

Abstract
Recent evidence has linked novel mutations in the arginine vasopressin receptor 2 gene (AVPR2) and aquaporin-2 gene (AQP2) present in Southeast Asian populations to congenital nephrogenic diabetes insipidus (NDI). To investigate mutations in 2 distinct Chinese pedigrees with NDI patients, clinical data, laboratory findings, and genomic DNA sequences from peripheral blood leukocytes were analyzed in two 5.5- and 8-year-old boys (proband 1 and 2, respectively) and their first-degree relatives. Water intake, urinary volume, body weight and medication use were recorded. Mutations in coding regions and intron-exon borders of both AQP2 and AVPR2 gene were sequenced. Three mutations in AQP2 were detected, including previously reported heterozygous frameshift mutation (c.127_128delCA, p.Gln43Aspfs ×63) inherited from the mother, a novel frameshift mutation (c.501_502insC, p.Val168Argfs ×30, inherited from the father) in proband 1 and a novel missense mutation (c. 643G>A, p. G215S), inherited from both parents in proband 2. In family 2 both parents and one sister were heterozygous carriers of the novel missense mutation. Neither pedigree exhibited mutation in the AVPR2 gene. The patient with truncated AQP2 may present with much more severe NDI manifestations. Identification of these novel AQP2 gene mutations expands the AQP2 genotypic spectrum and may contribute to etiological diagnosis and genetic counseling.
Keywords: Congenital, nephrogenic diabetes insipidus (NDI), aquaporin-2, mutation, autosomal recessive
Introduction
Congenital nephrogenic diabetes insipidus (NDI) represents a group of rare genetic disorders characterized by an inability of the kidney to concentrate urine in response to normal or elevated arginine vasopressin (AVP) levels [1]. Affected individuals generally present with characteristic symptoms within the first year of life including polyuria, polydipsia, vomiting, constipation, fever and hypernatremic dehydration that may also lead to severe and chronic symptoms [2,3]. While patients that receive early diagnosis and treatment can go on to lead long and healthy lives, patients that do not receive proper treatment early in life can develop chronic cognitive dysfunction and failure to thrive due to irreversible organ and tissue damage [4]. Thus, better understanding of the genetic basis of NDI may facilitate early and accurate diagnosis and optimal management for patients with congenital NDI.
Recent studies have consistently linked congenital NDI with mutations in the arginine vasopressin receptor 2 gene (AVRP2) and the aquaporin-2 gene (AQP2); however, several different mutations have been reported with variant inheritance modes [5,6]. Up to 90% of all NDI cases are X-linked recessive NDI (OMIM 304800) with mutations in the AVPR2 gene, while only approximately 10% of affected individuals exhibit defects in the AQP2 gene inherited by autosomal recessive or autosomal dominant modes (OMIM 125800) [7]. The human AVPR2 gene, located on chromosome Xq28 consists of 3 exons and 2 small introns. The sequence of human AVPR2 cDNA predicts a polypeptide of 371 amino acids with seven transmembrane, four extracellular and four cytoplasmic domains [6]. The human AQP2 gene is mapped to chromosome 12q12.13 and consists of 4 exons distributed over approximately 5 kb of genomic DNA [8]. The coding sequence is a 271 amino acid polypeptides that form a vasopressin-sensitive water channel protein designated as aquaporin-2 (AQP2), which contains six membrane-spanning helical domains with both amino and carboxyl terminals located in the cytoplasm [6,8]. In kidney epithelial cells, AQP2 is regulated by AVP and is linked to both short-term plasma membrane vesicle-mediated changes and long-term AQP2 gene expression-mediated changes in the reabsorption rate of water from urine in the kidneys [9]. Therefore, whilst different forms of inherited NDI exist due to varied mutations [8,10], the clinical presentations are virtually identical due to similar dysfunctions in the regulation of long-term urine concentration by AQP2 gene-mediated AQP2 expression.
Recently, many isolated cases were published reporting novel mutations associated with congenital NDI in Chinese, Korean and Japanese families [11-13]. It has been reported that it is possible for NDI probands to have normal stature and intelligence due to some residual activity and only partial insensitivity to antidiuretic hormones [12]. As a result, diagnosis is more challenging in these patients, potentially resulting in delayed treatment and greater risks for the patient. Consequently, this study was conducted to describe the mutations associated with NDI in two Chinese families with members exhibiting clinical characteristics of congenital NDI. The AVPR2 and AQP2 genes in these two pedigrees were sequenced, revealing novel mutations associated with congenital NDI.
Materials and methods
Ethics statement
The study was approved by the Medical Ethics Committee of the Peking Union Medical College Hospital. Written informed consent was obtained from all family members and from 50 healthy controls with the same ethnic backgrounds as the proband group (Han ethnicity).
Patients
Two Chinese Han male individuals with inherited NDI were identified and enrolled: proband 1 (II:1) in family 1 and proband 2 (V:3) in family 2. The diagnosis of NDI was based on clinical manifestations, including medical history, symptoms, physical examination and biochemistry results.
