

Abstract
Recognition of cytosolic DNA initiates a series of innate immune responses by inducing IFN-I production and subsequent triggering JAK1-STAT1 signaling which plays critical roles in the pathogenesis of infection, inflammation and autoimmune diseases through promoting B cell activation and antibody responses. The stimulator of interferon genes protein (STING) has been demonstrated to be a critical hub of type I IFN induction in cytosolic DNA-sensing pathways. However, it still remains unknown whether cytosolic DNA can directly activate the JAK1-STAT1 signaling or not. And the role of STING is also unclear in this response. In the present study, we found that dsDNA directly triggered the JAK1-STAT1 signaling by inducing phosphorylation of the Lyn kinase. Moreover, this response is not dependent on type I IFN receptors. Interestingly, STING could inhibit dsDNA-triggered activation of JAK1-STAT1 signaling by inducing SHP-1 and SHP-2 phosphorylation. In addition, compared with normal B cells, the expression of STING was significantly lower and the phosphorylation level of JAK1 was significantly higher in B cells from MRL/lpr lupus-prone mice, highlighting the close association between STING low-expression and JAK1-STAT1 signaling activation in B cells in autoimmune diseases. Our data provide a molecular insight into the novel role of STING in dsDNA-mediated inflammatory disorders.
Keywords: B cells, double-stranded DNA, JAK1-STAT1 signaling, SHP-1/2, Stimulator of interferon genes protein
INTRODUCTION
The presence of DNA in endosome or the cytosol is a danger signal for the innate immune system. These DNA molecules, including exogenous DNA derived from invading pathogens, endogenous inappropriately aggregated self-DNA and even possibly cellular retrotransposons, can be recognized by DNA sensing systems to initiate innate immune responses (Ahn et al., 2012; Burdette and Vance, 2013; Paludan and Bowie, 2013). Multiple studies have shown that recognition of cytosolic DNA induces IFN-I production and indirectly activates the JAK1-STAT1 signaling which plays critical roles in the pathogenesis of infection, inflammation and autoimmune diseases by promoting B cell activation and antibody responses (Ishii et al., 2006; Le Bon et al., 2001; Stetson and Medzhitov, 2006; Swanson et al., 2010).
A recently identified adaptor protein, the stimulator of interferon genes protein (STING), exhibits a vital role in DNA-triggered signaling pathways (Ishikawa and Barber, 2008; Sun et al., 2009; Zhong et al., 2008). STING can activate the innate immune responses via induction of IFN-I through a well-characterized pathway involving TBK1 and interferon regulatory factor 3 (IRF3) (Burdette and Vance, 2013). A series of studies have established the essential roles of STING in innate immune responses (Chen et al., 2011; Ishikawa and Barber, 2008; Watson et al., 2012). Although DNA can indirectly activate the JAK1-STAT1 signaling by inducing the production of type I IFN, it remains unknown whether DNA can directly activate the JAK1-STAT1 signaling and the role of STING plays in this process.
In the canonical JAK1-STAT1 signaling, binding of IFN-α to its receptor results in phosphorylation of IFN-α receptor 1 (IFNAR1) and subsequent association of phosphorylated receptor complex with signal transducer and activator of transcription (STATs) 1 and 2 (Velazquez et al., 1992; Yan et al., 1996). The phosphorylated STAT1-STAT2 heterodimer translocates to nucleus and complexes with IFN-regulatory factor 9 (IRF9) to form a specific transcription complex IFN-stimulated gene factor 3 (ISGF3) (Darnell et al., 1994). ISGF3 binds to a highly conserved cis-acting DNA sequence called IFN-stimulated response element (ISRE) sequence (AGTTTCNNTTTCNC, N can represent any nucleotide) in the promoters of IFN-stimulated genes (ISGs) (Platanias, 2005). Representative ISGs encodes proteins include interferon-induced proteins with tetratricopeptide repeats 1 (IFIT1), myxovirus resistance protein 1 (MX1), 2′,5′ -oligoadenylate synthetase 1 (OAS1), and interferon-induced protein 44 (IFI44). IFIT1 is involved in many processes of cellular functions, including regulation of transcription, translation and inflammatory response (Diamond and Farzan, 2013; Fensterl and Sen, 2011). MX1 antagonizes the replication process of several different RNA and DNA viruses (Haller et al., 2007). OAS1, as a member of the 2′,5′-oligoadenylate synthetase family, is the central in innate immune system detection of cytoplasmic double-stranded RNA (dsRNA) and promotion of host antiviral responses (Justesen et al., 2000; Vachon et al., 2015). Mutations in this gene have been associated with host susceptibility to viral infection. IFI44 contributes to the interferon-induced inhibition of bunyamwera orthobunyavirus replication (Carlton-Smith and Elliott, 2012; Hallen et al., 2007).
In particular, many proteins and tyrosine phosphatases, such as SHP-1, SHP-2 and Lyn, are implicated in the regulation of JAK1-STAT1 signaling (Alexander and Hilton, 2004; Biron et al., 1989; Bunde et al., 2005). SHP-1 has been shown to inhibit tyrosine phosphorylation of JAK kinases following their recruitment to receptor complexes (Klingmuller et al., 1995). SHP-2 can bind JAK1 and JAK2, and directly dephosphorylates JAKs (Yin et al., 1997). The Lyn kinase is able to influence the phosphorylation of JAK and STAT proteins (Al-Shami and Naccache, 1999; Simon et al., 1997).
As is well known, the activation of JAK1-STAT1 signaling plays a critical role in the pathogenesis of systemic lupus erythematosus (SLE), a typical autoimmune disease (Mathian et al., 2011; Uccellini et al., 2008). B cells from both patients with SLE and MRL/lpr mice display a higher activation level of JAK1-STAT1 signaling (Becker et al., 2013). Notably, dsDNA plays a vital role in the pathogenesis of SLE through triggering the innate immune activation and promoting the auto-reactive Ig production (Cohen et al., 2002; Frese and Diamond, 2011; Vinuesa and Goodnow, 2002). Interestingly, recent studies show that deletion of STING does not prevent the autoantibody production in DNaseII−/−/IFNAR−/− mice (Baum et al., 2015). Moreover, another study show that STING plays a negative role in the pathogenesis of SLE and STING deficiency leads to increased autoantibody production (Sharma et al., 2015). These findings hint that STING may play a negative role in regulating the antibody responses in B cells. Considering the important role of JAK1-STAT1 signaling in regulating antibody responses in B cells, it is essential to investigate the association between STING and the activation of JAK1-STAT1 signaling in B cells.
