

Abstract
MicroRNAs (miRNAs) have emerged as important regulators that potentially play critical roles in various biological processes. Previous studies have shown that miR-615 regulates proliferation and apoptosis in many types of cancers. The biological function of this microRNA in breast cancer remains largely unexplored. In the present study, we found that miR-615 expression was markedly downregulated in breast cancer tissues and breast cancer cells. The enforced expression of miR-615 was able to inhibite the proliferation and anchorage-independent growth of breast cancer cells, while miR-615-in showed the opposite effect. Bioinformatics analysis further revealed AKT2, a putative tumor promoter as a potential target of miR-615. Ectopic expression of miR-615 led to downregulation of AKT2 protein, which resulted in the upregulation of p27 and p21 and the downregulation of cyclin D1. In sum, these results suggest that miR-615 represents a potential anti-onco-miR and participates in breast cancer carcinogenesis by suppressing AKT2 expression.
Keywords: miR-615, breast cancer, AKT2, cell proliferation, cell cycle
Introduction
Breast cancer is the most common malignancy which leading the second leading cause of cancer-related mortality among females among the world [1,2]. In recent years, the incidence of breast cancer has been experiencing a significant rise, highlighting the requirement for therapies of breast cancer.
Growing evidence suggests that MicroRNAs (miRNAs) act as either tumor suppressors or promoters in the development of various cancers and play crucial roles in regulating posttranscriptional gene expression by binding to 3’-untranslated of the target mRNA [3-5]. Moreover, treatment by targeting miRNAs has attracted extensive research attention in various kinds of cancers, including breast cancer [6,7]. Recently, miR-615 was reported to be frequently downregulated and acted as a tumor suppressor involved in cellular function [8]. However, the relationship between breast cancer and the expression of miR-615 has not yet been elucidated. In this present study, we found that miR-615 expression was markedly downregulated in breast cancer tissues and breast cancer cells. Bioinformatics analysis indicated that miR-615 may directly target the 3’-UTR of AKT2. We demonstrated that miR-615 inhibited breast cancer cell proliferation by directly targeting the 3’-UTR of AKT2 mRNA, which subsequently reduced expression of the cell-cycle regulator cyclin D1 and upregulated the cyclin-dependent kinase (CDK) inhibitors, p27 and p21. Our data demonstrated that miR-615 may play an important role in the development and progression of breast cancer through the AKT2-mediated signaling pathway.
Materials and methods
Clinical specimens
Normal breast sample was collected from the mammoplasty material of a 34-year-old woman at Cangzhou Centerl Hospital (Cangzhou, Hebei Province, People’s Republic of China). Eight paired human breast cancer tissues and the matched adjacent non-tumor tissues (ANT) were obtained from breast cancer patients and histopathologically diagnosed at Cangzhou Centerl Hospital (Cangzhou, Hebei Province, People’s Republic of China). The study was approved by the ethics committee of Cangzhou Centerl Hospital (Cangzhou, Hebei Province, People’s Republic of China). Written informed consent was obtained from all patients.
Cell culture
Primary normal breast cell (NBEC), from mammoplasty material, were cultured in the Keratinocyte serum-free medium (Invitrogen, Carlsbad, CA) supplemented with epithelial growth factor, bovine pituitary extract and antibiotics (120 mg/ml streptomycin and 120 mg/ml penicillin). Human breast cancer cell lines BT549, MCF-7, Bcap37, MDA-MB231, ZR-75-30, SKBR3, MDA-MB435, MDA-MB453 and T47D were purchased from National Rodent Laboratory Animal Resource (Shanghai, People’s Republic of China). All cell lines were grown in were cultured in RPMI-1640 (Gbico, USA) supplemented with 10% fetal bovine serum (FBS, Sigma, St Louis, MI), 100units/ml of penicillin-streptomycin (Invitrogen, Carlsbad, CA), The media for BT-549 cells were additionally supplemented with 0.01 mg/ml insulin (Novo Nordisk A/S) and for T47D with 1% glucose. Cell lines were cultured in a humidified incubator at 37°C in an atmosphere of 5% CO2 and 95% air.