Molecular analysis of the AQP2 and AVPR2 genes
The entire coding regions and intron-exon borders of the AVPR2 and AQP2 genes were amplified from genomic DNA extracted from peripheral blood leukocytes and further identified by DNA sequencing. The primers used for polymerase chain reaction (PCR) were designed using Primer 3.0 (available at: http://frodo.wi.mit.edu/) (sequences are listed in Table 1). All PCR reactions were performed using 100 ng of genomic DNA template in a total volume of 50 μl containing 10 pmol of each primer, and 25 μl of 2× Easytaq mix (Tiangen Biotech, Beijing, China), according to standard PCR protocol and annealing temperatures (Table 1). The PCR products were sequenced using an ABI PRISM 3700 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), according to the protocol of manufacturer. Sequences generated from the patients and their relatives were compared with the published AQP2 (accession # DNA: NC_000012, cDNA: NM_000486, protein: NP_000477) and AVPR2 (accession # DNA: NC_000023, cDNA: NM_000054, protein: NP_000045) gene sequences. Once a missense mutation was found, both sense and antisense strands were sequenced to confirm the variant and the fifty controls were sequenced to exclude the possibility of a single nucleotide polymorphysm (SNP). If a frameshift mutation was detected, subclonal sequencing of this segment was performed and the purified PCR product of the mutational exon ligated into pGEM-T easy vector (Promega, Madison, WI, USA) and then transformed into E. coli strain DH-5a. Thereafter, positive subclones were sequenced using an ABI PRISM 3700 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The descriptions of sequence variants in DNA and protein levels were based on the mutation nomenclature [14].
Table 1.
PCR conditions for AQP2 and AVPR2 gene mutation analysis
| Gene | Exon | Primer sequence (5 ‘ to 3 ’) | AT (°C) | Size (bp) |
|---|---|---|---|---|
| AQP2 | Exon 1 | F: AGGTATTGGCCTCAACGACTC | 61 | 797 |
| R: TGAGGCTCTTCCTGCACGAC | ||||
| Exon 2+3 | F: GCCTCAGGCCCCAATCTAAT | 61 | 947 | |
| R: AGGAGGAGTGTGGAGGGTTT | ||||
| Exon 4 | F: GCTGGCGTTGTCGTTGTA | 62 | 793 | |
| R: AGAGAACTTGGGGATGAACACA | ||||
| AVPR2 | Exon 1 | F: ATCCTGGGTTCTGTGCATCCGT | 60 | 388 |
| R: CTCCCCTCCCCACTCATTG | ||||
| Exon 2A | F: CATGAGCCTGGGGTGTGTATCC | 63 | 696 | |
| R: CGCTGCCACCTTCCACGTTG | ||||
| Exon 2B | F: TGCCTCCTCCTACATGATCCTG | 62 | 677 | |
| R: GGCCAGCAACATGAGTAGCAC | ||||
| Exon 3 | F: GGCCAAGACTGTGAGGATGACG | 62 | 688 | |
| R: TGCAGCCCCTCCTACACC |
F, forward; R, reverse; AT, annealing temperature; bp, base pair.
Predicted effects of AQP2 mutations
The alignment of mutant proteins among various species was performed using Bioedit (http://www.mbio.ncsu.edu/bioedit/bioedit.html), with reference sequences derived from the NCBI protein database (http://www.ncbi.nlm.nih.gov/protein/). Furthermore, to predict the influence of amino acid substitution on protein function, an in silico analysis was performed by using ‘MutPred’ (http://mutpred.mutdb.org/cgi-bin/mutpred), ‘Mutation Taster’ (http://doro.charite.de/cgi-bin/MutationTaster) and ‘SNPs&Go’ (http://snps-and-go.biocomp.unibo.it/snps-and-go/).
Clinical and biochemistry assessments
Clinical characteristics, including body weight, length/height, symptoms (e.g., fever, vomiting, and polyuria) and volume of daily urine output and water intake, were recorded. Biochemical tests were conducted to determine hypernatremia, serum chloride level, serum osmolality and urinary osmolality. All drug use was recorded.
Imaging assessments
Magnetic resonance imaging (MRI) of the brain was conducted to confirm the hypothalamus and pituitary gland are normal in Proband 1.
Cognitive function assessment
Cognitive function was assessed in Proband 1 according to the Chinese Wechsler Young Children Scale of Intelligence (C-WYCSI), including full intelligence quotient (IQ), verbal IQ and performance IQ [15].
Results
Proband demographics
Proband 1 was a 5.5-year-old Chinese boy (Figure 1A; II:1), delivered by cesarean section due to macrosomia after an uncomplicated first-time pregnancy of non-consanguineous Chinese parents with no family history of NDI (birth weight 4.85 kg). Proband 2 was an 8-year-old male (Figure 2A; V: 3) delivered naturally after an uncomplicated third pregnancy of consanguineous Chinese parents with no family history of NDI (birth weight 2.9 kg).
Figure 1.
