

Abstract
Wilson’s disease (WD) is a rare inherited disorder of copper metabolism and the main manifestations are liver and brain disorders. Hemolytic anemia is an unusual complication of WD. We describe a 15-year-old girl who developed hemolytic anemia as the first manifestation of Wilson’s disease. An Arg952Lys mutation was found in exon 12 of the ATP7B gene, which is uncommon among Chinese Han individuals. From this case and reviews, we can achieve a better understanding of WD. Besides, we may conclude that the probable diagnosis of WD should be considered in young patients with unexplained hemolytic anemia, especially in patients with hepatic and/or neurologic disorder.
Keywords: Hemolytic anemia, Wilson’s disease, ATP7B
Introduction
Wilson’s disease (WD, hepatolenticular degeneration, HLD) is an autosomal recessive disorder of copper transport and is caused by mutations in the ATP7B gene, which is located on the long arm of chromosome 13 (13q14.1) consisting of 21 exons and encodes copper-transporting P-type adenosine triphosphatase [1,2]. WD is characterized by excessive amounts of copper in the liver, brain, eye and other body tissues, and the main clinical symptoms are usually due to hepatic (42%) or/and neurologic (34%) involvement [3]. Hemolytic anemia is a rare complication. Here we report a case of WD patient whose gene analysis revealed an uncommon mutation of ATP7B, and initial presentation of the disease was hemolytic anemia.
Case report
A 15-year-old girl presented with fatigue and red wine-like urine. A routine blood test showed a hemoglobin level of 89 g/L, and a bone marrow smear supported the diagnosis of hemolytic anemia, accompanied by increased reticulocytes and indirect bilirubin. Urobilinogen was positive. Blood biochemistry showed that liver function was normal with the exception of bilirubin. Blood examination of antinuclear antibodies, hepatitis virus and Coomb’s test were all negative. The blood and bone marrow expression of CD55 and CD59 on the surface of granulocytes and erythrocytes was normal. The patient was diagnosed with Coomb’s test negative autoimmune hemolytic anemia and received dexamethasone at a dose of 10 mg/day in a local hospital. Twelve days later, blood and urine examinations were normal. She was advised to take oral prednisone and the dose was reduced gradually.
One month later, she presented with fatigue and red wine-like urine again. At that time, the daily dose of prednisone was 25 mg. A routine blood test showed that the hemoglobin level was 88 g/L and decreased to 69 g/L within three days. The reticulocytes occupied 10% of erythrocytes, total bilirubin was 74 μmol/L and indirect bilirubin was 27 μmol/L, albumin level was 25.7 g/L, accompanied by coagulation dysfunction. The results of a repeat Coomb’s test, Ham’s test, and CD55 and CD59 tests were all normal. A physical examination revealed edema of the extremities without bleeding. Abdominal ultrasound showed splenomegaly and ascites. The blood red cell morphology examination showed 5% ragged red fibers. She was retreated with dexamethasone at a dose of 10 mg/day. A diuretic and hepatinica were also administered. One week later, she had abnormal spirit, however, her hemoglobin was normal. She initially presented with deliriousness, and then subsequent haziness of spirit/mind, dysarthria, and loss of alertness/cognition. An MR scan of the skull was normal. The patient showed hepatic and neurologic dysfunction. WD was considered, and the patient underwent copper metabolism assessment. Serum ceruloplasmin was 4.69 mg/dL urinary copper was 1236 μg/L, and physical eye examination revealed Kayser-Fleischer rings. According to the scoring system developed at the 8th International Meeting on Wilson’s Disease, Leipzig 2001, the Leipzig score was 9 [4]. She was diagnosed with WD and received penicillamine 1 g/day. One week later, her mental state became normal. Routine blood, liver function and coagulation examinations all returned to normal. During the diagnostic procedure, gene analysis was also performed and a mutation of ATP7B located at exon 12, Arg952Lys, was found (Figure 1). This is an uncommon mutation in Chinese Han individuals.
Figure 1.
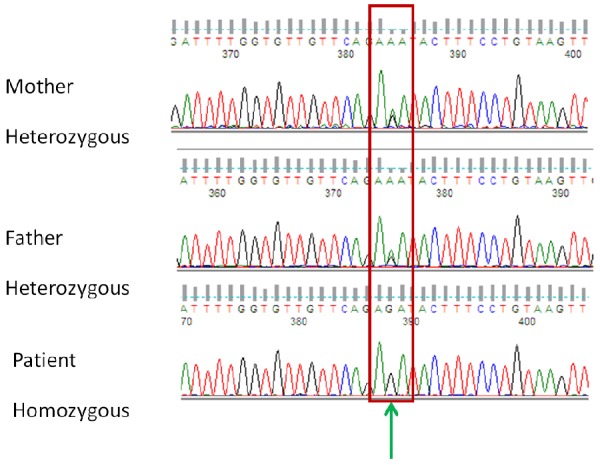
A Chinese patient with Wilson’s disease. A mutation in exon 12 of the ATP7B gene, AAT>AGT Arg952Lys was found, which is a high frequency mutation.