Plasmids, small interfering RNA and transfection
The 3’-UTR of the human AKT2 messenger RNA was cloned in between the Not1 and Xba1 sites of pGL-3 vector (Promega). MiR-615 mimic, miR-615 inhibitor, miR-615-mut and negative control were purchased from GeneCopoeia (Guangzhou, China) and transfected into breast cancer cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
RNA extraction and real-time quantitative PCR
Total RNA was extracted from the tumor tissues and cancer cells and using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction. The expression levels of miR-615 were quantified using the miRNA-specific TaqMan MiRNA Assay Kit (Applied Biosystems). The relative miR-615 expression levels after normalization to U6 small nuclear RNA were calculated using 2-[(Ct of miR-615) - (Ct of U6)]. Quantitative PCR was performed by SYBR Kit (Qiagen, China) using a Light Cycler system. The primers selected are as follows: Cyclin D1 forward 5’-TCCTCTCCAAAATGC CAGAG-3’ and Cyclin D1 reverse 5’-GGCGGATTGGAAATGAACTT-3’; p27 forward 5’-TGCAACCGACGATTCTTCTACTCAA-3’ and reverse 5’-CAAGCAGTGATGTATCTGATAAAC AAGGA-3’; p21 forward 5’-CATGGGTTCTGACGGA CAT-3’ and p21 reverse 5’-AGTCAGTTCCTTGTGGAGCC-3’. Expression data was normalized to the geometric mean of GAPDH to control the variability in expression levels (forward primer 5’-GACGGCCGCATCTTCTTGT-3’; reverse primer 3’-CACACC GA CCTTCACCATTTT-5’) and calculated as 2-[(Ct of CyclinD1, p27 and p21) – (Ct of GAPDH)].
Western blotting
Protein lysates were prepared, equal quantities of protein were s subjected to SDS-PAGE, gels were transferred onto PVDF membranes. The following anti-bodies were used as follows: anti-AKT2 (1:500; Abcam), anti-cyclin D1 (1:1000; Cell Signaling Technology), anti-p27 (1:1000; Cell Signaling Technology), anti-p21 (1:1000; Cell Signaling Technology), anti-retinoblastoma (anti-Rb,1:1000; Cell Signaling Technology) and anti-phosphorylated Rb (anti-p-Rb,1:1000; Cell Signaling Technology). To control sample loading, the blotting membranes were stripped and re-probed with an anti-a-tubulin antibody (Sigma, St Louis, MI). Immunocomplexes were visualized using the chemiluminesence (GE, USA) following the manufacturer’s protocol.
MTT assay
Cell growth was measured by MTT assay, MDA-MB231 cells were seeded in 96-well plates in medium containing 10% FBS at approximately 2,000 cells/well. For quantitation of cell viability, cultures were stained after 1, 2, 3 and 4 days and 100 μl MTT (Sigma) working solution was added into wells for 4 h at 37°C, followed by removal of the culture medium and addition of 150 μl DMSO. The absorbance at 490 nm was measured in a Thermo Scientific Multiskan (Thermo Fisher Scientific, USA).
Colony formation assay
MDA-MB231 cells were plated 6-well plates (1000 cells per well) and incubated for 10 days. Colonies were stained with 1.0% crystal violet for 30 s after fixation with 10% formaldehyde for 5 min. The number of colonies, defined as > 50 cells/colony were counted. Three independent experiments were performed and the data was calculated using paired t test.
Anchorage-independent growth assay
One thousand cells were trypsinized and suspended in 2 ml complete medium plus 0.3% agar (Sigma, St Louis, MI). The agar-cell mixture was plated on top of a bottom layer consisting of 1% agar in complete medium. Cells were incubated for 14 days at 37°C until colony formation and colonies were stained with 0.5% Crystal Violet for counting under microscope and cell colonies were photographed at an original magnification of 100 ×. Only cell colonies containing more than 50 cells were counted.