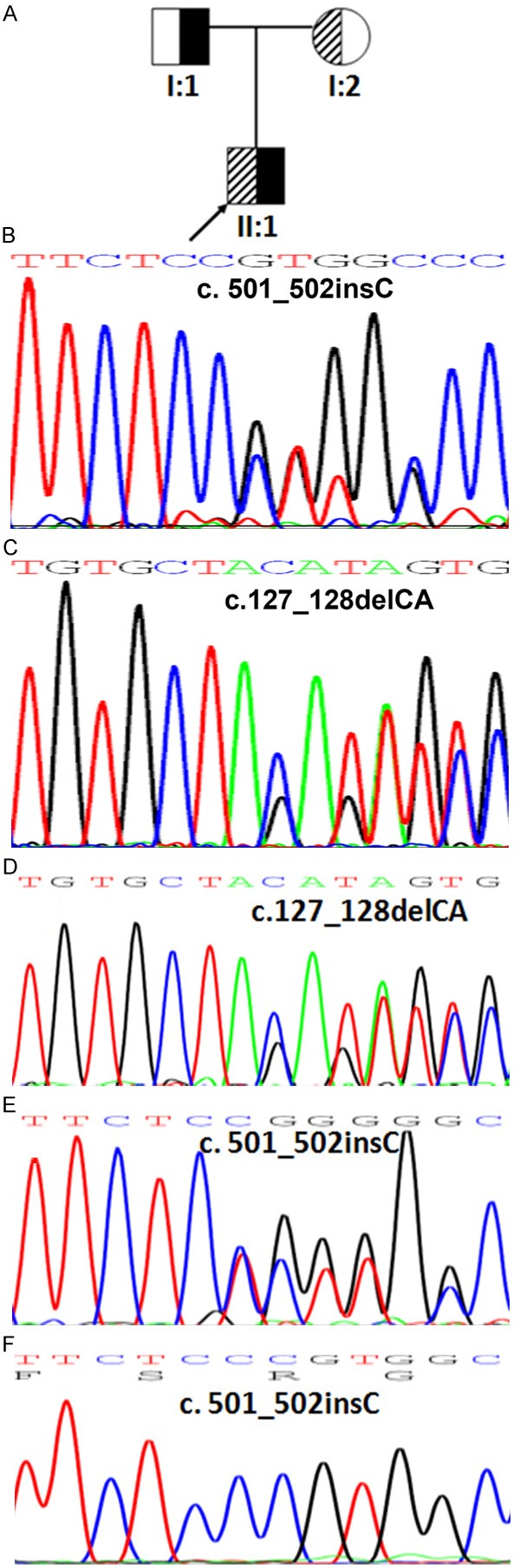
Mutations in AQP2 in Pedigree 1 and the genetic profile of the proband. A. The pedigree of a non-consanguineous Chinese family with NDI. B. Partial sequence of the AQP2 gene from proband 1’s father (I:1) showing a heterozygous insertion of cytosine at nucleotide 502 (c. 501_502 insC) in exon 2. C. Partial sequence of the AQP2 gene from proband 1’s mother (I:2) showing a heterozygous deletion of cytosine and adenine at nucleotide 127 and 128 (c.127_128delCA) in exon 1. D. Partial sequence of the AQP2 gene from proband 1 (II:1), showing a heterozygous insertion of cytosine at nucleotide 502 (c. 501_502 insC) in exon 2, leading to a truncated protein at the 30th amino acid downstream from the initiated mutation (p.Val168Argfs ×30). E. Partial sequence of the AQP2 gene from proband 1, showing a heterozygous deletion of cytosine and adenine at nucleotide 127 and 128 (c.127_128delCA) in exon 1, resulting in a premature stop codon at the 63rd amino acid downstream from the initiated mutation (p.Gln43Asp fs ×63). F. Subclonal sequencing of p.Val168Argfs ×30.
Figure 2.
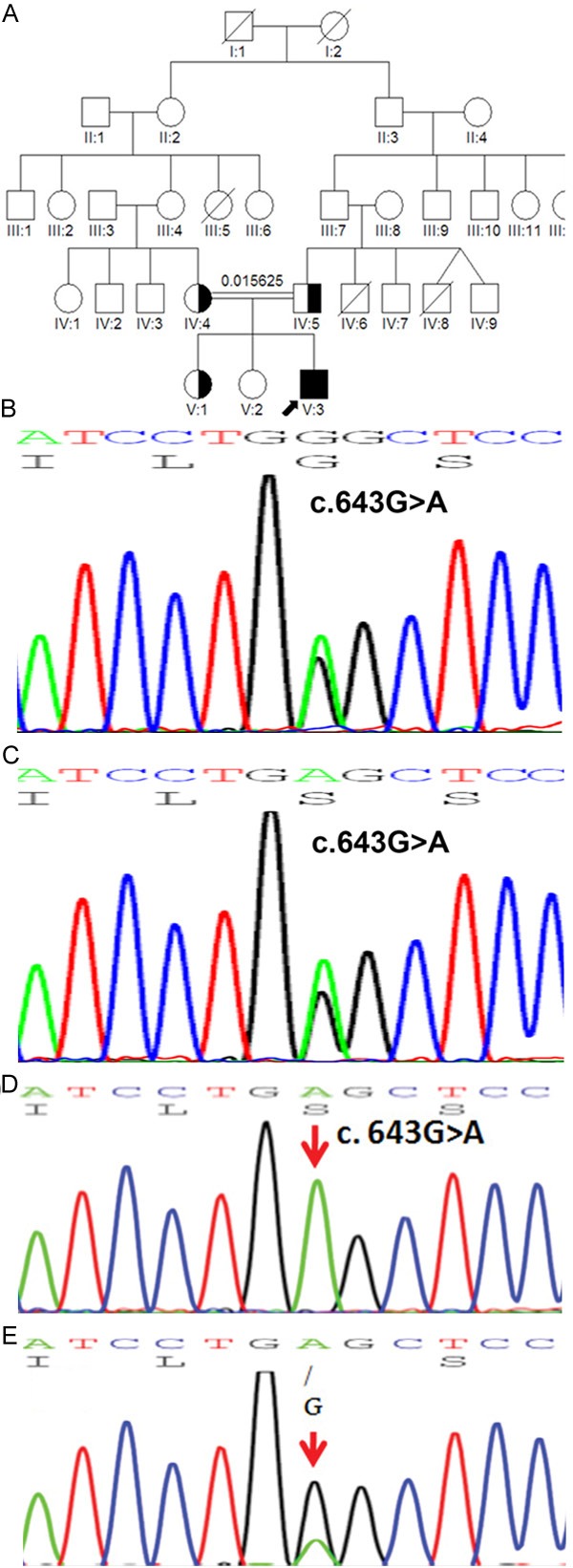
Mutations in AQP2 in Pedigree 2 and the genetic profile of the proband. A. The pedigree of a consanguineous Chinese family with NDI. B. Partial sequence of the AQP2 gene from proband 2’s mother (IV:4) showing a heterozygous guanine-to-adenine change at nucleotide position 643 (c.643G>A) in exon 4, causing a glycine-to-serine substitution at codon 215 (p.G215S). C. Partial sequence of the AQP2 gene from proband 2’s father (IV:5) showing a heterozygous guanine-to-adenine change at nucleotide position 643 (c.643G>A) in exon 4, causing a glycine-to-serine substitution at codon 215 (p.G215S). D. Partial sequence of the AQP2 gene from the proband 2’s older sister (V:1). E. Partial sequence of the AQP2 gene from proband 2 (V:3) showing a homozygous guanine-to-adenine change at nucleotide position 643 (c.643G>A) in exon 4 causing a glycine-to-serine substitution at codon 215 (p.G215S). Red arrows indicate the gene mutation.
NDI clinical signs and diagnosis