Discussion
WD is a rare inherited disorder of copper metabolism. The incidence of WD is approximately 1 in 35 000-100 000 live births, and the age at onset is usually between 5 and 35 years of age [5]. Hepatic manifestations of WD are more likely to occur in early childhood, while neurological symptoms are more commonly observed in adolescents [6]. Hemolytic anemia is an uncommon complication, especially as the first manifestation of WD. The initial clinical manifestations are hepatic in origin in 42% of cases, neurological in 34%, psychiatric in 10%, hematological and endocrinological in 12%, and renal in 1%. Approximately 25% of patients have evidence of the involvement of more than one organ at presentation [7].
Herein, we report an adolescent patient presenting with hemolytic anemia as the initial manifestation of WD. The patient improved with glucocorticoid therapy. However, this effect was not sustained. Hemolytic anemia recurred and further investigations revealed features of WD, including Kayser-Fleischer rings, low serum ceruloplasmin and high 24 h urinary copper. Based on the diagnostic criteria of AASLD [8], WD was diagnosed.
It is known that hemolysis in WD is due to a deficiency in ceruloplasmin, the copper transport protein which results in excessive inorganic copper in the blood circulation, much of which accumulates in red blood cells (RBCs). Although the exact mechanism is not known, increased copper accumulation in the RBCs may damage the cell membrane, accelerate oxidation of hemoglobin and inactivate enzymes of the pentose phosphate and glycolytic pathways. Coomb’s negative hemolytic anemia associated with WD is mainly due to acute liver failure. Low-grade hemolysis may be present when liver disease is not clinically evident, and may be the initial manifestation of the disease in 1%-11% of cases [8]. High-dose glucocorticoids can be used to protect against acute hemolysis.
WD is known to be caused by abnormalities in the copper-transporting protein encoding gene ATP7B. Over 500 mutations have now been reported in the ATP7B gene in all 21 exons according to the Wilson’s disease mutation database [9]. The most common mutation in European populations is an amino acid substitution in exon 14, p.His1069Gln, and another frequent mutation is located in exon 8 [2,10]. Analysis of eight exons (exons 2, 5, 8, 13, 14, 18, 19 and 20) detected 82% of the mutations found, and mutations in exon 12 were uncommon in the UK population [10]. In Chinese Han individuals, mutations were most frequently found in exon 8 in addition to exons 2, 12, 13, 16 and 18, and the most common mutation was Arg778Leu located in exon 8 [11-13]. In the present case, the patient’s family also underwent gene analysis and found an Arg952Lys mutation in exon 12 of ATP7B, which is an uncommon mutation in Chinese Han individuals. No definite genotype-phenotype correlations have been established so far. However, a few reports have suggested a possible relationship between age at onset or clinical course with a specific genotype. One of the most common mutations, p.H1069Q, may be associated with a later age (around 18 years) at presentation and milder disease phenotype [14,15]. Mutation T1288R may be related to the onset of hemolytic anemia [16]. Our patient with WD showed the genetic mutation, Arg952Lys of ATP7B, first presenting with Coomb’s-negative hemolytic anemia, and later with liver and brain involvement, accompanied by Kaiser-Fleischer rings, and a high response to penicillamine. Thus, there may be a relationship between Coomb’s-negative hemolytic anemia and the Arg952Lys mutation.
In conclusion, WD is a disorder of copper metabolism due to mutation of the ATP7B gene. Many mutations have been identified, and Arg952Lys is an uncommon mutation located in exon 12. Hemolytic anemia may be the presenting feature in some patients with WD, especially in young patients, accompanied by hepatic disorders. The differentiation between Coomb’s negative hemolytic anemia and WD is mainly by the persistent therapeutic effect of glucocorticoids. The treatment is aimed at WD, and glucocorticoids can help patients to get through the hemolytic crisis. More research on the relationship between WD and hematological disorders is needed, especially gene studies. The probable diagnosis of WD should be considered in young patients with unexplained hemolytic anemia, especially in patients with hepatic and/or neurologic disorder. For these patients, treatment with glucocorticoids is inappropriate, while chelation therapy with penicillamine or zinc is effective.
Disclosure of conflict of interest
None.