Flow cytometric analysis
Forty-eight hours after transfection, MDA-MB231 cells were harvested washed with PBS, and fixed with 70% ethanol. Fixed cells were then treated with 20 μg/mL of RNaseA and 50 μg/mL of PI (Sigma-Aldrich) for 30 minutes, and stained cells were immediately analyzed by a FACS can flow cytometer (BD Biosciences).
Luciferase assays
The pGL3 luciferase reporter gene plasmids pGL3-AKT2-3’-UTR, or the control-luciferase plasmid were cotransfected into the cells with 5ng pRL-TK Renilla plasmid (Promega) using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA). Luciferase and Renilla activities were assayed 48 hours after transfection using the dual luciferase assay kit to the manufacturer’s protocol. The levels of firefly luciferase activities were obtained by normalizing to Renilla luciferase activities.
Statistical analysis
All statistical analyses except for microarray data were performed using the SPSS 17.0 (SPSS, Inc., Chicago, IL, USA). Student’s t test was used to evaluate the significance of the differences between two groups of data in all the pertinent experiments. P < 0.05 was considered statistically significant.
Result
MiR-615 expression was elevated in breast cancer tissues and breast cancer cell lines
Firstly, expression level of miR-615 was determined using Real-time PCR analysis. As shown in Figure 1, our results demonstrated that miR-615 expression was consistently downregulated in the breast cancer tissues compared with the matched tumor adjacent tissues, and in all 9 tested breast cancer cell lines showed significantly downregulated expression of miR-615 compared to the normal breast cell (NBEC). Taken together, these results suggested that miR-615 was downregulated in breast cancer.
Figure 1.
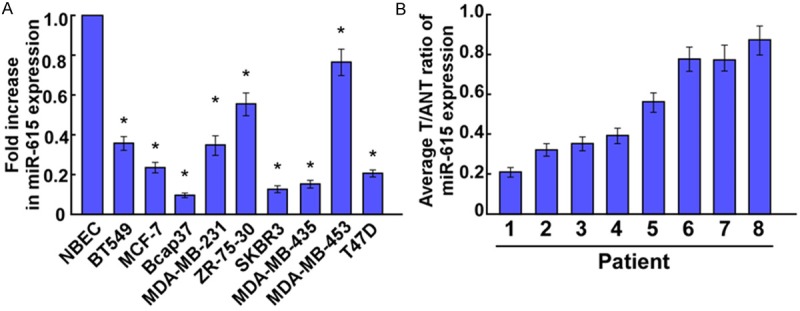
Expression of miR-615 in human breast cancer cell lines and tissues. (A) Real-time PCR analysis of miR-615 expression in normal breast cells (NBEC) and breast cancer cell lines, including BT549, MCF-7, Bcap37, MDA-MB231, ZR-75-30, SKBR3, MDA-MB435, MDA-MB453 and T47D. (B) Relative miR-615 mRNA expression levels in 8 paired primary breast cancer tissues (T) and the matched adjacent non-tumor tissues (ANT) from the same patient were detected by PCR analysis. Experiments were repeated at least three times (A and B). Each bar represents the mean of three independent experiments. *P < 0.05.
MiR-615 inhibited cell proliferation of breast cancer
In order to explore the effects of miR-615 on breast cancer cell growth, we transfected the MDA-MB-231 cells with miR-615 mimics, miR-615 inhibitor or the respective controls and the cell growth was examined. Relative miR-615 expression was verified using qRT-PCR (Figures 2A and 3A). MTT assay showed that miR-615 overexpression significantly decreased the proliferation rate of MDA-MB-231 cells (Figure 2B), and results of colony formation assay revealed that miR-615-transfected cells displayed fewer and smaller colonies compared with control NC transfectants (Figure 2C). Strikingly, we found that enforced expression of miR-615 in MDA-MB-231 cells significantly decreased their anchorage-independent growth ability (Figure 2D). In contrast, the cell growth rates and colony numbers of MDA-MB-231 cells transfected with miR-615 inhibition (miR-615-in) were significantly increased the cell growth rate than those transfected with NC (Figure 3B and 3C). In addition, the anchorage-independent growth ability of MDA-MB-231 cells was drastically increased in response to miR-615-in (Figure 3D). Taken together, MTT, colony formation, and anchorage-independent growth assay demonstrated that miR-615 was able to inhibit the proliferation of breast cancer cells in vitro.