Proband 1 displayed clinical symptoms of polyuria, polydipsia and unexplained recurrent fever since infancy. Proband 2 was breastfed until 3 months of age and exhibited poor weight gain and intermittent vomiting upon introduction of mixed feeding and was admitted to the hospital at 9 months of age as a result of an intermittent fever of an unknown cause for 5 months. Both patients exhibited febrile attacks that could be relieved by large intake of water. Clinical diagnosis of NDI in both proband 1 and 2 was made at the age of 5.5 and 8.0 years, respectively.
Clinical characteristics
Proband 1 had a body weight of 20.5 kg (50-75 percentile) and a height of 106.2 cm (-2 SD), while proband 2 had a body weight of 18.5 kg (<3 percentile) and a height of 118.6 cm (-2 SD) (Table 2). The discrepancy in weight between two probands was due to poor weight gain suffered by proband 2. In proband 1, laboratory studies revealed hypernatremia and elevated serum chloride, with increased serum osmolality yet low urinary osmolality. Proband 2, however, exhibited random serum of 307 and urine osmolality of 157 mOsm/(kg·H2O (Table 3). Due to the persistent short stature of proband 2, serum IGF-1 levels and thyroid function tests were conducted revealing normal hormone levels.
Table 2.
Clinical characteristics of two probands with congenital NDI from two unrelated families
| Characteristic | Proband 1 | Proband 2 |
|---|---|---|
| Sex | Male | Male |
| Age at presentation | <1 year old | 4 month old |
| Age at diagnosis (years) | 5.5 | 8.0 |
| Polydipsia | + | + |
| Polyuria (24 h urine output, ml) | ~5000 | 4000~5000 |
| Nocturia (times/night) | 2-3 | 3-4 |
| Intermittent fever | + | + |
| Failure to thrive | + | + |
| Dehydration presentation | - | - |
| Mental retardation | + | - |
| Growth Retardation | + | + |
| Height/SD (cm) | 106.0/-2SD | 118.6/-2SD |
| Weight (kg) | 20.5 (50-75 percentile) | 18.5 (<3 percentile) |
| Therapy | Hydrochlorothiazide and amiloride | Hydrochlorothiazide and potassium chloride supplementation |
| Treatment efficacy (urine output, ml/24 h) | No change | Reduced to ~2000 ml |
+, positive symptom; -, negative symptom; SD, standard deviation.
Table 3.
