

Abstract
In this study, chrysophanol, isolated from a marine fungus, was examined for its protective effects against inflammatory responses and oxidative stress in BV2 microglia. Chrysophanol was studied to assess its capabilities of protecting against lipopolysaccharide (LPS)-induced inflammatory responses in BV2 cells. It was found that chrysophanol reduced the level of nitric oxide (NO) and prostaglandin-E2 (PGE2) production by diminishing reducing the expression of inducible NO synthase (iNOS) and cyclooxygenase-2 (COX-2). Assessment of the inhibitory activities of chrysophanol on the generation of pro-inflammatory cytokines was also performed. Furthermore, Chrysophanol treatment significantly reduced intracellular reactive oxygen species (ROS)-mediated cell damage and inhibited DNA oxidation in BV2 cells. Moreover, antioxidative mechanisms by of chrysophanol were evaluated investigated by measuring the expression levels of antioxidative enzymes such superoxide dismutase (SOD) and glutathione (GSH). Therefore, results suggested that chrysophanol has potential antioxidant and anti-inflammatory activities in microglia and further might be a useful therapeutic agent for the treatment of neurodegenerative diseases.
Keywords: Chrysophanol, neuroprotection, microglial activation, BV2
Introduction
Microglia is resident immune cells in the central nervous system (CNS). They enter the CNS from the blood circulation early in an organism’s development and play a role in immune surveillance [1]. It has been demonstrated that activation of microglia and the resulting production of pro-inflammatory and neurotoxic factors are sufficient to induce neurodegeneration in a rat model [2]. Moreover, activation of microglia and excessive amounts of pro-inflammatory mediators released by microglia have been observed during the pathogenesis of Parkinson’s disease (PD), Alzheimer’s disease (AD), multiple sclerosis (MS), and AIDS dementia complex, as well as in post-neuronal death in cerebral stroke and traumatic brain injury [2,3]. Thus, understanding the mechanism that regulates microglia activation may have important therapeutic potential for the treatment of neurodegenerative diseases. BV2 immortalized murine microglia have been shown to exhibit phenotypic and functional properties comparable to those of primary microglia, and therefore, this cell line represents a suitable model for in vitro studies of activated microglial cells [4].
Furthermore, compared to other parts of the body, the CNS is more sensitive to oxidative stress due to its high oxygen consumption and lipid content. Increased oxidative stress in the CNS will further damage macromolecules such as lipids, DNA and proteins [5]. Antioxidants may have a positive effect because they can protect against CNS damage from uncontrolled generation of reactive oxygen species (ROS), which attack macromolecules leading to neurodegenerative diseases. Therefore, it is important to tightly regulate microglial activation and oxidative damage mediated by free radicals. For this reason, a neuroprotective agent with anti-inflammatory and antioxidant properties would be more effective than any neuroprotective agent with only anti-inflammatory or antioxidant properties.
It has been reported that marine fungi are enormous sources of natural products with unique chemical structures and interesting bioactivity such as antimicrobial, antifungal, antioxidant, anti-cancer, antiinflammatory, etc [2,3]. In the present study, we investigated the effects of chrysophanol, a bioactive constituent derived from Microsporum sp., on the production of pro-inflammatory mediators induced by lipopolysaccharides (LPS) and free radical-mediated oxidative damage in microglia.
Materials and methods
Materials
Cell culture medium [Dulbecco’s Modified Eagle’s Medium (DMEM)], penicillin/streptomycin, Fetal Bovine Serum (FBS) and the other materials required for culturing cells were purchased from Gibco BRL, Life Technologies (Grand Island, US). LPS from Escherichia coli 026:B6, Griess reagent, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St. Louis, US). TRIzol® RNA extraction was provided by Invitrogen Ltd. (Paisley, UK). RT-PCR reagents were purchased from Promega (Madison, US). Specific antibodies used for western blot analysis were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, US) and Amersham Pharmacia Biosciences (Piscataway, US); other chemical and reagents used in this study were of analytical grade.
Isolation, identification and culture of the marine-derived fungus Microsporum sp
The fungal strain (MFS-YL) was isolated from the surface of the marine red alga collected in the Guryongpo, Nam-gu, Pohang, Korea in 2009. The strain was cultured in 0.5% yeast extract, 0.5% peptone, 1% glucose, 60% seawater (YPG) and stored in the 10% glycerol YPG medium at 75°C. The further culture for investigation was completed on YPG medium from 20 ml to large scale [1]. The fungus was cultured (20 L) at 25°C pH 7.6 in YPG for 30 days. Based on cellular fatty acid composition, the strain was identified as Microsporum sp. (Korean Culture Center of Microorganisms, Seoul, Korea, similarity index 0.62).
Isolation and purification of secondary metabolites from the marine-derived fungus Microsporum sp
The culture broth and mycelium were separated and the resulted broth and mycelium were extracted with ethyl acetate to provide broth extract and the mycelium extract, respectively. Both extracts were combined and the total extract was fractionated by silica gel flash chromatography (n-hexane: EtOAc and CHCl3: MeOH) to generate thirteen fractions containing the active compounds, respectively. Final purification of each fraction by ODS column chromatography (H2O: MeOH), followed by HPLC (YMC ODS-A, MeOH).