Figure 2.
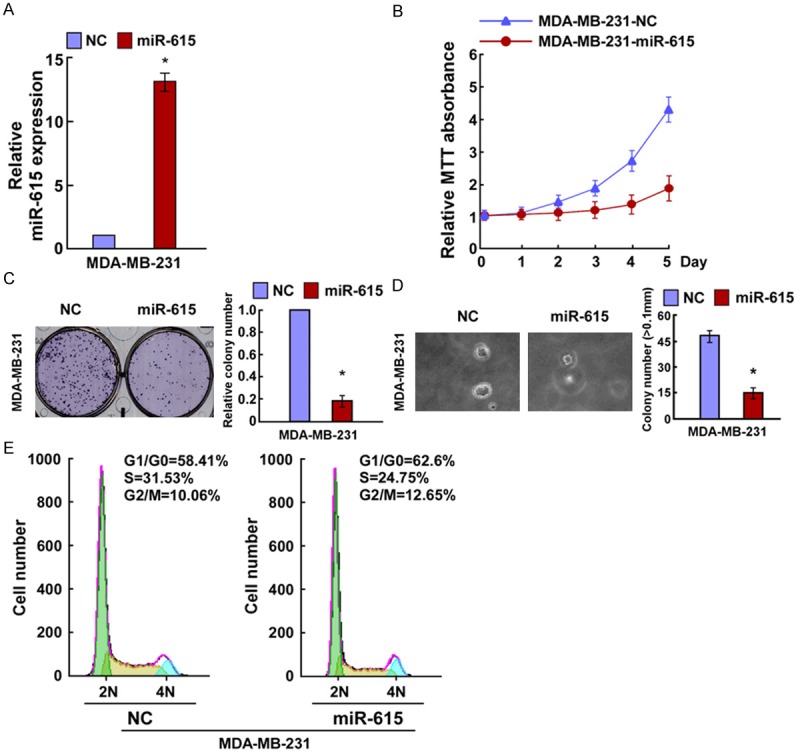
miR-615 upregulation inhibited bre-ast cancer cell proliferation. A. Validation of miR-615 expression levels after transfection by PCR analysis. B. MTT assays revealed that upregulation of miR-615 suppressed growth of MDA-MB231 cells. C. Representative micrographs (left) and quantification (right) of crystal violet-stained cell colonies. D. Upregulation of miR-615 inhibited the anchorage-independent growth of MDA-MB2310 cells. Representative micrographs (left) and quantification of colonies that were > 0.1 mm (right). E. Flow cytometric analysis of the indicated MDA-MB231 cells transfected with NC or miR-615. Each bar represents the mean of three independent experiments. *P < 0.05.
Figure 3.