Laboratory test and imaging results of two probands with congenital NDI
| Characteristics | Proband 1 | Proband 2 | Normal range |
|---|---|---|---|
| Osmolality (mOsm/Kg·H2O) in fluid restriction | |||
| Serum | 350 | 307 | 275-295 |
| Urine | 50 | 157 | >850 |
| Serum studies | |||
| Sodium (mmol/l) | 152 | 142 | 134-143 |
| Chloride (mmol/l) | 148 | 103 | 98-106 |
| Potassium (mmol/l) | 4.1 | 4.2 | 3.3-4.6 |
| Fasting blood glucose (mmol/l) | 5.2 | 4.7 | 3.3-5.5 |
| Total calcium (mmol/l) | 2.54 | 2.69 | 2.20-2.70 |
| Blood urine nitrogen (mg/dl) | 18 | 11 | 5-18 |
| Creatine (mg/dl) | 0.75 | 0.62 | 0.2-1.0 |
| Pituitary magnetic resonance image | Absent posterior pituitary bright spot (T1-weighted image) | N/A | — |
| Abdominal ultrasound | Normal | Normal | — |
Medical treatment
Proband 1 was treated with a large amount of fluids and subsequent administration of a combination of hydrochlorothiazide and amiloride that failed to increase urine osmolality and did not relieve NDI-related symptoms. Proband 2 was given the similar treatment with oral administration of hydrochlorothiazide (12.5 mg, bid) combined with potassium chloride (20 ml, bid~tid), and polyuria and polydipsia were partially improved (Table 2).
Urine output
Proband 1 exhibited a daily urine output of 4000-5000 ml balanced by approximately the same water intake volume, as confirmed by NDI by water deprivation and vasopressin stimulation tests (Table 4). Proband 2 had a 24 h urine volume relieved from 4000~5000 ml/day prior to medical treatment to almost 2000 ml/day with medical treatment. This reduction in urinary output successfully reduced nocturia to 1-2 times/night with medical treatment compared with that of the untreated status of 3-4 times/night. Treatment for proband 1 was less effective than that for proband 2 with regard to 24 h urinary output.
Table 4.
Results of water deprivation and vasopressin stimulation tests for proband 1
| Time | Urine output (ml/h) | Urine SG | Urine Osm (mOsm/Kg·H2O) | SBP/DBP | BW (Kg) |
|---|---|---|---|---|---|
| 06:00* | 50 | 1.000 | 100 | 105/70 | 20.5 |
| 07:00 | 200 | 1.002 | 125 | 20.5 | |
| 08:00 | 170 | 1.002 | 125 | 20.5 | |
| 09:00 | 95 | 1.002 | 125 | 106/60 | 20.3 |
| 10:00 | 130 | 1.002 | 125 | 20.0 | |
| 11:00 | 80 | 1.000 | 125 | 100/60 | 20.0 |
| 12:00† | 95 | 1.002 | 125 | 20.0 | |
| 13:00 | 275 | 1.000 | <50 | ||
| 14:00 | 310 | 1.000 | <50 |
SG, specific gravity; Osm, osmolality; SBP, Systolic blood pressure; DBP, Diastolic blood pressure; BW, body weight.
Water deprivation started at 06:00;
3u Pituitrin was given by intramuscular injection at 12:00.
MRI of the hypothalamus and pituitary gland and cognitive assessments
In proband 1 and 2, abdominal and renal ultrasound showed that abdominal and renal regions were normal; however the posterior pituitary bright spot was not seen by MRI in proband 1 (Figure 3). C-WYCSI full IQ, verbal IQ, and performance IQ for proband 1 were 35, 56, and 40, respectively. MRI and cognitive assessment of proband 2 were not performed.
Figure 3.
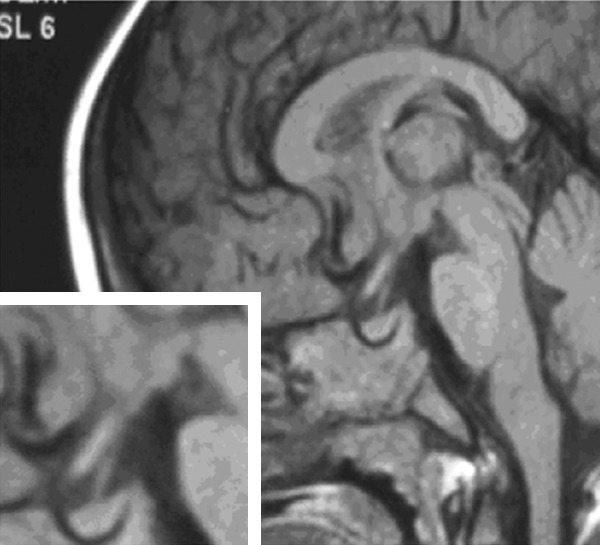
Brain MRI in proband 1. The bright spot of the posterior pituitary is missing.
Proband 1 mutations: comparisons with family and control samples
Direct sequencing results indicated that proband 1 harbored compound heterozygous mutations in the AQP2 gene (Figure 1D; Table 5). One allele displayed a two-base deletion (c.127_128delCA) in exon 1 (Figure 1D), inherited from the mother (Figure 1A and 1C) resulting in a premature stop codon at the 63rd amino acid downstream from the initiated mutation (p.Gln43Aspfs*63). In addition, a cytosine insertion occurred in exon 2 of the other allele (c. 501_502 insC) (Figure 1E and 1F) and this mutation encoded another truncated protein referred to as p.Val168Argfs*30. The father of proband 1 was a heterozygous carrier of the same insertion mutation (Figure 1A and 1B). Following identification of the compound heterozygous frameshift mutation, the change was confirmed on different alleles by subcloning. No mutation was detected in any other exon of the AQP2 gene or the AVPR2 gene in proband 1.