We report here that STING negatively regulates the activation of JAK1-STAT1 signaling directly triggered by dsDNA. We found that dsDNA could directly activate the JAK1-STAT1 signaling by inducing the phosphorylation of the Lyn kinase, whereas STING inhibited this response by phosphorylating SHP-1 and SHP-2. Furthermore, we demonstrated that STING expression in B cells from both patients with SLE and MRL/lpr mice was significantly lower than that from healthy donors and wild-type mice, respectively. These results reveal a critical role of STING in regulating dsDNA-triggered activation of the JAK1-STAT1 signaling in B cells and highlight the close associations of STING low-expression with JAK1-STAT1 signaling activation in SLE B cells.
MATERIAL AND METHODS
Isolation of human peripheral blood mononuclear cells
Whole blood was obtained with written informed consent from each patient and healthy subject. All SLE patients were diagnosed according to the criteria set out by American College of Rheumatology revised criteria in 1997. Disease activity was evaluated using the SLE Disease Activity Index (SLEDAI) with a cutoff of ≥ 8 that was used to define active disease. For flow cytometric analysis, 2 ml whole blood of each person were recruited from eight healthy subjects with a mean age of 28 ± 6 years and eight SLE patients with a mean age of 28 ± 7 years. For B cells culture, 200 ml whole blood of healthy subjects were recruited. Human peripheral blood mononuclear cells (PBMCs) were separated from plasma by Ficoll centrifugation (Lymphoprep, Nycomed, Oslo, Norway) according to the standard procedures. The study protocol was approved by the research ethics committee of Nanjing University.
Purification of human CD19+ B cells
B cells were purified from PBMCs by labeling cells with CD19 microBeads and positively selecting CD19+ B cells (Miltenyi Biotec, Germany). The purity of B cells was always above 97%. For in vitro experiments, isolated human CD19+ B cells were cultured in RPMI 1640 medium containing 10% FBS and stimulated with TLR7 ligand R848 (1 μg/ml, Enzo Life Sciences International), TLR9 ligand CpG-2006S (0.3 μM, Invitrogen), AffiniPure F(ab’)2 Fragment Goat Anti-human IgM (10 μg/ml, Jackson ImmunoResearch Laboratories) or human IFN-α (1000 U/ml, Millipore). The study protocol was approved by the research ethics committee of Nanjing University.
Mice
Female C57BL/6 mice, which were used to isolate B cells, were 6–8 weeks old and obtained from Model Animal Research Center at Nanjing University. Female C57BL/6 mice (n = 10) and MRL/lpr lupus-prone mice (n = 9), 6–8 weeks old, were also obtained from Model Animal Research Center at Nanjing University and sacrificed at 22–24 weeks old. All mice were maintained under specific pathogen-free conditions. All experiments were conducted in accordance with institutional guidelines for animal care and performed based on the Guide for the Animal Care Committee at Nanjing University.
Purification of murine splenic B cells
Lymphocytes of spleen were isolated by ficoll density centrifugation according to standard procedures. B cells were purified using the mouse B cell Isolation Kit (Miltenyi Biotec, USA) and the purity of obtained B cells was always above 92 %. Purified B cells were cultured in RPMI 1640 medium containing 10 % FBS and stimulated with TLR7 ligand R848 (1 μg/ml, Enzo Life Sciences International), TLR9 ligand CpG-1826 (0.3 μM, Invitrogen), AffiniPure F(ab′)2 Fragment Goat Anti-mouse IgM (10 μg/ml, Jackson ImmunoResearch Laboratories) or mouse IFN-α (1000 U/ml, eBioscience).
Cell culture
Human B cell line BJAB and Daudi, and human T cell line Jurkat were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum. Human HeLa cell line and the HEK293T cell line were cultured in DMEM (Gibco, USA) supplemented with 10% fetal bovine serum. Human mesenchymal stem cells were cultured in DMEM/F12 (Gibco, USA) supplemented with 10% fetal bovine serum. All the cells were maintained in 5% CO2 at 37°C.
Transfection
The nucleofector device (Amaxa, Germany) and cell line kit V (Amaxa Biosystems, USA) were used to transfect poly(dA:dT) (Sigma, USA), poly(dG:dC) (Invivogen, USA) or ISD (Invivogen, USA) into BJAB and Jurkat cells. Transfection efficiency, as determined by green fluorescent protein expression in parallel plates, varied between 50% and 60%. Transfection of HeLa, MSCs and HEK293T cells with poly(dA:dT), poly(dG:dC) or ISD was done using Lipofectamine 2000 (Invitrogen, USA) according to standard procedures.
Reverse transcription PCR (RT-PCR) and quantitative real-time PCR (qPCR) analysis
Total RNA was extracted using TRIZOL (Invitrogen, USA) according to the manufacturer’s instructions. Q-PCR assays of mRNA were carried out on a StepOne Plus real-time polymerase chain reaction system or ABI Vii 7 detection system (Applied Biosystems, USA) using SYBR Green PCR Master Mix. The 2−ΔΔCt method was used for real-time PCR gene expression analysis. All quantification data are presented as a ratio to the GAPDH level.
Western blot analysis
Proteins were extracted using lysis buffer, and the concentration of each sample was detected using the BCA protein assay kit (Pierce Chemical, USA). Appropriate amount of proteins (60 μg) were electrophoresed on SDS polyacrylamide gels with tris-glycine running buffer and electrically transferred onto polyvinylidene difluoride membranes (MilliporeCorp, USA). After blocking with 3% (w/v) bovine serum albumin (BSA) in tris buffered saline (TBS)/Tween-20 (Bio-Rad Laboratories, USA) for 1 h, the membranes were washed and then incubated with primary antibodies against phos-STAT1, total STAT1, phos-JAK1, total JAK1, Lyn, phos-SHP-1, total SHP-1, phos-SHP-2, total SHP-2 and STING (Cell Signaling Technology, USA, all dilution at 1:1,000) over night at 4°C. After washing, the membranes were incubated with the secondary antibody AffiniPure Goat Anti-Rabbit IgG(H+L) purchased from Beyotime Institute of Biotechnology (China) (dilution at 1:3,000) for 1 h at room temperature. The ECL Plus western blotting detection reagents (Millipore, USA) were used to visualize protein expressions. The antibody to the control protein GAPDH (dilution at 1:1,000) was purchased from Cell Signaling Technology (USA). Integrated density values were then calculated using an AlphaI-mager 3400 (Alpha Innotech).