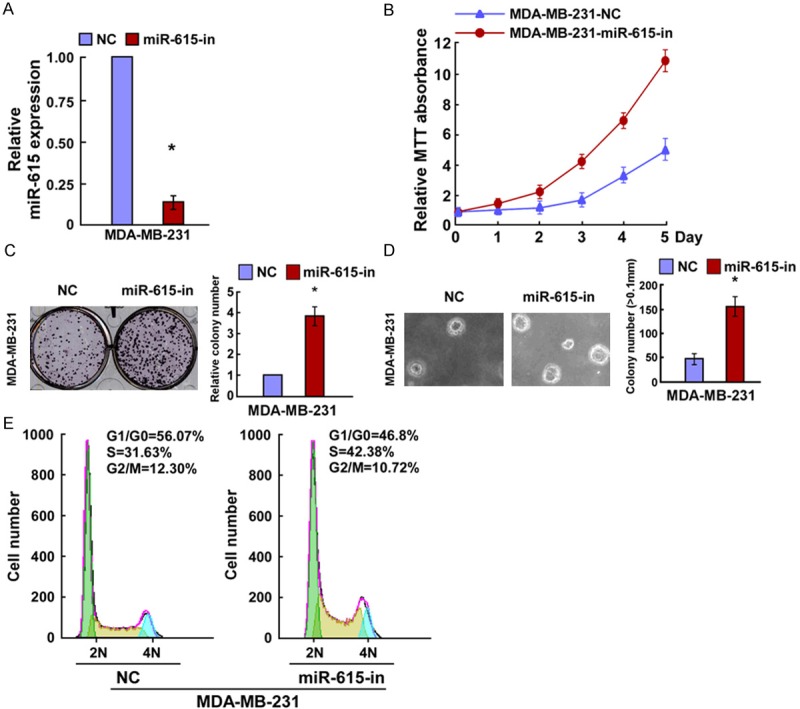
Inhibition of miR-615 promoted breast cancer cell proliferation. A. Validation of miR-615 expression levels after transfection by PCR analysis. B. MTT assays revealed that Inhibition of miR-615 promoted growth of MDA-MB231 cells. C. Representative micrographs (left) and quantification (right) of crystal violet-stained cell colonies. D. Inhibition of miR-615 promoted the anchorage-independent growth of MDA-MB231 cells. Representative micrographs (left) and quantification of colonies that were > 0.1 mm (right). E. Flow cytometric analysis of the indicated MDA-MB231 cells transfected with NC or miR-615-in. Each bar represents the mean of three independent experiments. *P < 0.05.
To further investigate the mechanisms by which miR-615 inhibits breast cancer cells proliferation, we predicted whether miR-615-induced inhibition of cell proliferation resulted from a blocked cell-cycle checkpoint. To confirm this prediction, we analyzed the cell cycle distribution. We found that the percentage of G0/G1 phase was increased in miR-615 overexpressed cells and decreased in its depleted cells, whereas the percentage of S phase was decreased in cells with miR-615 transfection and increased in cells with miR-615-in (Figures 2E and 3E). These results suggested that the miR-615 would initiate its antiproliferation potential at G1-S phase in breast cancer cells, thus preventing further malignancy progression.
MiR-615 inhibits breast cancer cell proliferation and cell cycle via directly targeting AKT2
To uncover the mechanisms by which miR-615 induced G1-S arrest, we searched for the target genes of miR-615. Potential target of miR-615 was predicted using TargetScan 6.2, we found that AKT2 was a potential target of miR-615 (Figure 4A). To test whether miR-615 expression affected endogenous AKT2 expression, expression of AKT2 were detected in the MDA-MB-231 cells, which were transfected with miR-615 mimics, miR-615-in or the respective controls. As predicted, western blotting analysis showed that miR-615 mimics markedly suppressed AKT2 protein levels in MDA-MB-231 cells (Figure 4B), while miR-615-in clearly promoted AKT2 protein expression. To validate whether AKT2 was the direct target gene of miR-615, a Dual-Luciferase Reporter System containing wild-type 3’-UTR of AKT2 was used. The luciferase assay showed that miR-615 significantly led to the suppression of luciferase activity (Figure 4C), indicating that miR-615 directly bound to its predicted binding site on AKT2. Meanwhile, miR-615-mut had no effect on the luciferase activity of AKT2 3’-UTR wild type. These results, taken together, demonstrated that AKT2 is a bona fide target of miR-615.
Figure 4.
