducts develop. The phenotype is ambiguous and varies from mild hypospadias to complete failure of masculinisation. Variable virilisation occurs at puberty. Fertility has not been reported except with 5α-reductase enzyme deficiency [24,25]. The gonads are at risk of malignancy, especially with 17-β-hydroxysteroid dehydrogenase deficiency. The sex assignment depends on the phenotype. In patients raised as female a gonadectomy should be performed before puberty.
In the syndrome of partial androgen resistance (46,XY DSD), the gonads are testes bilaterally. The Wolffian duct structures remain rudimentary and the Müllerian structures regress. The external genitalia are ambiguous, classically with perineoscrotal hypospadias, pseudovagina and cryptorchidism. At puberty, gynaecomastia and infertility develop, with a normal endocrine profile. Characterisation of the androgen-receptor gene in serum DNA by PCR is helpful in distinguishing between complete and partial androgen-receptor resistance [26]. The gonads are at risk of malignancy. Virilisation of the external genitalia serves as a guide for androgen imprinting of the brain, but the phallus remains small.
CAIS occurs at a frequency of 1 in 20,000 to 1 in 64,000 male births. The karyotype is 46,XY and the gonads are symmetrical and exclusively testicular tissue (seminiferous tubules with no spermatogenesis and increased numbers of Leydig cells). The Wolffian ducts develop in half of these cases, while all Müllerian structures regress. Phenotypically, patients are normal, tall and hairless females with feminine external genitalia and a very short and shallow utricle (Figure 4). At the time of puberty, normal breasts develop due to peripheral conversion of testosterone to oestrogen. Axillary and pubic hair is absent or scanty, with slight vulval hair development. Amenorrhoea is the rule. Patients might present prenatally when there is a female gender with a 46,XY karyotype. In the prepubertal period patients might present with an inguinal hernia. After puberty the condition can be diagnosed during the evaluation of primary amenorrhoea. The diagnosis after puberty can be made by the presence of a male androgen and gonadotrophin profile in a phenotypically female patient. A prepubertal diagnosis is more difficult and requires an hCG stimulation test and PCR characterisation of the androgen-receptor gene in DNA obtained from a venous blood sample [26]. The gonads are at low risk of malignancy because of the cryptorchidism (2%). As the risk starts after puberty a gonadectomy at puberty allows the spontaneous onset of puberty to be followed by oestrogen therapy. A prepubertal gonadectomy can be done if the condition is causing discomfort or hernia formation, or if the gene mutation has not been characterised, to avoid virilisation at the time of puberty.
Figure 4.
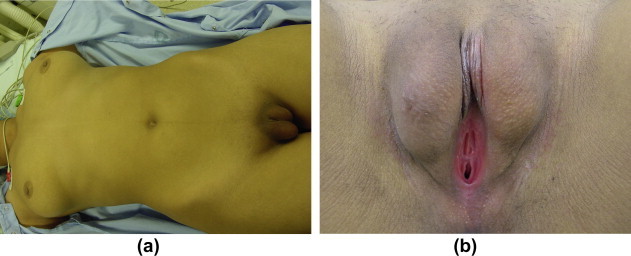
(a) A patient with CAIS. Note a normal, tall and hairless female with normal breast development and scanty pubic hair and (b) note the feminine external genitalia, a very short and shallow utricle and two testicles in the labia.
‘Bilateral vanishing testes’ implies absent gonads that could be due to mutation, exposure to a teratogen, or bilateral torsion. The karyotype is typically 46,XY. The phenotype shows as male with undescended testes. Laparoscopy can detect rudimentary cord structures with no recognisable testicular tissue histologically. These patients will require androgen replacement and testicular prostheses. Embryonic testicular regression syndrome [27] is a variant of bilateral vanishing testes in which the testicular function is lost early in embryonic life. This results in individuals with ambiguous genitalia.
Persistent Müllerian duct syndrome (PMDS) or hernia uterine inguinale results from either a failure of secretion of MIS or a failure of the Müllerian duct to respond to its secretion [28]. Affected individuals have a normal male phenotype with symmetrically or asymmetrically undescended testes or inguinal hernias. Both Wolffian and Müllerian ducts develop. A primary or staged orchidopexy is all that is required. Surgical removal of the Müllerian duct derivatives is controversial, as malignancy in retained Müllerian structures been reported [29] and the potential fertility can be compromised by a surgical attempt to remove the Müllerian derivatives. Abnormalities in the attachment of the epididymis to the testis are also reported.