Table 5.
AQP2 gene mutations identified in two probands from two distinct pedigrees
| Index | Sex | Nucleotide change | Homozygous/heterozygous | Amino acid Change | Mutation type | Encoding region | Protein location | Parent origin | Consanguinity | Family history | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | c.127_128delCA | Heterozygous | G43Dfs*63 | Frame-shift | Exon1 | TMD-2 | Mother | No | Negative | [20] No in vitro study Novel |
| c.501_502 insC | V168Rfs*30 | Frame-shift | Exon2 | TMD-5 | Father | ||||||
| 2 | M | c. 643G>A | Homozygous | G215S | Missense | Exon 4 | TMD-6 | Mother/father | Yes | Negative | Novel |
M, male; TMD, transmembrane domain.
Proband 2 mutations: comparisons with family and control samples
Gene mutation analysis of both the AQP2 gene and the AVPR2 gene in proband 2 revealed a homozygous mutation (c. 643G>A) in the AQP2 gene (Figure 2E; Table 5), which resulted in amino acid substitution at exon 4 (p.G215S) located at transmembrane 6 of the water channel acquaporin-2. No other mutation was detected in the AVPR2 gene. The parents (IV:4 and IV:5) (Figure 2B and 2C) and one older sister (V:1) (Figure 2D) of proband 2 were heterozygous for this mutation, while no mutations were found in the other sister (V:2) (Figure 2A). Fifty control samples from healthy volunteers of similar ethnic origin (Han Chinese) were sequenced to detect <5% polymorphism with 95% power (23). This sequence variation was not present in any of the 50 control samples, indicating a true genetic mutation rather than a polymorphism.
Protein alignment and in silico analysis
Within AQP2, the glycine at codon 215 is located at a highly conserved transmembrane domain (Figure 4). Both the ‘SNPs&Go’ and ‘Mutation Taster’ software predicted that G215S was a disease-causing mutation. Likewise, the ‘MutPred’ software placed the mutation in the ‘Actionable Hypothesis’ [16] class, the MutPred general score >0.5 and the property score P<0.05, in which the score obtained for this new substitution was 0.733 (P=0.007).
Figure 4.
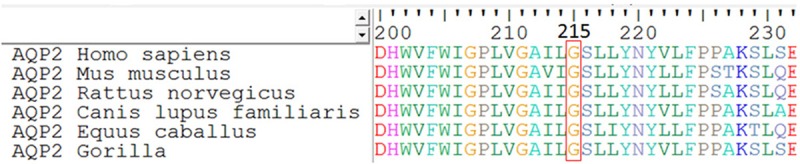
Sequence alignment of the AQP2 proteins from different species. Codon 215 is framed in a red box and shows a 100%-conservation across six different species.
Discussion
In this study, two Chinese male patients (proband 1 and proband 2) with polyuria, polydipsia and serum hyperosmolality with unsuitable decreased levels of urinary osmolality were diagnosed with congenital NDI. Further genetic testing revealed that proband 1 was carrying two mutations within AQP2: the previously reported (c.127_128delCA, p.Gln43Aspfs ×63) and novel (c.501_502insC, p.Val168Argfs ×30). The former was inherited from the mother, while the novel mutation was inherited from the father. Proband 2 was a homozygote with a novel missense mutation in AQP2 (p. G215S).
The AQP2 gene encodes the AQP2 protein, a vasopressin-regulated water channel that determines the permeability of kidney collecting ducts to water and plays an important role in water homeostasis. The AQP2 monomer consists of six transmembrane domains that are connected by five loops (A through E) with intracellular N- and C-terminus. The highly-conserved, membrane-embedded asparagine-proline-alanine (NPA) motifs of Loops B and E are positioned in the proximity of the cell membrane’s surface and it is postulated that they are critical for formation of the water-selective pore [17,18]. To date, at least 53 putative disease-causing mutations in the AQP2 gene have been reported, most of which are missense variants, with a recessive inheritance pattern [10,13,19].
In this study, we have identified three mutations: the previously reported (p.Gln43Aspfs ×63) and novel (p.Val168Argfs ×30 and G215S).mutations. Whilst we did not assess the potential contribution of other genes or variants to the phenotype, and cannot exclude the influence of additional genes, it is likely these mutations are responsible for these patients’ symptoms. The reported p.Gln43Aspfs ×63 mutation in exon 1 found in proband 1 was first described by Tajima in a Japanese boy [20]. Whilst the mutation reported by Tajima was described as 197_198 del CA, in this study the mutation was named c.127_128 del CA according the nomenclature used by den Dunnen and Antonarakris, Sasaki et al. and Park et al. [14]. The position of the mutated amino acid was exactly the same for both mutations (both 197_198 del CA and c.127_128 del CA) and therefore either nomenclature can be used to describe this mutation. It is possible that either this mutation arose in diverse ethnic backgrounds, or that it is an ancient founder mutation in East Asia populations, however due to lack of data, this assumption cannot be confirmed. This mutation is expected to produce a truncated protein lacking the amino acid sequence of both the conserved NPA motifs in loop B and E, transmembrane domains 2-6 and C-terminal. The p.Val168Argfs ×30 mutation encodes a truncated protein, affecting the NPA motif in loop E, transmembrane domains 5-6 and C-terminus. Therefore, it is possible that such defects may impair the normal physiological function of aquaporin for water reabsorption resulting in NDI.