Cell culture and viability assay
The murine BV2 cells were a kind gift of Prof. Il Whan Choi (Inje University, Korea). BV2 cells were grown in T-75 tissue culture cell flasks SPL Life Science (Seoul, Republic of Korea) at 5% CO2 and 37°C humidified atmosphere using appropriate media supplemented with 5% FBS, 2 mM glutamine and 100 μg/ml penicillin-streptomycin. DMEM were used as the culture mediums. For viability assay, cells were seeded into 96-well plates at a density of 2 × 104 cells/well and incubated with serum free medium for 24 h, followed by treatment with different concentration of chrysophanol. After incubation for 24 h, 100 µl of MTT (0.5 mg/ml final concentration) was added and incubation was continued for another 4 h. MTT is used as an indicator of cell viability through its mitochondrial reduction to formazan. Mitochondrial succinate dehydrogenase in live cells converts MTT into visible formazan crystals during incubation. The formazan crystals were then solubilized in DMSO and the absorbance was measured at 540 nm by using enzyme-linked immunosorbent assay (ELISA) microplate reader, Tecan GmbH (Grödig, Austria). Relative cell viability was calculated compared with the absorbance of the untreated control group.
Determination of nitric oxide (NO) production
NO levels in the culture supernatants were measured by the Griess reaction [6]. In brief, BV2 cells were pre incubated overnight in 24-well plates using DMEM without phenol red at a density 2 × 105 cells/ml, followed by the treatment with chrysophanol. After treated with chrysophanol for 2 h, the NO production was stimulated by adding LPS (1 µg/ml final concentration) and incubated for further 24 h. Then 50 µl of culture supernatants from each samples was mixed with the same volume of the Griess reagent [1% sulfanilamide/0.1% N-1-naphthyl]-ethylenediamine dihydrochloride/2.5% phosphoric acid] following the incubation for 15 min. Absorbance values were read at 540 nm using ELISA microplate reader. The values obtained were compared with those of standards concentrations of sodium nitrite dissolved in DMEM, and the concentration of nitrite in conditioned media of samples treated cells were calculated.
Determination of prostaglandin-E2 (PGE2) production
Assessment of PGE2 synthesis was performed by enzyme immunoassay without prior extraction or purification using commercially available PGE2 enzyme immunometric assay kit, R&D Systems Inc. (Minneapolis, USA). BV2 cells were treated with different concentration of chrysophanol for 2 h and stimulated with LPS (1 µg/ml) for 24 h. The conditioned media was collected to perform PGE2 enzyme immuno-metric assay (PGE2-EIA) according to the instructions of the manufacturer. The concentration of PGE2 was calculated according to the equation obtained from the standard curve plot using PGE2 standard solution in the EIA kit.
Measurement of pro-inflammatory cytokines production
The inhibitory effects of chrysophanol on the production of pro-inflammatory cytokines were determined by an ELISA. Cells were treated with different concentration of chrysophanol for 2 h and stimulated with LPS (1 µg/ml) for 24 h. The conditioned medium were collected and analyzed as per the manufacturer’s instruction with Quantikine mouse tumor necrosis factor-α (TNF-α), (interleukin-6) IL-6, and interleukin-1β (IL-1β) Immunoassay (R&D Systems Inc, Minneapolis, USA). The concentration of TNF-α, IL-6 and IL-1β was calculated according to the equation obtained from the standard curve plot using TNF-α, IL-6 and IL-1β standard solution in the ELISA kit.
Measurement of intracellular ROS by 2’, 7’-dichlorofluorescein diacetate (DCFH-DA) assay
Intracellular formation of ROS was assessed according to a method described previously by employing oxidation sensitive dye DCFH-DA as the substrate. BV2 cells were grown in black microtiter 96-well plates and were labeled with 20 µM DCFH-DA in Hank’s balanced salt solution (HBSS) and kept for 20 min in the dark. The non-fluorescent DCFH-DA dye which freely penetrates into cells was then hydrolyzed by intracellular esterase to 2’, 7’-dichlorodihydrofluorescein (DCFH), and trapped inside the cells. Cells were then treated with different concentrations of chrysophanol and incubated for 1 h. After washing cells for three times with PBS, 500 µM H2O2 (in HBSS) were added. The formation of fluorescent dichlorofluorescein (DCF) due to oxidation of DCFH in the presence of various ROS was read after every 30 min at the excitation wavelength of 485 nm and the emission wavelength of 528 nm (GENios microplate reader, Tecan Austria GmbH, Grodig/Salzburg, Austria). Dose-dependent and time-dependent effects of treatments were plotted and compared with fluorescence intensity of the control group in which samples were not treated.