Summary
Great advances have been made over recent years in understanding the genetics and pathology of DSD. The newly proposed terminology and classifications have eliminated some confusion for both the patient and the family, as well as for health professionals. However, the available data remain far from sufficient. An improved molecular diagnosis might help to predict the long-term risks to quality of life, gender dysphoria, and/or tumour formation.
Conflict of interest
None
Source of funding
None
Footnotes
Peer review under responsibility of Arab Association of Urology.

References
- 1.Achermann J.C., Meeks J.J., Jameson J.L. Phenotypic spectrum of mutations in DAX-1 and SF-1. Mol Cellular Endocrinol. 2001;185:17–25. doi: 10.1016/s0303-7207(01)00619-0. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed S.F., Hughes I.A. The genetics of male undermasculinization. Clin Endocrinol (Oxf) 2002;56:1–18. doi: 10.1046/j.1365-2265.2002.01430.x. [DOI] [PubMed] [Google Scholar]
- 3.Clarkson M.J., Harley V.R. Sex with two SOX on. SRY and SOX9 in testis development. Trends Endocrinol Metabolism. 2002;13:106–111. doi: 10.1016/s1043-2760(01)00541-0. [DOI] [PubMed] [Google Scholar]
- 4.Niaudet P., Gubler M.C. WT1 and glomerular diseases. Pediatr Nephrol. 2006;21:1653–1660. doi: 10.1007/s00467-006-0208-1. [DOI] [PubMed] [Google Scholar]
- 5.Klamt B., Koziell A., Poulat F., Wieacker P., Scambler P., Berta P., Gessler M. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/–KTS splice isoforms. Hum Mol Genet. 1998;7:709–714. doi: 10.1093/hmg/7.4.709. [DOI] [PubMed] [Google Scholar]
- 6.Lee M.M., Donahoe P.K., Hasegawa T., Silverman B., Crist G.B., Best S. Mullerian inhibiting substance in humans: normal levels from infancy adulthood. J Clin Endocrinol Metab. 1996;81:571–576. doi: 10.1210/jcem.81.2.8636269. [DOI] [PubMed] [Google Scholar]
- 7.Zucker K.J. Intersexuality and gender identity differentiation. J Ped Adolescent Gynecol. 2002;15:3–13. doi: 10.1016/s1083-3188(01)00133-4. [DOI] [PubMed] [Google Scholar]
- 8.Nordenstrom A. Sex-typed toy play behavior correlates with the degree of prenatal androgen exposure assessed by CYP21 genotype in girls with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87:5119–5124. doi: 10.1210/jc.2001-011531. [DOI] [PubMed] [Google Scholar]
- 9.Cohen-Bendahan C.C., van de Beek C., Berenbaum S.A. Prenatal sex hormone effects on child and adult sex-typed behavior: methods and findings. Neurosci Biobehav Rev. 2005;29:353–384. doi: 10.1016/j.neubiorev.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Barthold J.S. Disorders of sex differentiation: a pediatric urologist’s perspective of new terminology and recommendations. J Urol. 2011;185:393–400. doi: 10.1016/j.juro.2010.09.083. [DOI] [PubMed] [Google Scholar]
- 11.Meyer-Bahlburg H.F.L. Gender identity outcome in female-raised 46, XY persons with penile agenesis, cloacal exstrophy of the bladder, or penile ablation. Arch Sex Behav. 2005;34:423–438. doi: 10.1007/s10508-005-4342-9. [DOI] [PubMed] [Google Scholar]
- 12.Warne G. Support groups for CAH AIS. Endocrinologist. 2003;13:175–178. [Google Scholar]
- 13.MacLaughlin D.T., Donahoe P.K. Mechanisms of disease: sex determination and differentiation. N Engl J Med. 2004;350:367–378. doi: 10.1056/NEJMra022784. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed S.F., Cheng A., Dovey L., Hawkins J.R., Martin H., Rowland J. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab. 2000;85:658–665. doi: 10.1210/jcem.85.2.6337. [DOI] [PubMed] [Google Scholar]