Phenotype and intracellular protein analysis by flow cytometry
For phenotype staining, cells were washed twice with phosphate buffered saline (PBS) containing 1% FBS and 0.1% NaN3. Then cells were incubated for 30 min at 4°C with human CD19-APC antibody (Miltenyi Biotec, USA) according to the standard procedure. For intracellular staining, cells were washed twice with PBS containing 1% FBS and 0.1% NaN3. Then cells were resuspended with pulse vortex and 100 μl IC Fixation Buffer (eBioscience, USA) was added. After incubating at 4°C for 20 min in the dark, cells were washed twice with 2 ml Permeabilization Buffer (eBioscience, USA) and centrifuged. Next, intracellular staining was performed in 100 μl of Permeabilization Buffer by using STING Ab (dilution at 1:100, Abcam). After incubating for 20 min in the dark at room temperature, cells were incubated with FITC-conjugated F(ab′)2 Donkey Anti-mouse IgG H&L antibody (dilution at 1:2,000, Miltenyi Biotec, USA) for 20 min in dark at room temperature. After washing for twice with 2 ml Permeabilization Buffer, cells were analyzed by FACS Calibur (Becton Dickinson). An isotype control was used for each antibody. Data analysis was performed using the FlowJo (TreeStar, USA).
Luciferase assay
BJAB cells were co-transfected with ISRE luciferase reporter plasimd (2 μg, Promega, USA) and renilla luciferase reporter plasmids (0.2 μg, Promega, USA) for 24 h and then transfected with poly(dA:dT), poly(dG:dC) or ISD. The cells were harvested in luciferase assay lysis buffer 18 h later, and the luciferase activity was measured according to the manufacturer’s protocol (Promega, USA). Values for luciferase activity were normalized to renilla luciferase activity. In all cases, data represent the average values of triplicate samples normalized to co-transfected renilla luciferase activity and are expressed as a percentage of the control stimulated sample value.
Plasmid construct and transfection
Human STING cDNA was PCR-amplified using the primer pair: FP: 5-CCCAAGCTTGATGCCCCACTCCAGCCT-3, RP: 5-CCGCTCGAGCTCAAGAAATCCGTGC-3. The product was cloned into the BamH1/HindIII sites of pcDNA3.1-Myc-His (Invitrogen, USA), resulting in STING-containing pcDNA3.1. Then STING-containing pcDNA3.1 plasmid (2 μg) was transfected into BJAB cells using the nucleofector device (Amaxa, Germany) and cell line kit V (Amaxa Biosystems, USA). Empty vector were transfected as matched control. At 36th h after transfection, cells were harvested and used for subsequent experiments.
Small interfering RNA (siRNA)
The siRNA sequences used for silencing STING, Lyn, AIM2, cGAS, LRRFIP1 and ZBP1 were purchased from RIBOBIO (China). The nucleofector device (Amaxa, Germany) and cell line kit V (Amaxa Biosystems, USA) were used to transfect siRNA duplexes into BJAB cells. Negative control of siRNA was transfected as matched control. Cells were harvested at 36th hour after transfection and used for subsequent experiments.
Statistical analysis
All values in the graphs were given as means plus or minus standard error of the mean (SEM). Significance was assessed by using the Student Neuman-Keuls multiple comparison tests for unequal replications. Student’s t-test was used to compare the two groups. P values < 0.05 were considered significant.
RESULTS
dsDNA directly activates the JAK1-STAT1 signaling
Previous studies have shown that dsDNA indirectly activates the JAK1-STAT1 signaling by promoting the production of type I IFN (Burdette and Vance, 2013). To explore whether dsDNA can directly activate the JAK1-STAT1 signaling in B cells, we transfected the synthetic dsDNA analogs, including poly(dA:dT) (B-form DNA), poly(dG:dC) (Z-form DNA) and ISD (interferon stimulatory DNA), into the human B cell line BJAB cells for 6 h. As shown in Fig. 1A, transfection with poly(dA:dT), poly(dG:dC) or ISD could induce considerable expression of IFIT1, MX1, OAS1 and IFI44. In addition, dsDNA also induced the expression of IFIT1, MX1, OAS1 and IFI44 in another human B cells line Daudi cell (Data not shown). To investigate whether this phenomenon is widespread in different cell types, we transfected poly(dA:dT), poly(dG:dC) and ISD into Jurkat, hMSC, HEK293T and HeLa cells, and detected the expression of IFIT1, MX1, OAS1 and IFI44. As shown in Supplementary Fig. S1, transfection with poly(dA:dT), poly(dG:dC) or ISD could also significantly induce considerable expression of downstream genes of JAK1-STAT1 signaling pathway in Jurkat, hMSC, HEK293T and HeLa cells. These results suggest that dsDNA can indeed induce the expression of JAK1-STAT1 signaling pathway downstream genes in different types of cells.
Fig. 1.
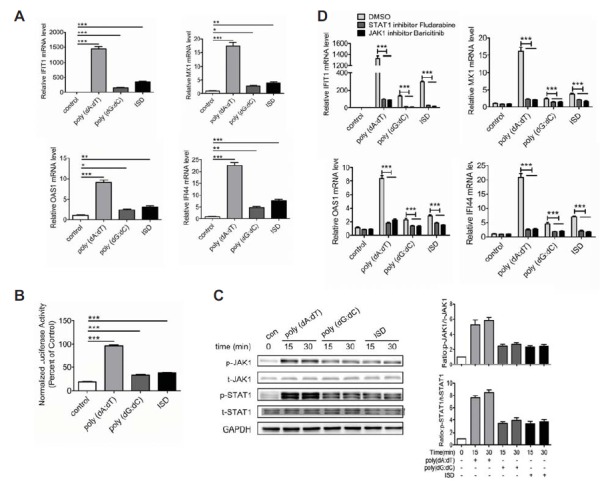
dsDNA directly triggers the activation of JAK1-STAT1 signaling. (A) BJAB cells were tansfected with poly(dA:dT) (2 μg/ml), poly(dG:dC) (2 μg/ml) or ISD (2 μg/ml) for 6 h and the expression levels of IFIT1, IFI44, MX1 and OAS1 were detected by qPCR. (B) BJAB cells were tansfected with ISRE-luciferase reporter for 36 h and then tansfected with poly(dA:dT), poly(dG:dC) or ISD for 18 h. Luciferase assay analysis of the luciferase reporter gene activity. (C) BJAB cells were tansfected with poly(dA:dT), poly(dG:dC) or ISD for 15 min and 30 min. The phosphorylation of JAK1 and STAT1 were detected by Western blot. (D) BJAB cells were treated with JAK1 inhibitor Baricitinib or STAT1 inhibitor Fludarabine for 1 h prior to tansfection with poly(dA:dT), poly(dG:dC) or ISD for 6 h. The expression levels of IFIT1, IFI44, MX1 and OAS1 were detected by qPCR. The data shown represent the means of three independent experiments, and the error bars represent the s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001; ns denotes p > 0.05.