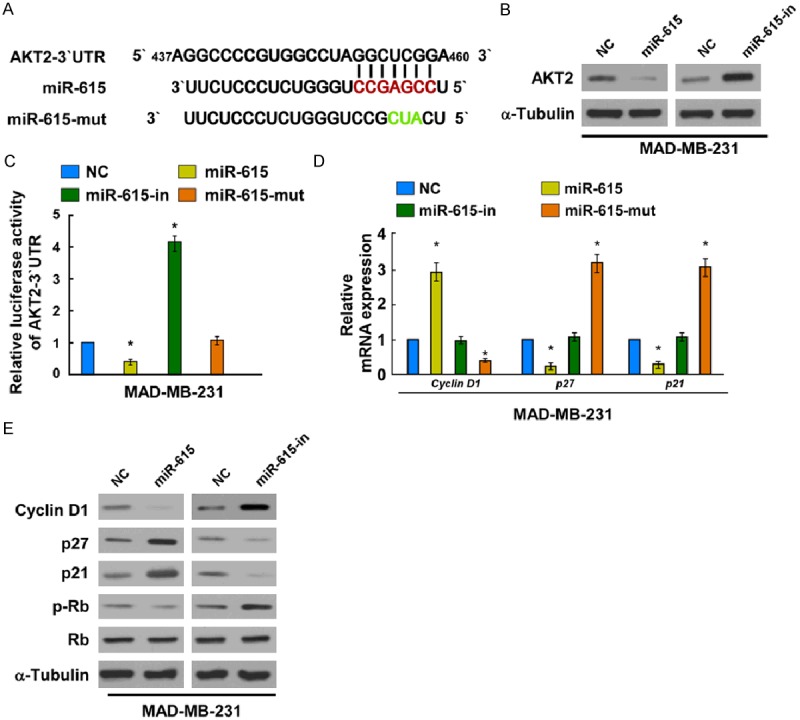
MiR-615 suppresses AKT2 expression by directly targeting the AKT2 3’-UTR and altered levels of proteins related to cell proliferation and cell cycle in MDA-MB231 cells. A. Predicted miR-615 target sequence in the 3’-UTR of AKT2 (AKT2-3’-UTR) and positions of three mutated nucleotides in the 3’-UTR of miR-615 (miR-615-mut). B. AKT2 protein expression in MDA-MB231 cells transfected with miR-615 or the miR-615 inhibitor were detected by Western blotting analysis. α-Tubulin served as the loading control. C. Luciferase reporter assay of MDA-MB231 cells transfected with the pGL3-AKT2-3’-UTR reporter and miR-615 or miR-615-in or miR-615-mut or NC with oligonucleotides. D. Real-time PCR analysis of expression of Cyclin D1, p27 and p21 in MDA-MB231 cells. E. Cyclin D1, p27, p21, phosphorylated retinoblastoma (p-Rb) and Rb were measured by western blot in MDA-MB231 cells. α-Tubulin was used to serve as the loading control. *P < 0.05.
MiR-615 altered levels of proteins related to cell proliferation and cell cycle in breast cancer
It was reported previously that AKT2 is closely correlated with cell proliferation and cell cycle that remarkably regulates the expression of its downstream genes [9-11]. As miR-615 inhibited cell proliferation of breast cancer, we examined its functions on expression of the genes which regulate the G1/S transition, including the CDK inhibitors p21 and p27, the CDK regulator Cyclin D1, Rb and p-Rb. Our results suggested that p21 and p27 were strikingly upregulated at both the protein and mRNA levels while cyclin D1 levels were downregulated in miR-615-overexpressing MDA-MB-231 cells, compared to NC transfected cells (Figure 4D and 4E). Cyclin D1 exerts its functions by binding cyclin-dependent kinases (CDKs) subunit 4 and 6 to form a complex, which results in successive phosphorylation of the Rb protein [12]. The phosphorylation of retinoblastoma (pRb) levels was decreased in miR-615-overexpressing cells while increased in the miR-615-inhibited cells (Figure 4E). These results provides further evidence that miR-615 plays an important role in breast cancer cell proliferation. Altogether, our results indicated that miR-615 functionally modulates cell cycle regulators, p21, p27, cyclin D1, Rb and p-Rb, thus relevant to cell proliferation and cell cycle.
Discussion
Accumulating evidences have demonstrated that miRNAs has been shown to be a common event that can control cell proliferation in development and progression of breast cancer [7,13,14]. In the current study, we found that miR-615 expression was markedly downregulated in both breast cancer tissues and cell lines. Furthermore, ectopic expression of miR-615 inhibited breast cancer cells proliferation by blocking the G1-S transition in vitro, probably through directly targeting AKT2-related cell-cycle signaling. Taken together, our data provided a more comprehensive understanding of the tumor suppressor role of miR-615 during development of breast cancer.