Genomic DNA isolation
Genomic high molecular weight DNA was extracted from BV2 cells using standard phenol/proteinase K procedure with slight modifications [7]. Briefly, cells cultured in 10 cm culture dishes were washed twice with PBS and scraped into 1 ml of PBS containing 10 mM EDTA. After centrifugation cells were dissolved in RNase (0.03 mg/ml), NaOAC (0.175 M), proteinase K (0.25 mg/ml) and SDS (0.6%). The mixture was then incubated for 30 min at 37°C and 1 h at 55°C. Following incubation, phenol/chloroform/isoamyl alcohol was added at a 1:1 (v/v) ratio and the mixture was centrifuged at 6000 g for 5 min at 4°C. Following centrifugation, the supernatant was mixed with 100% cold ethanol at a 1:1.5 (v/v) ratio and kept for 15 min at -20°C. After centrifugation at 12,000 g for 5 min, the pellet was dissolved in 10 mM TE buffer (pH 7.5) and the purity of DNA was determined using spectrophotometer at 260/280 nm. Further, the quality of isolated DNA was evaluated using 1% agarose gel electrophoresis in 0.04 M Tris-acetate, 0.001 M EDTA buffer (pH 7.5).
Determination of radical-mediated DNA damage
Hydrogen peroxide mediated DNA oxidation was determined as described previously [8]. Briefly, 40 µl of DNA reaction mixture were prepared by adding pre-determined concentrations of test sample (or same volume of distilled water as control), 100 µM final concentration of FeSO4, 0.1 mM final concentration of H2O2 and 5 µl of isolated genomic DNA in the same order. Then the mixture was incubated at room temperature for 10 min and the reaction was terminated by adding 10 mM final concentration of EDTA. An Aliquot (20 µl) of the reaction mixture containing about 1 µg of DNA was electrophoresed on a 1% agarose gel for 40 min at 100 V. Gels were then stained with 1 mg/ml ethidium bromide and visualized under UV light using AlphaEase gel image analysis software (Alpha Innotech, St. San Leandro, CA, USA).
RNA extraction and reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from BV2 cells treated with LPS or H2O2 in the presence or absence of chrysophanol using TRIzol® reagent (Invitrogen, UK), as reported in the manufacturer’s manual. Equal amount of RNA (2 µg) was used for each cDNA synthesis reaction using a reverse transcription system (Promega, Madison, WI, USA). Single stranded cDNA was amplified by PCR with specific primers. The following PCR conditions were applied for all amplifications: 27 cycles of denaturation at 94°C for 30 s, annealing at 57°C for 30 s, and extended at 72°C for 30 s. The resulting cDNA were separated by electrophoresis on 1% agarose gel for 15 minutes at 100 V, followed visualization under UV light after ethidium bromide staining. Band intensities were quantifies with Multi gauge Software (Fujifilm Life Science, Tokyo, Japan) and band of specific genes were normalized using Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as references.
Western blot analysis
Standard procedures were used for western blotting, where BV2 cells treated with different concentration of chrysophanol followed by LPS or H2O2 treatment for 24 h were lysed in RIPA buffer (Invitrogen, UK). Cell debris was removed by centrifugation followed by quick freezing of the supernatants. The protein concentrations were determined according to the Bicinchoninic acid (BCA) assay. Cell lysates were separated by SDS-PAGE gel electrophoresis and electro-blotted onto a nitrocellulose membrane. The membrane were blocked with 5% bovine serum albumin (BSA) and the incubated with different antibodies which were used to detect respective proteins using a chemiluminescent ECL assay kit, according to the manufacturer instructions. Western blots were visualized using a LAS3000® Luminescent image analyzer (Fujifilm Life Science, Tokyo, Japan) and protein expression was quantified by Multi gauge V3.0 software (Fujifilm Life Science, Tokyo, Japan).
Statistical analysis
The data were presented as means ± SD (n = 3). Differences between the means of the individual groups were assessed by one-way ANOVA with Duncan’s multiple range tests. Differences were considered significant at P<0.05. The statistical software package, SPSS v.16 (SPSS Inc., Chicago, Illinois, USA) was used for the analysis.
Results
Effect of chrysophanol on nitric oxide (NO) and prostaglandin-E2 (PGE2) production in lipopolysaccharide (LPS)-stimulated BV2 cells
Initially, we determined the potential anti-inflammatory properties of chrysophanol on LPS-induced NO and PGE2 production in BV2 cells. The cells were treated with chrysophanol (0, 5, 10 and 50 µM) for 2 h followed by 24-h treatment with LPS (1 µg/ml). NO and PGE2 were measured in the culture supernatants using the Griess reaction assay and enzyme-linked immunsorbent assay (ELISA), respectively. Stimulation with LPS markedly induced the production of NO and PGE2 compared to not stimulating with LPS (Figure 1A, 1B). NO levels after treatment with 5, 10 and 50 µM of chrysophanol were 18.07 ± 2.14 µM, 14.42 ± 1.06 µM and 8.21 ± 0.64 µM, respectively. Furthermore, the ELISA result clearly demonstrated that chrysophanol significantly (P<0.05) suppressed LPS-induced PGE2 production even at the lowest concentration tested (5 µM).