To investigate whether the induction of JAK1-STAT1 signaling downstream genes by dsDNA is related to the activation of JAK1-STAT1 signaling, we next detected the effect of poly(dA:dT), poly(dG:dC) and ISD on ISRE-luciferase reporter gene activity. BJAB cells were transfected with ISRE-luciferase activity reporter for 36 h and then transfected with poly(dA:dT), poly(dG:dC) or ISD. The ISRE-luciferase reporter gene activity was detected at 18th hour. Expectedly, the ISRE-luciferase reporter gene activity was significantly enhanced by transfection with poly(dA:dT), poly(dG:dC) or ISD (Fig. 1B). Moreover, we detected the phosphorylation levels of JAK1 and STAT1 in the cells transfected with poly(dA:dT), poly(dG:dC) or ISD. Strikingly, the phosphorylation levels of JAK1 and STAT1 increased rapidly at 15 min and 30 min (Fig. 1C), indicating that dsDNA can directly trigger the activation of the JAK1-STAT1 signaling in B cells.
To further confirm this phenomenon, BJAB cells were treated with STAT1 inhibitor Fludarabine and JAK1 inhibitor Baricitinib for 1 h prior to transfection with poly(dA:dT), poly(dG:dC) or ISD. Intriguingly, both of the STAT1 inhibitor Fludarabine and the JAK1 inhibitor Baricitinib significantly reduced dsDNA-induced expression of IFIT1, MX1, OAS1 and IFI44 (Fig. 1D). All of these results suggest that dsDNA can directly activate the JAK1-STAT1 signaling in B cells.
dsDNA activates the JAK1-STAT1 signaling through Lyn and in an interferon α/β receptors-independent manner
Considering that poly(dA:dT) shows a better effect on activating the JAK1-STAT1 signaling compared with poly(dG:dC) and ISD, we focused on exploring the mechanism of poly(dA:dT)-activated JAK1-STAT1 signaling in this study. As is known, type I IFN binds to the interferon α/β receptors (IFNAR) and stimulates JAK1 and STAT1 tyrosine phosphorylation (Darnell, 1997; Platanias, 2005). To evaluate the effect of transactivation by IFNAR on poly(dA:dT)-activated JAK1-STAT1 signaling, BJAB cells were pre-incubated with the anti-IFNAR neutralizing antibody for 30 min prior to transfected with poly(dA:dT). As shown in Supplementary Fig. S2, the neutralizing antibody could significantly inhibit IFN-α-induced expression of IFIT1, MX1, OAS1 and IFI44, while it didn’t affect poly(dA:dT)-induced expression of IFIT1, MX1, OAS1 and IFI44 (Fig. 2A). Moreover, IFN-α-induced phosphorylation of JAK1 was abrogated in cells treated with the neutralizing antibody, whereas there was no inhibitory effect of the antibody on poly(dA:dT)-induced phosphorylation of JAK1 (Fig. 2B). These data indicate that poly(dA:dT)-triggered activation of JAK1-STAT1 signaling is independent of IFNAR.
Fig. 2.
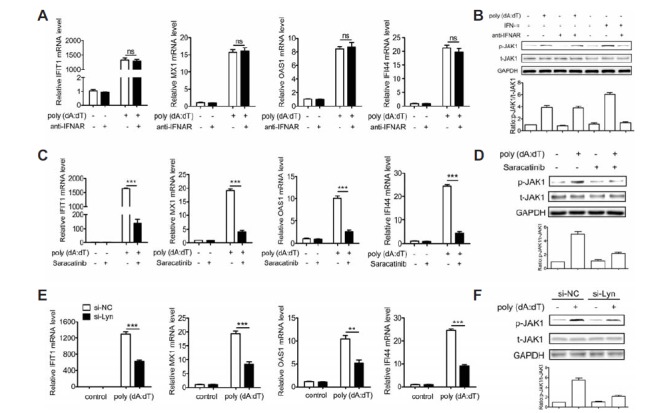
dsDNA-induced activation of the JAK1-STAT1 signaling requires Lyn and is independent of IFNAR. (A, B) BJAB cells were treated with the neutralizing antibody for the IFN-α/β receptor (Merck & Millipore) for 30 min prior to transfection with poly(dA:dT) or stimulation of IFN-α for 30 min or 6 h. The expression levels of IFIT1, MX1, OAS1 and IFI44 were detected at 6 h by qPCR (A). The phosphorylation of JAK1 was detected at 30 min by Western blot (B). (C, D) BJAB cells were treated with Lyn inhibitor Saracatinib for 1 h prior to tansfection with poly(dA:dT) for 30 min or 6 h. The expression levels of IFIT1, IFI44, MX1 and OAS1 were detected at 6 h by qPCR (C). The phosphorylation of JAK1 was detected at 30 min by Western blot (D). (E, F) BJAB cells were tansfected with si-Lyn or si-NC for 36 h and then tansfected with poly(dA:dT) for 30 min or 6 h. The expression levels of IFIT1, MX1, OAS1 and IFI44 were detected at 6 h by qPCR (E). The phosphorylation of JAK1 and STAT1 were detected at 30 min by Western blot (F). The data shown represent the means of three independent experiments, and the error bars represent the s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001; ns denotes p > 0.05.
To investigate the mechanism of poly(dA:dT) on activating the JAK1-STAT1 signaling, we focused on the Src family kinase Lyn, which has been shown to influence the phosphorylation of the JAK and STAT proteins (Simon et al., 1997). BJAB cells were pretreated with Lyn inhibitor Saracatinib for 1 h and then transfected with poly(dA:dT). As shown in Fig. 2C, the expression of IFIT1, MX1, OAS1 and IFI44 induced by poly(dA:dT) was significantly inhibited by Saracatinib. Moreover, poly(dA:dT)-induced phosphorylation of JAK1 was also significantly inhibited by Saracatinib (Fig. 2D). To further confirm the role of Lyn in regulating poly(dA:dT)-activated JAK1-STAT1 signaling, the expression of Lyn was down-regulated using siRNA in BJAB cells (Supplementary Fig. S3). BJAB cells were transfected with si-Lyn or si-NC for 36 h and then transfected with poly(dA:dT). Strikingly, Lyn knockdown could not only significantly inhibit poly(dA:dT)-induced expression of IFIT1, IFI44, MX1 and OAS1 but also significantly inhibit the phosphorylation of JAK1 triggered by poly(dA:dT) (Figs. 2E and 2F). Together with the results presented in Fig. 1, these results provide evidence that poly(dA:dT)-triggered activation of JAK1-STAT1 signaling requires Lyn and is independent of IFNAR.
STING inhibits the activation of JAK1-STAT1 signaling triggered by dsDNA
STING is known to control the induction of cytokines by aberrant cytosolic DNA species (Burdette and Vance, 2013; Ishikawa and Barber, 2008). Considering the importance of STING in mediating DNA recognition, we next queried the role of STING in dsDNA-triggered activation of the JAK1-STAT1 signaling. We down-regulated STING expression in BJAB cells using siRNA against STING (Supplementary Figs. S4A and S4B). BJAB cells were transfected with si-STING or si-NC for 36 h and then transfected with poly(dA:dT). Interestingly, STING knockdown significantly promoted the expression of IFIT1, IFI44, MX1 and OAS1 (Fig. 3A), and phosphorylation of JAK1(Fig. 3B) triggered by poly(dA:dT), relative to the response in control cells.