Recently, deregulation of miRNAs in cancer cells and their roles in tumorigenesis have been increasingly investigated. However, the underlying mechanism responsible for decreased expression of miR-615 in breast cancer is still unknown. Thus, in this study, our study revealed that miR-615 was significantly downregulated in breast cancer cell lines and breast carcinoma tissues. And ectopic expression of miR-615 in MDA-MB-231 cells inhibited cell proliferation, clonogenicity, and also induced G1-S arrest in vitro, suggesting miR-615 as a tumor suppressor in breast cancer development.
AKT2 is a serine/threonine kinase, has been found to be associated with numerous other cancers, including colorectal, liver and breast cancers [15-18]. Moreover, previous studies have reported that Akt2 plays a critical role in cell proliferation, survival and apoptosis in various kinds of cancers, including breast cancer [19,20]. AKT2 is essential for progression from G0-G1 to S-phase by inducing the regulator of G1-S transition, including cyclin D1, p21 and p27, during cell-cycle progression [11,21]. However, precise details of the mechanisms which regulate AKT2 remain elusive. Our experimental results further proved that AKT2 was direct targets of miR-615 in breast cancer cells. The activities of AKT2 3’UTR luciferase reporter were responsive to miR-615 overexpression. Endogenous AKT2 protein expression decreased in miR-615-transfected breast cancer cells. The cell regulators were detected by PCR and Western Blots. Our results suggested that both the protein and mRNA levels of p21 and p27 were strikingly upregulated while cyclin D1 levels were downregulated in miR-615-overexpressing MDA-MB-231 cells, compared to NC transfected cells. Our findings suggest that miR-615 may function as a cell-cycle suppressor by targeting AKT2 in breast cancer. Taken together, our results suggested that miR-615 suppressed AKT2 expression by binding directly to the 3’-UTR of AKT2, and thus inducing cell cycle.
In conclusion, the current study revealed that the roles of miR-615 and its targeted gene, AKT2, in their control of the cell cycle and their potential implication in development of breast cancer. Our results revealed that miR-615 may be a novel tumor suppressor that blocks the growth of breast cancer cells through targeting AKT2-related cell-cycle progression signaling pathways. Based on all data, it was inspiring to speculate that miR-615 could be a significant diagnostic and prognostic biomarker for breast cancer.
Acknowledgements
This work was supported by Department of Surgical Oncology, Cangzhou Centerl Hospital. All authors designed the study together, performed the experiment together, analyzed the data and wrote the paper; all authors approved the final manuscript.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics, 2011. CA Cancer J Clin. 2011;61:409–418. doi: 10.3322/caac.20134. [DOI] [PubMed] [Google Scholar]
- 3.Dalmay T. Mechanism of miRNA-mediated repression of mRNA translation. Essays Biochem. 2013;54:29–38. doi: 10.1042/bse0540029. [DOI] [PubMed] [Google Scholar]
- 4.van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 5.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 6.Zhang ZG, Chen WX, Wu YH, Liang HF, Zhang BX. MiR-132 prohibits proliferation, invasion, migration, and metastasis in breast cancer by targeting HN1. Biochem Biophys Res Commun. 2014;454:109–114. doi: 10.1016/j.bbrc.2014.10.049. [DOI] [PubMed] [Google Scholar]
- 7.Shen F, Cai WS, Feng Z, Li JL, Chen JW, Cao J, Xu B. MiR-492 contributes to cell proliferation and cell cycle of human breast cancer cells by suppressing SOX7 expression. Tumour Biol. 2014;36:1913–21. doi: 10.1007/s13277-014-2794-z. [DOI] [PubMed] [Google Scholar]