Figure 1.
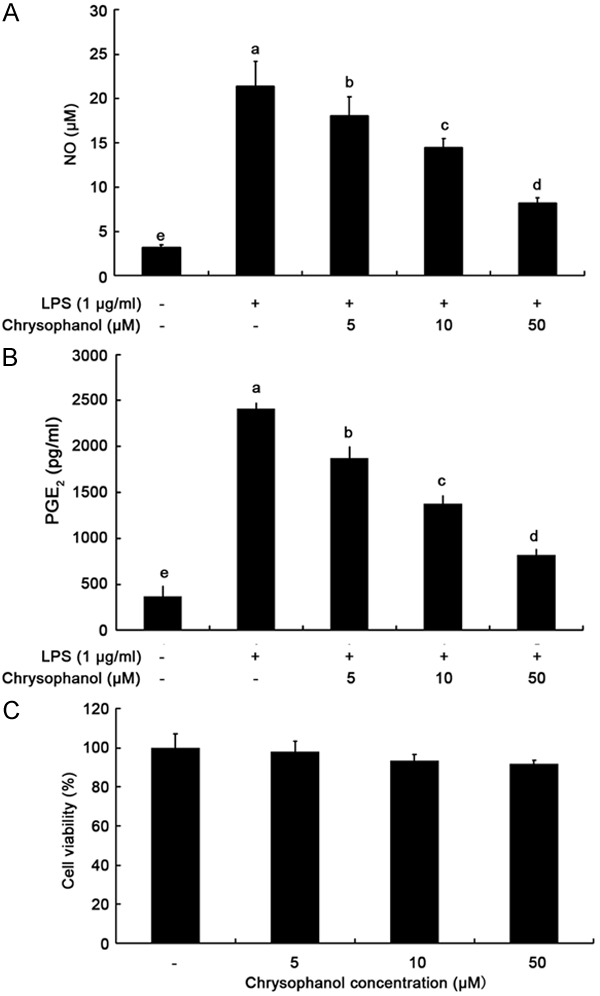
Effects of chrysophanol on LPS-induced NO and PGE2 production and cell viability in BV2 microglia. Culture supernatant was isolated and analyzed; the NO amounts were determined using Griess reagent (A) and the PGE2 amounts were determined using ELISA (B). Cell viability was assessed by MTT assay (C). Stimulation with LPS markedly induced the production of NO and PGE2 compared to not stimulating with LPS (A, B). Chrysophanol exerted no significant toxic effect on BV2 cells under tested concentrations after 24-h treatment (C). Values correspond to the mean ± SD from three independent experiments. a-eIndicate P<0.05 by Duncan’s multiple range test.
To exclude the possibility that the inhibition of NO and PGE2 production was due to cytotoxicity caused by chrysophanol, we tested toxicity of chrysophanol at various concentrations using MTT assays. As presented in Figure 1C, chrysophanol exerted no significant toxic effect on BV2 cells under tested concentrations after 24-h treatment (P>0.05). Thus, the inhibitory effect of chrysophanol on LPS-stimulated NO and PGE2 production was not due to cytotoxic action of chrysophanol.
Effects of chrysophanol on inducible NO synthase (iNOS) and cyclooxygenase-2 (COX-2) expression in LPS-induced BV2 cells
Next, to investigate the molecular mechanism involved in the inhibition of NO and PGE2 production, the effects of chrysophanol on iNOS and COX-2 mRNA and protein expression were analyzed by polymerase chain reaction (PCR) and western blot analysis, respectively. Figure 2 shows that chrysophanol elicited inhibition of LPS-induced iNOS and COX-2 mRNA and protein expression. The result indicated that chrysophanol suppressed LPS-induced NO and PGE2 synthesis through down-regulation of iNOS and COX-2 mRNA and protein expression.
Figure 2.
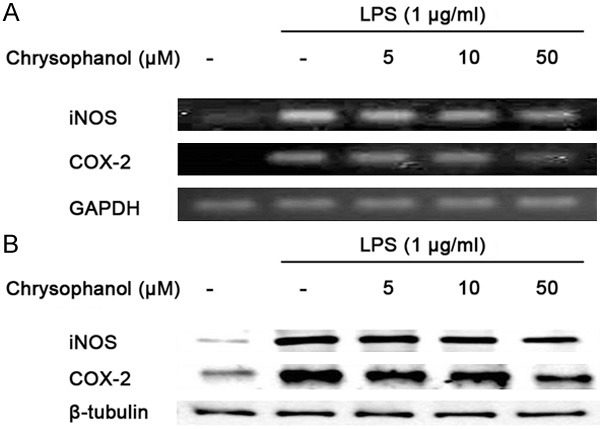
Effects chrysophanol on LPS-induced iNOS, COX-2 mRNA and protein expression in BV2 microglia. The expression of iNOS and COX-2 mRNA was measured by RT-PCR (A); the expression of iNOS and COX-2 protein was measured by western blot analysis (B). Chrysophanol elicited inhibition of LPS-induced iNOS and COX-2 mRNA and protein expression. GAPDH and β-tubulin were used as internal controls for PCR and western blot analysis, respectively.