Fig. 3.
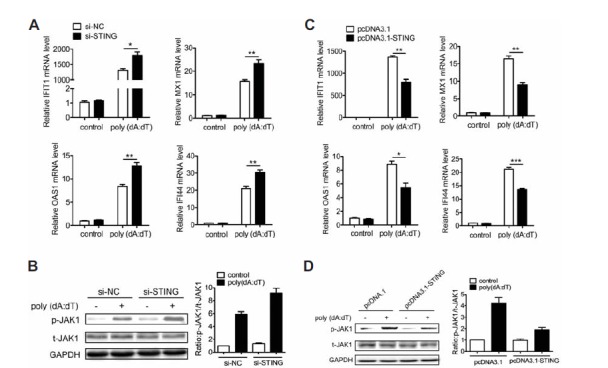
STING inhibits dsDNA-induced activation of the JAK1-STAT1 signaling. (A, B) BJAB cells were tansfected with si-STING or si-NC for 36 h and then tansfected with poly(dA:dT) for 30 min or 6 h. The expression levels of IFIT1, MX1, OAS1 and IFI44 were detected at 6 h by qPCR (A). The phosphorylation of JAK1 was detected at 30 min by Western blot (B). (C, D) BJAB cells were tansfected with pcDNA3.1-STING or control plasmid for 36 h and then tansfected with poly(dA:dT) for 30 min or 6 h. The expression levels of IFIT1, IFI44, MX1 and OAS1 were detected at 6 h by qPCR (C). The phosphorylation of JAK1 and STAT1 were detected at 30 min by western blot (D). The data shown represent the means of three independent experiments, and the error bars represent the s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001; ns denotes p > 0.05.
To further investigate the role of STING in regulating dsDNA-activated JAK1-STAT1 signaling, we over-expressed STING in BJAB cells by transfecting the pcDNA3.1-STING plasmid into the cells (Supplementary Figs. S4C and S4D). BJAB cells were transfected with pcDNA3.1-STING plasmid or control for 36 h and then transfected with poly(dA:dT). Consistently, over-expression of STING could not only significantly inhibited poly(dA:dT)-induced expression of IFIT1, IFI44, MX1 and OAS1 but also inhibited poly(dA:dT)-induced phosphorylation of JAK1, relative to the response in control cells (Figs. 3C and 3D). Since several DNA sensors, such as AIM2, cGAS, LRRFIP1 and ZBP1, can recognize poly(dA:dT) and activate STING (Chiu et al., 2009; Hornung et al., 2009; Takaoka et al., 2007; Yang et al., 2010), we next investigated the role of AIM2, cGAS, LRRFIP1 and ZBP1 in poly(dA:dT)-triggered activation of JAK1-STAT1 signaling. The expression of AIM2, cGAS, LRRFIP1 and ZBP1 were down-regulated by siRNA (Supplementary Fig. S5A). To investigate the effect of AIM2, cGAS, LRRFIP1 and ZBP1 knockdown on poly(dA:dT)-triggered activation of JAK1-STAT1 signaling, BJAB cells were transfected with siRNA against AIM2, cGAS, LRRFIP1 or ZBP1 for 48 h prior to transfection with poly(dA:dT). Expectedly, knockdown of AIM2, cGAS, LRRFIP1 or ZBP1 promoted poly(dA:dT)-induced expression of IFIT1, IFI44, MX1 and OAS1, relative to the response in control cells (Supplementary Fig. S5B). Moreover, phosphorylation of JAK1 triggered by poly(dA:dT) was also promoted by knockdown of AIM2, cGAS, LRRFIP1 or ZBP1, relative to the response in control cells (Supplementary Fig. S5C). All of these results suggest that STING plays a negative role in regulating the activation of JAK1-STAT1 signaling triggered by dsDNA.
STING inhibits dsDNA-activated JAK1-STAT1 signaling by inducing SHP-1 and SHP-2 phosphorylation
Since SHP-1 and SHP-2 have been shown to directly dephosphorylate JAKs (Yin et al., 1997), we next explored the role of SHP-1 and SHP-2 in STING-mediated inhibition of the activation of JAK1-STAT1 signaling. BJAB cells were transfected with siRNA against STING or control for 36 h and then transfected with poly(dA:dT) for 15 min. Intriguingly, poly(dA:dT) significantly induced the phosphorylation of SHP-1 and SHP-2, whereas STING knockdown inhibited the phosphorylation of SHP-1 and SHP-2 induced by poly(dA:dT) (Fig. 4A). These results indicate that STING can promote the phosphorylation of SHP-1 and SHP-2 triggered by poly(dA:dT).
Fig. 4.
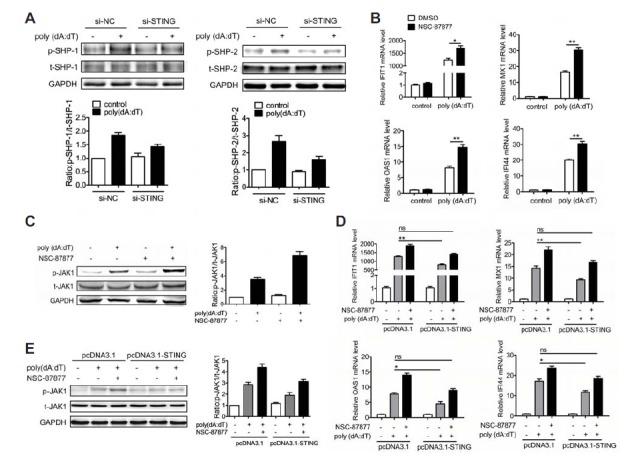
STING inhibits dsDNA-activated JAK1-STAT1 signaling by phosphorylation of SHP-1/2. (A) BJAB cells were tansfected with si-STING or si-NC for 36 h and then tansfected with poly(dA:dT) for 15 min. The phosphorylation of SHP-1/2 was detected by Western blot. (B, C) BJAB cells were treated with SHP-1/2 inhibitor for 30 min prior to tansfection with poly(dA:dT). The expression levels of IFIT1, MX1, OAS1 and IFI44 were detected at 6 h by qPCR (B). The phosphorylation of JAK1 was detected at 30 min by Western blot (C). (D, E) BJAB cells were tansfected with pcDNA3.1-STING or control plasmid for 36 h. Then the cells were treated with SHP-1/2 inhibitor NSC-87877 for 30 min prior to tansfection with poly(dA:dT). The expression levels of IFIT1, MX1, OAS1 and IFI44 were detected at 6 h by qPCR (D). The phosphorylation of JAK1 was detected at 30 min by Western blot (E). The data shown represent the means of three independent experiments, and the error bars represent the s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001; ns denotes p > 0.05.