- 8.Wang XY, Wu MH, Liu F, Li Y, Li N, Li GY, Shen SR. Differential miRNA expression and their target genes between NGX6-positive and negative colon cancer cells. Mol Cell Biochem. 2010;345:283–290. doi: 10.1007/s11010-010-0582-7. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Yao J, Zhang X, Guo B, Le X, Cubberly M, Li Z, Nan K, Song T, Huang C. miRNA-302b suppresses human hepatocellular carcinoma by targeting AKT2. Mol Cancer Res. 2014;12:190–202. doi: 10.1158/1541-7786.MCR-13-0411. [DOI] [PubMed] [Google Scholar]
- 10.Ding Z, Xu F, Li G, Tang J, Tang Z, Jiang P, Wu H. Knockdown of Akt2 Expression by ShRNA Inhibits Proliferation, Enhances Apoptosis, and Increases Chemosensitivity to Paclitaxel in Human Colorectal Cancer Cells. Cell Biochem Biophys. 2014;71:383–8. doi: 10.1007/s12013-014-0209-9. [DOI] [PubMed] [Google Scholar]
- 11.Chou Y, Lin HC, Chen KC, Chang CC, Lee WS, Juan SH. Molecular mechanisms underlying the anti-proliferative and anti-migratory effects of folate on homocysteine-challenged rat aortic smooth muscle cells. Br J Pharmacol. 2013;169:1447–1460. doi: 10.1111/bph.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajabi HN, Takahashi C, Ewen ME. Retinoblastoma Protein and MyoD Function Together to Effect the Repression of Fra-1 and in Turn Cyclin D1 during Terminal Cell Cycle Arrest Associated with Myogenesis. J Biol Chem. 2014;289:23417–23427. doi: 10.1074/jbc.M113.532572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng Y, Zou Q, Liu T, Cai X, Huang Y, Pan J. microRNA335 inhibits proliferation, cellcycle progression, colony formation, and invasion via targeting PAX6 in breast cancer cells. Mol Med Rep. 2015;11:379–385. doi: 10.3892/mmr.2014.2684. [DOI] [PubMed] [Google Scholar]
- 14.Fu P, Du F, Yao M, Lv K, Liu Y. MicroRNA-185 inhibits proliferation by targeting c-Met in human breast cancer cells. Exp Ther Med. 2014;8:1879–1883. doi: 10.3892/etm.2014.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao HJ, Ren LL, Wang ZH, Sun TT, Yu YN, Wang YC, Yan TT, Zou W, He J, Zhang Y, Hong J, Fang JY. MiR-194 deregulation contributes to colorectal carcinogenesis via targeting AKT2 pathway. Theranostics. 2014;4:1193–1208. doi: 10.7150/thno.8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Guo X, Xiong L, Yu L, Li Z, Guo Q, Li Z, Li B, Lin N. Comprehensive analysis of microRNA-regulated protein interaction network reveals the tumor suppressive role of microRNA- 149 in human hepatocellular carcinoma via targeting AKT-mTOR pathway. Mol Cancer. 2014;13:253. doi: 10.1186/1476-4598-13-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009;2:ra62. doi: 10.1126/scisignal.2000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fohlin H, Perez-Tenorio G, Fornander T, Skoog L, Nordenskjold B, Carstensen J, Stal O. Akt2 expression is associated with good long-term prognosis in oestrogen receptor positive breast cancer. Eur J Cancer. 2013;49:1196–1204. doi: 10.1016/j.ejca.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Clark AR, Toker A. Signalling specificity in the Akt pathway in breast cancer. Biochem Soc Trans. 2014;42:1349–1355. doi: 10.1042/BST20140160. [DOI] [PubMed] [Google Scholar]
- 20.Chin YR, Yuan X, Balk SP, Toker A. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer Discov. 2014;4:942–955. doi: 10.1158/2159-8290.CD-13-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu YH, Chang CC, Yang NJ, Lee YH, Juan SH. RhoA-mediated inhibition of vascular endothelial cell mobility: positive feedback through reduced cytosolic p21 and p27. J Cell Physiol. 2014;229:1455–1465. doi: 10.1002/jcp.24583. [DOI] [PubMed] [Google Scholar]