- 15.Lee P.A., Houk C.P., Ahmed S.F., Hughes I.A. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118:e488–e500. doi: 10.1542/peds.2006-0738. [DOI] [PubMed] [Google Scholar]
- 16.Conn J., Gillam L., Conway G.S. Revealing the diagnosis of androgen insensitivity syndrome in adulthood. BMJ. 2005;331:628–630. doi: 10.1136/bmj.331.7517.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aaronson I.A., Aaronson A.J. How should we classify intersex disorders? J Pediatric Urol. 2010;6:443–446. doi: 10.1016/j.jpurol.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Rorth M., Rajpert-De Meyts E., Andersson L., Dieckmann K.P., Fossa S.D., Grigor K.M. Carcinoma in situ in the testis. Scand J Urol Nephrol Suppl. 2000;205:166–186. doi: 10.1080/00365590050509896. [DOI] [PubMed] [Google Scholar]
- 19.Wiersma R. The clinical spectrum and treatment of ovotesticular disorder of sexual development. Adv Exp Med Biol. 2011;707:101–103. doi: 10.1007/978-1-4419-8002-1_21. [DOI] [PubMed] [Google Scholar]
- 20.Ocal G., Berberoglu M., Siklar Z., Ruhi H.I., Tukun A., Camtosun E. The clinical and genetic heterogeneity of mixed gonadal dysgenesis: does ‘disorders of sexual development (DSD) ’ classification based on new Chicago consensus cover all sex chromosome DSD? Eur J Pediatr. 2012;171:1497–1502. doi: 10.1007/s00431-012-1754-0. [DOI] [PubMed] [Google Scholar]
- 21.Trapp C.M., Speiser P.W., Oberfield S.E. Congenital adrenal hyperplasia: an update in children. Curr Opin Endocrinol Diabetes Obes. 2011;18:166–170. doi: 10.1097/MED.0b013e328346938c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laue L., Rennert O.M. Congenital adrenal hyperplasia. Molecular genetics and alternative approaches to treatment. Adv Pediatrics. 1995;42:113–143. [PubMed] [Google Scholar]
- 23.Hughes I. Congenital adrenal hyperplasia. Phenotype and genotype. Journal of pediatric endocrinology & metabolism. JPEM. 2002;15(5):1329–1340. [PubMed] [Google Scholar]
- 24.Migeon C.J., Wisniewski A.B., Gearhart J.P., Meyer-Bahlburg H.F.L., Rock J.A., Brown T.R. Ambiguous genitalia with perineoscrotal hypospadias in 46, XY individuals: Long-term medical, surgical, and psychosexual outcome. Pediatrics. 2002;110 doi: 10.1542/peds.110.3.e31. e31-e31. [DOI] [PubMed] [Google Scholar]
- 25.Cohen-Kettenis P.T. Gender change in 46, XY persons with 5 alpha-reductase-2 deficiency and 17 beta-hydroxysteroid dehydrogenase-3 deficiency. Arch Sex Behav. 2005;34:399–410. doi: 10.1007/s10508-005-4339-4. [DOI] [PubMed] [Google Scholar]
- 26.Cheikhelard A., Morel Y., Thibaud E., Lortat-Jacob S., Jaubert F., Polak M. Long-term followup and comparison between genotype and phenotype in 29 cases of complete androgen insensitivity syndrome. J Urol. 2008;180:1496–1501. doi: 10.1016/j.juro.2008.06.045. [DOI] [PubMed] [Google Scholar]
- 27.Marcantonio S.M., Fechner P.Y., Migeon C.J., Perlman E.J., Berkovitz G.D. Embryonic testicular regression sequence. A part of the clinical spectrum of 46, XY gonadal dysgenesis. Am J Med Genet. 1994;49:1–5. doi: 10.1002/ajmg.1320490102. [DOI] [PubMed] [Google Scholar]
- 28.Clarnette T.D., Sugita Y., Hutson J.M. Genital anomalies in human and animal models reveal the mechanisms and hormones governing testicular descent. Br J Urol. 1997;79:99–112. doi: 10.1046/j.1464-410x.1997.25622.x. [DOI] [PubMed] [Google Scholar]
- 29.Shinmura Y., Yokoi T., Tsutsui Y. A case of clear cell adenocarcinoma of the Mullerian duct in persistent Mullerian duct syndrome: the first reported case. Am J Surg Pathol. 2002;26:1231–1234. doi: 10.1097/00000478-200209000-00014. [DOI] [PubMed] [Google Scholar]