Effects of chrysophanol on LPS-induced tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and IL-1β
In order to determine the effect of chrysophanol on the production of pro-inflammatory cytokines such as TNF-α, IL-6 and IL-1β, BV2 cells were incubated with chrysophanol (0, 5, 10 and 50 µM) in the presence or absence of LPS (1 µg/ml) for 24 h, and the cytokine levels were measured by ELISA. As presented in Table 1, TNF-α, IL-6 and IL-1β levels were decreased in a concentration-dependent manner by treatment with chrysophanol. Furthermore, Chrysophanol significantly attenuated TNF-α, IL-6, and IL-1β expression in a concentration-dependent manner at the mRNA level (Figure 3A).
Table 1.
Effects of chrysophanol on the pro-inflammatory cytokines production in LPS-stimulated BV2 microglia
| Pro-inflammatory cytokines concentration (pg/ml) | |||
|---|---|---|---|
|
|
|||
| TNF-α | IL-6 | IL-1β | |
| Blank | 52.56 ± 6.45e | 128.83 ± 21.92e | 105.38 ± 10.96d |
| Control | 1257.44 ± 86a | 1719.17 ± 59.16a | 482.13 ± 21.74a |
| Chrysophanol 5 µM | 835.12 ± 82.2b | 1190.17 ± 94.05b | 435 ± 15.91b |
| Chrysophanol 10 µM | 594.5 ± 24.22c | 891.67 ± 79.2c | 401 ± 10.96b |
| Chrysophanol 50 µM | 378.6 ± 27.4d | 458 ± 75.9d | 350.81 ± 26.6c |
Footnotes: Values correspond to means ± SD from three independent experiments.
Symbolize that the different letters in each sample are significantly different (P<0.05) by Duncan’s multiple range test;
Symbolize that the different letters in each sample are significantly different (P<0.05) by Duncan’s multiple range test;
Symbolize that the different letters in each sample are significantly different (P<0.05) by Duncan’s multiple range test;
Symbolize that the different letters in each sample are significantly different (P<0.05) by Duncan’s multiple range test;
Symbolize that the different letters in each sample are significantly different (P<0.05) by Duncan’s multiple range test.
Figure 3.
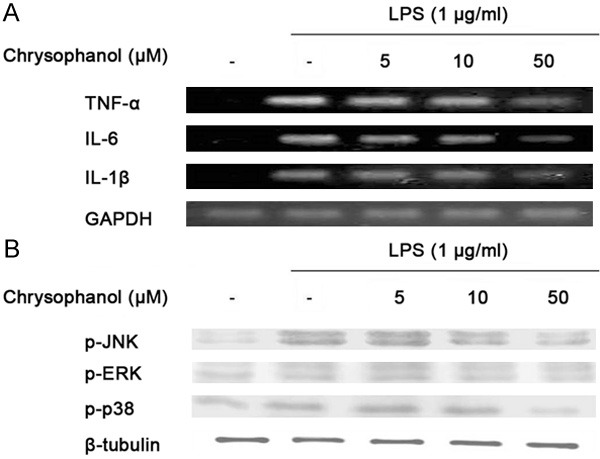
Effects chrysophanol on LPS-induced pro-inflammatory cytokines production in BV2 microglia. The expressions of TNF-α, IL-6 and IL-1β mRNA were measured by RT-PCR. GAPDH was used as an internal control for PCR (A). Chrysophanol significantly attenuated TNF-α, IL-6, and IL-1β expression in a concentration-dependent manner. The expressions of p-ERK, p-JNK and p-38 MAPK protein were measured by western blot analysis (B). At a concentration of 50 μg/ml, chrysophanol markedly inhibited ERK, p38 and JNK activation. GAPDH and β-tubulin were used as internal controls for PCR and western blot analysis, respectively.
Chrysophanol inhibits LPS-induced phosphorylation of mitogen-activated protein kinases (MAPK) in BV2 cells
An experiment was designed to elucidate the signaling cascades that induce the expression of pro-inflammatory genes in BV2 cells in response to LPS stimulation. The MAPK pathway plays a key role in the regulation of cell growth, differentiation and cellular responses to cytokines and stresses. Therefore, to investigate whether down-regulation of pro-inflammatory mediators by chrysophanol was mediated by the MAPK pathway, we examined the effect of chrysophanol on LPS-induced phosphorylation of extracellular signal-regulated kinases (ERK), p38 and JNK using western blot analysis (Figure 3B). We found that these proteins were phosphorylated following stimulation with LPS. Next, the effects of chrysophanol on LPS-induced activation of ERK, p38 and JNK were examined. At a concentration of 50 µg/ml, chrysophanol markedly inhibited ERK, p38 and JNK activation. These results suggest that phosphorylation of ERK, p38 and JNK are involved in the inhibitory effect of chrysophanol on LPS-induced pro-inflammatory mediators in BV2 cells.