The other novel missense mutation identified in the current study, a guanine-to-adenine mutation at nucleotide position 643 (c.643G>A) in exon 4, results in a substitution of glycine with serine at codon 215 (p.G215S). At the same nucleotide position, another mutation, a guanine-to-thymine mutation resulting in p.G215C, was reported in a boy with NDI [21]. The functional analysis of the missense mutation p.G215C indicated that the AQP2-215C mutant caused retention of AQP2 in the endoplasmic reticulum, and thus recessive NDI. This was corroborated by the absence of normal AQP2 protein in the patient’s urine, whilst normal AQP2 proteins were easily detected in the urine of the patient’s healthy parents [21]. Further evidence indicating that this variant acts as a causative mutation includes the high level of conservation of the amino acid G215. This position contains an identical amino acid in six different species present in the National Centre for Biotechnology Information database. Additionally ‘SNPs&Go’ and ‘Mutation Taster’ software predicted that G215S is a disease-causing mutation. This prediction was further replicated by the software ‘MutPred’, which placed the mutation in the ‘Actionable Hypothesis’ class.
Several in vitro studies have investigated the mechanisms of autosomal recessive or dominant NDI [5,8]. Recessive NDI is often caused by mutations in the transmembrane regions or NPA motifs of the AQP2 protein, leading to misfolding of the mutant AQP2, its retention in the endoplasmic reticulum and its rapid degradation. Dominant NDI results from mutations in the AQP2 C-terminus, whereby the AQP2 mutant forms heterotetramers with wild-type AQP2. These heterotetramers are missorted to other cellular organelles and not present at the apical plasma membrane. Thus, according to the position of the mutations observed in this study, Gln43Aspfs ×63, Val168Argfs ×30 and G215S, at the second, fifth and sixth transmembrane region respectively, indicates that such mutations result in NDI via the recessive inheritance mode. In addition, the parents of these patients were heterozygous for these mutations did not report symptoms of NDI suggesting autosomal recessive inherence. Further in vitro expression studies are required to ascertain the biological significance and functional consequences of the frameshift mutations and the novel missense mutation found in this study.
Both affected individuals in two non-relative families were male with negative family history of NDI. With regards to different clinical characteristics and laboratory results, the proband 1 may present much more severe clinical manifestations and related complications than proband 2, such as therapeutic effect based on 24 h output urine as well as the laboratory test results including serum osmolality, urine osmolarity and serum sodium. Thus, we can infer that the symptoms of proband 1 due to the compound heterozygous frameshift mutations coding for two different truncated proteins are much more severe than that of proband 2 with homozygous missense mutation.
The conclusions we can draw are, however, somewhat limited by the nature of this study, in particular the limited number of patients studied. We cannot exclude the role of additional or alternative mutations in the patients’ symptoms. Other genes or variants could be modifiers of the phenotype. For example, inactivating mutations in genes that encode the membrane proteins of the thick ascending limb of the loop of Henle can cause a complex polyuro-polydipsic syndrome with loss of water, sodium, chloride, calcium, magnesium, and potassium [4,22]. Also, the Kidd antigen (UT-B protein), encoded by SLC14A1, confers a mild form of partial congenital NDI. These patients cannot concentrate urine above 800 mmol/kg, even after overnight water deprivation and administration of exogenous vasopressin [23].
We also cannot comment on the prevalence of these mutations in Chinese or global population. Due to the potential impact of autosomal recessive NDI on early clinical diagnosis and treatment, this study indicates that further work is required to identify and characterize the multiple forms of congenital NDI. Further research is required to assess the genotype-phenotype correlation between these mutations and increased or reduced NDI severity.
In summary, two novel mutations in the AQP2 gene were identified in this report expanding the AQP2 genotypic spectrum. In addition, for the first time, compound heterozygous frameshift mutations in patients with nephrogenic diabetes were reported, and we observed that a patient with truncated AQP2 may present with much more severe NDI manifestations. Since there is no hot spot for mutations in the AQP2 and AVPR2 gene, mutation analysis is useful for genetic counseling and early diagnosis to avert the irreversible damage and severe complications occurring in patients with NDI. However, functional analysis about two novel mutations will be further studied.
Acknowledgements
We are sincerely indebted to the patients and clinicians involved in this study for their cooperation and collaboration. This work was supported by grants from (1) the National Natural Science Foundation of China (NO. 81170724) and (2) the National Key Program of Clinical Science.
Disclosure of conflict of interest
None.