Next we evaluated the effect of SHP-1 and SHP-2 on regulating poly(dA:dT)-activated JAK1-STAT1 signaling. BJAB cells were treated with SHP-1/2 inhibitor NSC-87877 for 1 h prior to transfection with poly(dA:dT). Strikingly, NSC-87877 not only promoted poly(dA:dT)-induced expression of IFIT1, MX1, OAS1 and IFI44 (Fig. 4B) but also increased the phosphorylation level of JAK1 induced by poly(dA:dT) (Fig. 4C), indicating that SHP-1/2 is involved in dsDNA-triggered activation of JAK1-STAT1 signaling.
Since STING can promote the phosphorylation of SHP-1/2 which are involved in poly(dA:dT)-activated JAK1-STAT1 signaling, we next investigated whether the function of STING on inhibiting the activation of JAK1-STAT1 signaling depends on SHP-1/2. BJAB cells were transfected with pcDNA3.1-STING plasmid or control for 36 h and then treated with NSC087877 for 1 h prior to transfection with poly(dA:dT). As shown in Fig. 4D, NSC-87877 markedly reversed the inhibition function of STING on poly(dA:dT)-induced expression of IFIT1, MX1, OAS1 and IFI44. Furthermore, NSC-87877 also significantly reversed the inhibition function of STING on poly(dA:dT)-induced phosphorylation of JAK1 (Fig. 4E). All of these results indicate that SHP-1/2 are required for STING to inhibit the activation of JAK1-STAT1 signaling triggered by poly(dA:dT).
STING is low-expressed in B cells from patients with SLE and MRL/lpr mice
Since STING plays a negative role in dsDNA-triggered activation of JAK1-STAT1 signaling in B cells, we next detected the expression of STING in B cells from patients with SLE and MRL/lpr mice. PBMCs from patients with SLE and healthy donors were collected and the expression of STING in CD19+ B cells was detected by intracellular FACS analysis. As shown in Fig. 5A, patients with SLE showed a lower expression level of STING in B cells compared with healthy donors. We also detected the expression of STING in splenic B cells from MRL/lpr mice and wild-type mice by qPCR. Consistently, MRL/lpr mice showed a lower expression level of STING (Fig. 5B). These results demonstrate that the expression of STING is down-regulated in B cells from patients with SLE and MRL/lpr mice. Furthermore, we detected the phosphorylation of JAK1, SHP-1 and SHP-2 in B cells isolated from spleen of MRL/lpr mice and wild-type mice. Intriguingly, compared with wild-type mice, MRL/lpr mice showed a significantly higher level of p-JAK1 and lower phosphorylation levels of SHP-1 and SHP-2 in B cells (Fig. 5C).
Fig. 5.
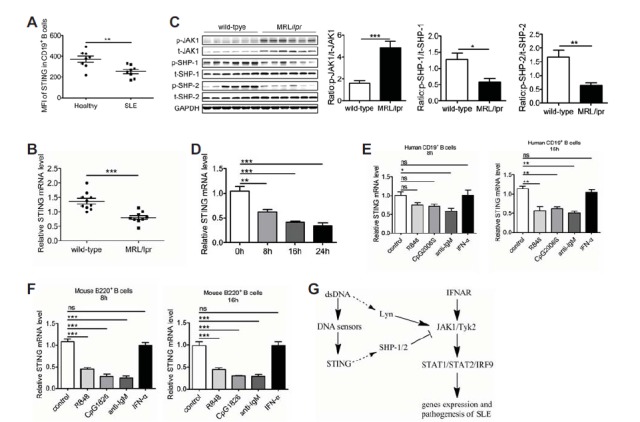
Low-expression of STING in B cells from patients with SLE and MRL/lpr mice. (A) FACS analysis of STING expression in B cells from patients with SLE (n = 8) and healthy donors (n = 8). (B) qPCR analysis of the expression of STING in B cells from MRL/lpr mice (n = 9) and wild-type mice (n = 10). (C) B cells were isolated from spleen of MRL/lpr mice (n = 6) and wild-type mice (n = 6). Western blot analysis of the phosphorylation levels of SHP-1/2 and JAK1. (D) BJAB cells were transfected with poly(dA:dT) for 8 h,16 h and 24 h. The expression of STING was analyzed by qPCR. (E) Isolated human CD19+ B cells were stimulated with TLR7 ligand R848 (1 μg/ml), TLR9 ligand CpG2006S (0.5 μM), human anti-IgM (10 μg/ml) and human IFN-α (1,000 U/ml) for 8 h and 16 h. The expression of STING was detected by qPCR. (F) Isolated murine splenic B cells were stimulated with TLR7 ligand R848 (1 μg/ml), TLR9 ligand CpG1826 (0.5 μM), mouse anti-IgM (10 μg/ml) and mouse IFN-α (1,000 U/ml) for 8 h and 16 h. The expression of STING was detected by qPCR. (G) Proposed regulatory pathway activated by dsDNA in B cells. The data shown represent the means of three independent experiments, and the error bars represent the s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001; ns denotes p > 0.05.
To identify the molecular events leading to STING down-regulation in SLE B cells, we first detected the effect of dsDNA on STING expression. BJAB cells were transfected with poly(dA:dT) for different times and then STING expression was detected. As shown in Fig. 5D, transfection with poly(dA:dT) could significantly down-regulate the expression of STING. TLR7-, TLR9-, BCR- and IFN-α-mediated signaling pathways were aberrantly activated in B cells from patients with SLE and lupus-prone mice (Baechler et al., 2003; Bennett et al., 2003; Christensen et al., 2006; Lau et al., 2005), we then detected the effect of TLR7-, TLR9-, BCR- and IFN-α-mediated signaling pathways on STING expression. Human CD19+ B cells were stimulated with TLR7 ligand R848, TLR9 ligand CpG2006S, BCR ligand anti-human IgM F(ab’)2 fragments and human IFN-α, and then the expression of STING was detected using qPCR. As shown in Fig. 5E, the expression of STING was significantly down-regulated by R848, CpG2006S and anti-human IgM F(ab’)2 fragments rather than IFN-α at 16 h. What’s more, we also detected the effect of TLR7-, TLR9-, BCR- and IFN-α-mediated signaling pathways on regulating STING expression in murine B cells. Murine B cells were stimulated with R848, CpG1826, anti-mouse IgM F(ab’)2 fragments and mouse IFN-α, and then STING expression was detected using qPCR. Consistently, STING was significantly down-regulated by R848, CpG1826 and anti-mouse IgM F(ab’)2 fragments rather than IFN-α (Fig. 5F). These findings indicate that dsDNA, TLR7-, TLR9- and BCR-mediated signaling pathways maybe related to the low-expression of STING in B cells from patients with SLE and MRL/lpr mice. These results may highlight the close associations of STING low-expression with the JAK1-STAT1 signaling in B cells and the pathogenesis of systemic lupus erythematosus. Taken together, dsDNA directly activate the JAK1-STAT1 signaling via Lyn, while STING inhibits dsDNA-activated JAK1-STAT1 signaling by phosphorylating SHP-1/2 (Fig. 5G). Low-expression of STING in B cells from patients with SLE and MRL/lpr mice may contribute to the activation of JAK1-STAT1 signaling in B cells and the pathogenesis of SLE.