Chrysophanol inhibits DNA oxidation in BV2 cells
The highly reactive hydroxyl radicals (*OH) react with all components of the DNA molecule, including bases (purine and pyrimidine) and also deoxyribose units, and damage DNA. This damage increases incrementally with the free radical attacks on cellular DNA, which is a concern with regard to cancer, mutagenesis and aging. In this study, oxidation of genomic DNA isolated from BV2 cells was determined by the combined effect of 2 mM H2O2 and 200 μM Fe (II) on DNA integrity. The protective effect of chrysophanol against DNA oxidative damage was assessed by DNA electrophoresis. After 10 min of reaction, almost all DNA was degraded in the control group treated with H2O2-Fe (II) alone (Figure 4A). Chrysophanol showed a dose-dependent inhibitory effect on radical-mediated oxidative DNA damage. At 50 μg/ml of chrysophanol, DNA damage was inhibited more than 90% as determined based on the intensity of DNA bands.
Figure 4.
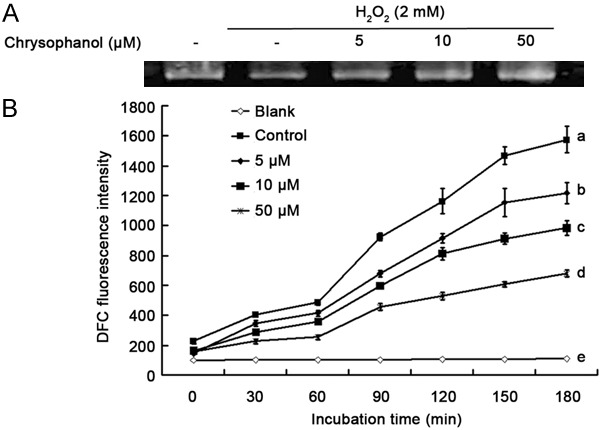
DNA oxidative protection by chrysophanol in BV2 microglia. Chrysophanol showed a dose-dependent inhibitory effect on radical-mediated oxidative DNA damage (A). However, pre-treatment with chrysophanol reduced DCF fluorescence in a dose- and time-dependent manner. Values correspond to means ± SD from three independent experiments. a-eSymbolize that different letters in each sample are significantly different (P<0.05) by Duncan’s multiple range test (B).
Cellular radical scavenging effect of chrysophanol
The cellular direct free radical scavenging effect of chrysophanol was employed on BV2 cells since these cells are able to produce high amounts of ROS following stimulation. To accomplish this, the cells were labeled with DCFH-DA as described in the Materials and Methods section. During labeling, non-fluorescent DCFH-DA dye that easily diffuses through cell membrane is hydrolyzed by esterase to become DCFH, and this compound is trapped inside the cells. Fluorescence is emitted by DCF following H2O2-mediated oxidation of DCFH. The monitoring of DCF fluorescence intensities was performed every 30 min for the 3 h duration that radical-mediated oxidation increased with the incubation time. However, pre-treatment with chrysophanol reduced DCF fluorescence in a dose- and time-dependent manner (Figure 4B). Therefore, we could confirm that chrysophanol scavenged free radicals and inhibited radical-mediated oxidation in BV2 cells.
Effect on H2O2-induced antioxidant enzyme expression
In order to determine whether chrysophanol affects the gene and protein expression levels of antioxidative enzymes, BV2 cells were treated with chrysophanol and 50 μM H2O2. The mRNA and protein expression levels of antioxidative enzymes were determined by RT-PCR (Figure 5A) and western blot analysis (Figure 5B), respectively. Superoxide dismutase (SOD) is one of the major antioxidant enzymes which convert dismutase superoxide radicals into hydrogen peroxide and molecular oxygen. In that reaction, the formation of intracellular H2O2 might decrease SOD expression to maintain the redox balance. However, treatment with chrysophanol up-regulated SOD expression dose-dependently. Furthermore, GSH plays an important role in regulating intracellular redox status. The increase in GSH level protects cells against cell death either by removing free radicals or by conjugating with toxicants. The mRNA and protein expression levels of GSH were up-regulated in the presence of chrysophanol. The results showed that chrysophanol can increase the expression levels of antioxidant enzymes in BV2 cells through scavenging free radicals.
Figure 5.