References
- 1.Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, Brzustowicz LM, Cayanis E, Vitale E, Russo JJ, Matseoane D, Boukhgalter B, Wasco W, Figus AL, Loudianos J, Cao A, Sternlieb I, Evgrafov O, Parano E, Pavone L, Warburton D, Ott J, Penchaszadeh GK, Scheinberg IH, Gilliam TC. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993;5:338–343. doi: 10.1038/ng1293-338. [DOI] [PubMed] [Google Scholar]
- 2.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM, Devoto M, Peppercorn J, Bush AI, Sternlieb I, Pirastu M, Gusella JF, Evgrafov O, Penchaszadeh GK, Honig B, Edelman IS, Soares MB, Scheinberg IH, Gilliam TC. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 3.Grudeva-Popova JG, Spasova MI, Chepileva KG, Zaprianov ZH. Acute hemolytic anemia as an initial clinical manifesta-tion of Wilson’s disease. Folia Med (Plovdiv) 2000;42:42–46. [PubMed] [Google Scholar]
- 4.Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 5.Roberts EA, Schilsky ML. Division of Gastroenterology and Nutrition, Hospital for Sick Children, Toronto, Ontario, Canada. A practical guidelines on Wilson’s disease. Hepatolog. 2003;37:1475–1492. [Google Scholar]
- 6.Kalra V, Khurana D, Mittal R. Wilson’s disease-Early onset and lessons from a pediatric cohort in India. Indian Pediatr. 2000;37:595–601. [PubMed] [Google Scholar]
- 7.Sternlieb I, Schienberg IH. In: Wilson’s disease: Diseases of the Liver. Schiff L, Schiff ER, editors. Philadelphia: Lippincott Company; 1992. pp. 659–668. [Google Scholar]
- 8.Roberts EA, Schilsky ML. American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47:2089–2111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- 9.Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat. 2007;28:1171–1177. doi: 10.1002/humu.20586. [DOI] [PubMed] [Google Scholar]
- 10.Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J, Lo C, Klaffke S, Joyce CJ, Dhawan A, Hadzic N, Mieli-Vergani G, Kirk R, Elizabeth Allen K, Nicholl D, Wong S, Griffiths W, Smithson S, Giffin N, Taha A, Connolly S, Gillett GT, Tanner S, Bonham J, Sharrack B, Palotie A, Rattray M, Dalton A, Bandmann O. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013;136:1476–1487. doi: 10.1093/brain/awt035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu YH, Kodama H, Du SL, Gu QJ, Sun HJ, Ushijima H. Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson’s disease. Clin Genet. 2003;64:479–484. doi: 10.1046/j.1399-0004.2003.00179.x. [DOI] [PubMed] [Google Scholar]
- 12.Mak CM, Lam CW, Tam S, Lai CL, Chan LY, Fan ST, Lau YL, Lai ST, Yuen P, Hui J, Fu CC, Wong KS, Mak WL, Tze K, Tong SF, Lau A, Leung N, Hui A, Cheung KM, Ko CH, Chan YK, Ma O, Chau TN, Chiu A, Chan YW. Mutational analysis of 65 Wilson disease patients in Hong Kong Chinese: identification of 17 novel mutations and its genetic heterogeneity. J Hum Genet. 2008;53:55–63. doi: 10.1007/s10038-007-0218-2. [DOI] [PubMed] [Google Scholar]
- 13.Li K, Zhang WM, Lin S, Wen L, Wang ZF, Xie D, Wei M, Qiu ZQ, Dai Y, Lin MC, Kung HF, Yao FX. Mutational analysis of ATP7B in north Chinese patients with Wilson disease. J Hum Genet. 2013;58:67–72. doi: 10.1038/jhg.2012.134. [DOI] [PubMed] [Google Scholar]
- 14.Panagiotakaki E, Tzetis M, Manolaki N, Loudianos G, Papatheodorou A, Manesis E, Nousia-Arvanitakis S, Syriopoulou V, Kanavakis E. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B) Am J Med Genet A. 2004;131:168–173. doi: 10.1002/ajmg.a.30345. [DOI] [PubMed] [Google Scholar]
- 15.Cocoş R, Şendroiu A, Schipor S, Bohîlţea LC, Şendroiu I, Raicu F. Genotype-phenotype correlations in a mountain population community with high prevalence of Wilson’s disease: genetic and clinical homogeneity. PLoS One. 2014;9:e98520. doi: 10.1371/journal.pone.0098520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leggio L, Addolorato G, Loudianos G, Abenavoli L, Lepori MB, Vecchio FM, Rapaccini GL, De Virgiliis S, Gasbarrini G. A new mutation of Wilson’s disease P-type ATPase gene in a patient with cirrhosis and coombs-positive hemolytic anemia. Dig Dis Sci. 2006;51:34–38. doi: 10.1007/s10620-006-3080-8. [DOI] [PubMed] [Google Scholar]