References
- 1.Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27:2183–2204. doi: 10.1007/s00467-012-2118-8. [DOI] [PubMed] [Google Scholar]
- 2.Verbalis JG. Posterior pituary. In: Goldman L, Schafer A, editors. Cecil medicine. 24th. Philadelphia: Saunders Elsevier; 2011. [Google Scholar]
- 3.van Lieburg AF, Knoers NV, Monnens LA. Clinical presentation and follow-up of 30 patients with congenital nephrogenic diabetes insipidus. J Am Soc Nephrol. 1999;10:1958–1964. doi: 10.1681/ASN.V1091958. [DOI] [PubMed] [Google Scholar]
- 4.Hoekstra JA, van Lieburg AF, Monnens LA, Hulstijn-Dirkmaat GM, Knoers VV. Cognitive and psychosocial functioning of patients with congenital nephrogenic diabetes insipidus. Am J Med Genet. 1996;61:81–88. doi: 10.1002/(SICI)1096-8628(19960102)61:1<81::AID-AJMG17>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 5.Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–270. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- 6.Fujiwara TM, Bichet DG. Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol. 2005;16:2836–2846. doi: 10.1681/ASN.2005040371. [DOI] [PubMed] [Google Scholar]
- 7.Knoers N, van den Ouweland A, Dreesen J, Verdijk M, Monnens LA, van Oost BA. Nephrogenic diabetes insipidus: identification of the genetic defect. Pediatr Nephrol. 1993;7:685–688. doi: 10.1007/BF00852579. [DOI] [PubMed] [Google Scholar]
- 8.Noda Y, Sohara E, Ohta E, Sasaki S. Aquaporins in kidney pathophysiology. Nat Rev Nephrol. 2010;6:168–178. doi: 10.1038/nrneph.2009.231. [DOI] [PubMed] [Google Scholar]
- 9.Bouley R, Hasler U, Lu HA, Nunes P, Brown D. Bypassing vasopressin receptor signaling pathways in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28:266–278. doi: 10.1016/j.semnephrol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spanakis E, Milord E, Gragnoli C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J Cell Physiol. 2008;217:605–617. doi: 10.1002/jcp.21552. [DOI] [PubMed] [Google Scholar]
- 11.Huang L, Li W, Tang W, Lu G. A novel AVPR2 missense mutation in a Chinese boy with severe inherited nephrogenic diabetes insipidus. J Pediatr Endocrinol Metab. 2011;24:807–809. doi: 10.1515/jpem.2011.302. [DOI] [PubMed] [Google Scholar]
- 12.Moon SD, Kim JH, Shim JY, Lim DJ, Cha BY, Han JH. Analysis of a novel AVPR2 mutation in a family with nephrogenic diabetes insipidus. Int J Clin Exp Med. 2011;4:1–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki S, Chiga M, Kikuchi E, Rai T, Uchida S. Hereditary nephrogenic diabetes insipidus in Japanese patients: analysis of 78 families and report of 22 new mutations in AVPR2 and AQP2. Clin Exp Nephrol. 2013;17:338–344. doi: 10.1007/s10157-012-0726-z. [DOI] [PubMed] [Google Scholar]
- 14.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Guo B, Aveyard P, Dai X. The Chinese Inte-lligence Scale for Young Children. International Journal of Epidemiology. 2013;42:160–171. [Google Scholar]
- 16.Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laski ME. Structure-function relationships in aquaporins. Semin Nephrol. 2006;26:189–199. doi: 10.1016/j.semnephrol.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Gonen T, Walz T. The structure of aquaporins. Q Rev Biophys. 2006;39:361–396. doi: 10.1017/S0033583506004458. [DOI] [PubMed] [Google Scholar]
- 19.Loonen AJ, Knoers NV, van Os CH, Deen PM. Aquaporin 2 mutations in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28:252–265. doi: 10.1016/j.semnephrol.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Tajima T, Okuhara K, Satoh K, Nakae J, Fujieda K. Two novel aquaporin-2 mutations in a sporadic Japanese patient with autosomal recessive nephrogenic diabetes insipidus. Endocr J. 2003;50:473–476. doi: 10.1507/endocrj.50.473. [DOI] [PubMed] [Google Scholar]
- 21.Iolascon A, Aglio V, Tamma G, D’Apolito M, Addabbo F, Procino G, Simonetti MC, Montini G, Gesualdo L, Debler EW, Svelto M, Valenti G. Characterization of two novel missense mutations in the AQP2 gene causing nephrogenic diabetes insipidus. Nephron Physiol. 2007;105:p33–41. doi: 10.1159/000098136. [DOI] [PubMed] [Google Scholar]
- 22.Shida Y, Matsuoka H, Chiga M, Uchida S, Sasaki S, Sugihara S. Characterization of AQP-2 gene mutation (R254Q) in a family with dominant nephrogenic DI. Pediatr Int. 2013;55:105–107. doi: 10.1111/j.1442-200X.2012.03614.x. [DOI] [PubMed] [Google Scholar]
- 23.Sands JM, Gargus JJ, Frohlich O, Gunn RB, Kokko JP. Urinary concentrating ability in patients with Jk(a-b-) blood type who lack carrier-mediated urea transport. J Am Soc Nephrol. 1992;2:1689–1696. doi: 10.1681/ASN.V2121689. [DOI] [PubMed] [Google Scholar]