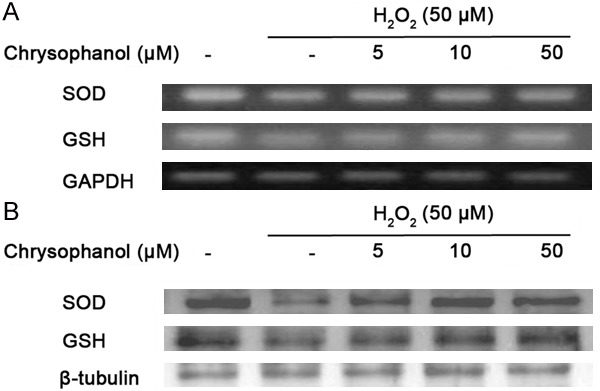
Effect of chrysophanol on H2O2-induced antioxidant enzyme in BV2 microglia. The expressions of SOD and GSH mRNA were measured by RT-PCR (A); the expressions of SOD and GSH protein were measured by western blot analysis (B). Treatment with chrysophanol up-regulated SOD and GSH expression dose-dependently. GAPDH and β-tubulin was used as an internal control for PCR and western blot analysis, respectively.
Discussion
Chrysophanol (1,8-dihydroxy-3-methylanthracenedione) is an anthraquinone compound with active functional groups such as hydroxyl and methyl groups attached to it. Chrysophanol has been reported to possess several biological activities including antimicrobial, anti-inflammatory, and hepatoprotective [6]. The present study was undertaken to examine the pharmacological and biological effects of chrysophanol on the inflammatory and oxidative responses of murine BV2 cells. BV2 cells treated with chrysophanol (5 to 50 μM) behaved indistinguishably from controls in terms of cell viability. Therefore, chrysophanol is cyto-compatible and did not adversely interfere with the other assay methods used in this study.
Microglia is immune cells responsible for homeostatic regulation and defense against injuries [7]. However, excessive activation of microglia under pathological conditions can destroy surrounding healthy neurons as well as damage neurons by releasing sizeable quantities of pro-inflammatory mediators. NO, PGE2, and pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β are known to be important mediators in the process of neuro-inflammation [8]. These pro-inflammatory mediators are thought to be responsible for some of the harmful effects of brain injuries and diseases, including ischemia, AD and neural death [9,10]. The results of this study indicate that chrysophanol effectively inhibits LPS-induced production of NO, PGE2, TNF-α, IL-6, and IL-1β through a blockade of the MAPK pathway in BV2 cells. Therefore, chrysophanol is regarded as a protective agent against neurodegeneration via suppression of the activity of pro-inflammatory mediators, causing a reduction in the level of inflammation in BV2 cells.
In addition, there is accumulating evidence suggesting a key role played by oxidative stress in the pathophysiology of neurodegenerative diseases. Because of its high metabolic rate and relatively reduced capacity for cellular regeneration compared with other organs, the brain is believed to be particularly susceptible to the damaging effects of ROS [11,12]. Therefore, it is important to tightly regulate microglial activation and oxidative damage. ROS are commonly produced during the inflammatory process, and the overproduction of ROS can lead to oxidative damage in the brain in PD, as shown by increased lipid peroxidation and DNA damage in the substantia nigra [13,14]. In this study, chrysophanol was assessed in a BV2 cell system to evaluate the intracellular radical scavenging effect. Intracellular ROS formation was measured using an oxidation sensitive dye, DCFH-DA. Within the cell, the acetate groups of DCFH-DA are enzymatically hydrolyzed by intracellular esterase activity and trapped within the cell. The amount of intracellular ROS in BV2 cells was significantly decreased in a dose-dependent manner by chrysophanol. The ROS scavenging effect of chrysophanol was investigated by studying the enzymes related to ROS production such as SOD and GSH. It has been hypothesized that alterations in antioxidant systems, including SOD, are implicated in neurodegenerative diseases. Mutation of SOD has been implicated in some cases of familial ALS diseases [15]. SOD is a metalloenzyme that removes oxygen radicals and protects against oxidative injury. SOD yielding H2O2 and OO2, and, subsequent removal of H2O2 is facilitated by catalase and GSH [16]. SOD activity can protect against oxidative stress produced by oxygen radicals and may represent a first line of defense against oxidative damage mediated by oxygen radicals. SOD is present in the brain. In addition, GSH has been reported to block mitochondrial apoptosis in neurons [17]. A novel observation suggested that Bcl-2 binds directly to GSH in vitro. Defining the molecular mechanisms by which Bcl-2 influences the mitochondrial GSH pool may reveal that GSH can be targeted to prevent neuronal apoptosis and subsequent neurodegenerative disease. Collectively, the results of the present study demonstrated that chrysophanol treatment positively regulated the expression level of SOD and GSH, suggesting that chrysophanol reduces oxidative stress in BV2 cells through up-regulation of the expression level of anti-oxidant enzymes.
Marine microorganisms have been known as an important source of naturally bioactive secondary metabolites including phenols and polyphenols with unique linkages (ether and/or phenyl) [18,19]. In this study, chrysophanol isolated from Microsporum sp. was analyzed for its capacity to suppress neuro-inflammatory and oxidative responses. The active focus of the chrysophanol components is attributable to the number and arrangement of hydroxyl and methyl groups in its structure. Interestingly, scientific evidence confirms that the functional groups have the ability to scavenge neurotoxic radical species by donating electrons. The combination of structural features of chrysophanol generates the basis of its anti-inflammatory and antioxidative activity.