DISCUSSION
dsDNA can trigger the production of type I IFN through STING and subsequently activate the JAK1-STAT1 signaling indirectly (Barber, 2011; Ishikawa et al., 2009). However, it remains unknown whether dsDNA can directly activate the JAK1-STAT1 signaling and the role of STING plays in this response. The data presented here address a negative role of STING in regulating the activation of JAK1-STAT1 signaling directly triggered by dsDNA. dsDNA can directly activate the JAK1-STAT1 signaling via Lyn, whereas STING inhibits this response by phosphorylating SHP-1/2.
The JAK1-STAT1 signaling is involved in a wide variety of intracellular responses, including cell differentiation and cell death, in addition to innate immunity (Bromberg et al., 1998; Wang et al., 2010). Thus, the regulation of JAK1-STAT1 signaling is complicated and remains to be elucidated. Given the importance of the JAK1-STAT1 signaling and dsDNA in the pathogenesis of SLE, we asked if dsDNA can directly activate the JAK1-STAT1 signaling. In this study, we transfected synthetic dsDNA poly(dA:dT), poly(dG:dC) and ISD into the BJAB cells and found that dsDNA could significantly induce the phosphorylation of JAK1 and STAT1 at 15 min and 30 min, indicating that dsDNA can directly trigger the activation of JAK1-STAT1 signaling in B cells.
In this study, dsDNA was transfected into B cells. As is known, genetic and environmental factors appear to contribute to the pathogenesis of SLE. They include a myriad of protagonists such as ultraviolet light (UVB), drugs, pollutants, vaccinations, smoking, vitamin D deficiency and microbial agents (Dedeoglu, 2009; Javierre et al., 2011; Sarzi-Puttini et al., 2005; Shoenfeld et al., 2009). Multiple bacterial and viral infections, a major cause of morbidity and mortality in SLE, have been documented in SLE both at presentation and during the course of the illness (Gourley and Miller, 2007; Perez-Mercado and Vila-Perez, 2010; Sfriso et al., 2010). In common, they may use mechanisms that interact or modify innate and acquired immune responses that could facilitate autoimmunity. It is plausible that chronic infection by microorganisms or self-DNA released from necrotic or incompletely apoptosed cells, or possibly even cellular retrotransposons, may activate innate immune pathways which cause inflammation-aggravated autoimmunity (Ahn et al., 2014; Crow, 2007; Tsokos, 2011). The presence of self or foreign DNA in the cytoplasm is sensed by eukaryotic cells as a danger signal or a sign of foreign invasion (Barbalat et al., 2011). DNA can be introduced into the cytoplasm by bacterial or viral infection, transfection, or ‘leakage’ from the nucleus or mitochondria under some pathological conditions that cause autoimmune diseases such as lupus (Wu et al., 2013). Thus, we used the method that transfecting dsDNA directly into B cells in this study.
We have detected the expression of type I interferon production in BJAB cells upon dsDNA transfection. Transfection with poly(dA:dT) could not induce expression of IFN-α and IFN-β in BJAB cells, while it could induce the expression of IFN-α and IFN-β in human mesenchymal stem cell. Considering that BJAB cells are the human B-lymphoma cell line, we think that it is more adequate to explore the effect of dsDNA on type I interferon production in primary B cells. However, transfection efficiency of primary B cells is too low to explore the effect of dsDNA on type I interferon production.
With the use of Lyn inhibitor Saracatinib and siRNA against Lyn, we showed that poly(dA:dT)-induced activation of the JAK1-STAT1 signaling requires Lyn. However, further studies are needed to do to explore the mechanism of phosphorylation of Lyn triggered by dsDNA. To investigate whether dsDNA-triggered activation of the JAK1-STAT1 signaling is dependent on type I IFN receptors, we added a neutralizing antibody targeting type I IFN receptors to the culture medium. The blockade of type I IFN receptors resulted in suppression of IFN-α-mediated JAK1 phosphorylation, suggesting that the neutralizing antibody targeting IFNAR had sufficient activity to suppress IFNAR-dependent signaling. The neutralizing antibody targeting type I IFN receptor had no effect on dsDNA-activated JAK1-STAT1 signaling indicating that dsDNA directly activated the JAK1-STAT1 signaling in an IFNAR-independent manner.
Previous studies have shown that STING can indirectly promote the activation of JAK1-STAT1 signaling by promoting the production of type I IFN (Burdette and Vance, 2013). Interestingly, we found that STING inhibited the activation of JAK1-STAT1 signaling triggered by dsDNA. What’s more, the silencing of DNA sensors, including AIM2, cGAS, LRRFIP1 and ZBP1, could promote the activation of JAK1-STAT1 signaling triggered by dsDNA. These results indicate that dsDNA-induced activation of the JAK1-STAT1 signaling does not depend on the DNA sensor-STING pathway which plays a negative role in dsDNA-activated JAK1-STAT1 signaling. This reveals dichotomous roles for STING in either stimulating or suppressing autoimmunity.
It has been shown that STING participates in the pathogenesis of many inflammatory diseases such as carcinogenesis, embryonic death and experimental autoimmune encephalitis (Ahn et al., 2014; Lemos et al., 2014). Here, we first reported that STING was low-expressed in B cell from patients with SLE and MRL/lpr mice. Given the negative role of STING in the activation of JAK1-STAT1 signaling triggered by dsDNA in B cells, low-expression of STING in B cells from patients with SLE and MRL/lpr mice may contribute to the pathogenesis of SLE by augmenting the activation of the JAK1-STAT1 signaling. What’s more, we also found that BCR-, TLR7- and TLR9-mediated signaling pathways could down-regulate the expression of STING in human and murine primary B cells. As is known, the BCR-, TLR7- and TLR9- mediated signaling pathways are highly activated in SLE B cells (Baechler et al., 2003; Bennett et al., 2003; Christensen et al., 2006; Lau et al., 2005), we think that these signaling pathways may contribute to the activation of JAK1-STAT1 signaling by down-regulating STING expression.