In summary, our results showed that chrysophanol treatment decreased the release of pro-inflammatory mediators following LPS stimulation in BV2 cells. Chrysophanol significantly attenuated the release of iNOS, PGE2, TNF-α, IL-6, and IL-1β in a concentration-dependent manner. The inhibitory effect of chrysophanol was mediated by inhibition of p38, ERK and JNK activation in activated microglia. These data indicate that chrysophanol targets p38, ERK and JNK in cells and inhibits the expression of pro-inflammatory mediators. Moreover, we also found that chrysophanol positively regulated antioxidant enzymes in BV2 cells. Given the fact that microglia contributes to the pathogenesis of neurodegenerative human brain diseases, chrysophanol may be considered as a potential therapeutic agent for various neurodegenerative diseases.
Acknowledgements
This study was supported by grants from National Science Foundation of China (No. 81202550), China Postdoctoral Science Foundation (2013M541548) and STCSM (12ZR1423400).
Disclosure of conflict of interest
None.
References
- 1.Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- 2.Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann N Y Acad Sci. 2002;962:318–331. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- 3.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–365. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bocchini V, Mazzolla R, Barluzzi R, Blasi E, Sick P, Kettenmann H. An immortalized cell line expresses properties of activated microglial cells. J Neurosci Res. 1992;31:616–621. doi: 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- 5.Akyol O, Herken H, Uz E, Fadillioglu E, Unal S, Sogut S, Ozyurt H, Savas HA. The indices of endogenous oxidative and antioxidative processes in plasma from schizophrenic patients. The possible role of oxidant/antioxidant imbalance. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:995–1005. doi: 10.1016/s0278-5846(02)00220-8. [DOI] [PubMed] [Google Scholar]
- 6.Karlina GS, Ninibe VA, Petra L, Luis M P. Chrysophanol, an Antimicrobial Anthraquinone from the Root Extract of Colubrina greggii . J Mex Chem Soc. 2006;50:76–78. [Google Scholar]
- 7.Pangestuti R, Bak SS, Kim SK. Attenuation of pro-inflammatory mediators in LPS-stimulated BV2 microglia by chitooligosaccharides via the MAPK signaling pathway. Int J Biol Macromol. 2011;49:599–606. doi: 10.1016/j.ijbiomac.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Pangestuti R, Kim SK. Neuroprotective properties of chitosan and its derivatives. Mar Drugs. 2010;8:2117–2128. doi: 10.3390/md8072117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung WK, Heo SJ, Jeon YJ, Lee CM, Park YM, Byun HG, Choi YH, Park SG, Choi IW. Inhibitory effects and molecular mechanism of dieckol isolated from marine brown alga on COX-2 and iNOS in microglial cells. J Agric Food Chem. 2009;57:4439–4446. doi: 10.1021/jf9003913. [DOI] [PubMed] [Google Scholar]
- 10.Seifert HA, Collier LA, Chapman CB, Benkovic SA, Willing AE, Pennypacker KR. Pro-inflammatory interferon gamma signaling is directly associated with stroke induced neurodegeneration. J Neuroimmune Pharmacol. 2014;9:679–689. doi: 10.1007/s11481-014-9560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer’s disease brain: new insights from redox proteomics. Eur J Pharmacol. 2006;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 12.Lavrentiadou SN, Tsantarliotou MP, Zervos IA, Nikolaidis E, Georgiadis MP, Taitzoglou IA. CCl4 induces tissue-type plasminogen activator in rat brain; protective effects of oregano, rosemary or vitamin E. Food Chem Toxicol. 2013;61:196–202. doi: 10.1016/j.fct.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 13.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zugno AI, Chipindo HL, Volpato AM, Budni J, Steckert AV, de Oliveira MB, Heylmann AS, da Rosa Silveira F, Mastella GA, Maravai SG, Wessler PG, Binatti AR, Panizzutti B, Schuck PF, Quevedo J, Gama CS. Omega-3 prevents behavior response and brain oxidative damage in the ketamine model of schizophrenia. Neuroscience. 2014;259:223–231. doi: 10.1016/j.neuroscience.2013.11.049. [DOI] [PubMed] [Google Scholar]
- 15.Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci U S A. 1995;92:954–958. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JA, Kong CS, Kim SK. Effect of Sargassum thunbergii on ROS mediated oxidative damage and identification of polyunsaturated fatty acid components. Food Chem Toxicol. 2010;48:1243–1249. doi: 10.1016/j.fct.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 17.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 18.Manivasagan P, Kang KH, Sivakumar K, Li-Chan EC, Oh HM, Kim SK. Marine actinobacteria: an important source of bioactive natural products. Environ Toxicol Pharmacol. 2014;38:172–188. doi: 10.1016/j.etap.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 19.Zhang C, Kim SK. Research and application of marine microbial enzymes: status and prospects. Mar Drugs. 2010;8:1920–1934. doi: 10.3390/md8061920. [DOI] [PMC free article] [PubMed] [Google Scholar]