In conclusion, we identified the role of STING in regulating the activation of JAK1-STAT1 signaling directly triggered by dsDNA. This may be useful in underscoring the role of STING in the pathogenesis of SLE and identifying novel targets for SLE therapy.
Supplemental Data
Acknowledgments
This work was supported by grants from the National Natural Science Foundation (Number: 31370899), the ministry of science and China postdoctoral science foundation (Number: 2013M531331) and the Scientific Research Foundation of Graduate School of Nanjing University (Number: 2013CL11).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. USA. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J. Immunol. 2014a;193:4634–4642. doi: 10.4049/jimmunol.1401337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun. 2014b;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shami A, Naccache PH. Granulocyte-macrophage colony-stimulating factor-activated signaling pathways in human neutrophils. Involvement of Jak2 in the stimulation of phosphatidylinositol 3-kinase. J. Biol. Chem. 1999;274:5333–5338. doi: 10.1074/jbc.274.9.5333. [DOI] [PubMed] [Google Scholar]
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 2011;23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, Marshak-Rothstein A, Gravallese EM. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase iI-deficient mice. J. Immunol. 2015;194:873–877. doi: 10.4049/jimmunol.1402573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, Li QZ, Lian Y, Wu T, Reimold AM, et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS One. 2013;8:e67003. doi: 10.1371/journal.pone.0067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Fan Z, Brown C, Mendelsohn J, Darnell JE., Jr Epidermal growth factor-induced growth inhibition requires Stat1 activation. Cell Growth Differ. 1998;9:505–512. [PubMed] [Google Scholar]
- Bunde T, Kirchner A, Hoffmeister B, Habedank D, Hetzer R, Cherepnev G, Proesch S, Reinke P, Volk HD, Lehmkuhl H, et al. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J. Exp. Med. 2005;201:1031–1036. doi: 10.1084/jem.20042384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- Carlton-Smith C, Elliott RM. Viperin, MTAP44, and protein kinase R contribute to the interferon-induced inhibition of Bunyamwera Orthobunyavirus replication. J. Virol. 2012;86:11548–11557. doi: 10.1128/JVI.01773-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y, et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011;147:436–446. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MK. Type I interferon in systemic lupus erythematosus. Curr. Top. Microbiol. Immunol. 2007;316:359–386. doi: 10.1007/978-3-540-71329-6_17. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Dedeoglu F. Drug-induced autoimmunity. Curr. Opin. Rheumatol. 2009;21:547–551. doi: 10.1097/BOR.0b013e32832f13db. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013;13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterl V, Sen GC. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 2011;31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese S, Diamond B. Structural modification of DNA--a therapeutic option in SLE? Nat. Rev. Rheumatol. 2011;7:733–738. doi: 10.1038/nrrheum.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley M, Miller FW. Mechanisms of disease: Environmental factors in the pathogenesis of rheumatic disease. Nat. Clin. Pract. Rheumatol. 2007;3:172–180. doi: 10.1038/ncprheum0435. [DOI] [PubMed] [Google Scholar]
- Hallen LC, Burki Y, Ebeling M, Broger C, Siegrist F, Oroszlan-Szovik K, Bohrmann B, Certa U, Foser S. Antiproliferative activity of the human IFN-alpha-inducible protein IFI44. J. Interferon Cytokine Res. 2007;27:675–680. doi: 10.1089/jir.2007.0021. [DOI] [PubMed] [Google Scholar]
- Haller O, Kochs G, Weber F. Interferon, Mx, and viral countermeasures. Cytokine Growth Factor Rev. 2007;18:425–433. doi: 10.1016/j.cytogfr.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discov. Med. 2011;12:535–545. [PubMed] [Google Scholar]
- Justesen J, Hartmann R, Kjeldgaard NO. Gene structure and function of the 2′-5′-oligoadenylate synthetase family. Cell. Mol. Life Sci. 2000;57:1593–1612. doi: 10.1007/PL00000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001;14:461–470. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, Pacholczyk G, Barber GN, Hayakawa Y, Munn DH, et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J. Immunol. 2014;192:5571–5578. doi: 10.4049/jimmunol.1303258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathian A, Gallegos M, Pascual V, Banchereau J, Koutouzov S. Interferon-alpha induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZBxNZW)F1 mice but not in BALB/c mice. Eur. J. Immunol. 2011;41:863–872. doi: 10.1002/eji.201040649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mercado AE, Vila-Perez S. Cytomegalovirus as a trigger for systemic lupus erythematosus. J. Clin. Rheumatol. 2010;16:335–337. doi: 10.1097/RHU.0b013e3181f4cf52. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Sarzi-Puttini P, Atzeni F, Iaccarino L, Doria A. Environment and systemic lupus erythematosus: an overview. Autoimmunity. 2005;38:465–472. doi: 10.1080/08916930500285394. [DOI] [PubMed] [Google Scholar]
- Sfriso P, Ghirardello A, Botsios C, Tonon M, Zen M, Bassi N, Bassetto F, Doria A. Infections and autoimmunity: the multifaceted relationship. J. Leukoc. Biol. 2010;87:385–395. doi: 10.1189/jlb.0709517. [DOI] [PubMed] [Google Scholar]
- Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, Huyler AH, Nundel K, Mohan C, Berg LJ, et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc. Natl. Acad. Sci. USA. 2015;112:E710–E717. doi: 10.1073/pnas.1420217112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoenfeld N, Amital H, Shoenfeld Y. The effect of melanism and vitamin D synthesis on the incidence of autoimmune disease. Nat. Clin. Pract. Rheumatol. 2009;5:99–105. doi: 10.1038/ncprheum0989. [DOI] [PubMed] [Google Scholar]
- Simon HU, Yousefi S, Dibbert B, Levi-Schaffer F, Blaser K. Anti-apoptotic signals of granulocyte-macrophage colony-stimulating factor are transduced via Jak2 tyrosine kinase in eosinophils. Eur. J. Immunol. 1997;27:3536–3539. doi: 10.1002/eji.1830271256. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CL, Wilson TJ, Strauch P, Colonna M, Pelanda R, Torres RM. Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response. J. Exp. Med. 2010;207:1485–1500. doi: 10.1084/jem.20092695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Tsokos GC. Systemic lupus erythematosus. N. Engl. J. Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- Uccellini MB, Busconi L, Green NM, Busto P, Christensen SR, Shlomchik MJ, Marshak-Rothstein A, Viglianti GA. Autoreactive B cells discriminate CpG-rich and CpG-poor DNA and this response is modulated by IFN-alpha. J. Immunol. 2008;181:5875–5884. doi: 10.4049/jimmunol.181.9.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon VK, Calderon BM, Conn GL. A novel RNA molecular signature for activation of 2′-5′ oligoadenylate synthetase-1. Nucleic Acids Res. 2015;43:544–552. doi: 10.1093/nar/gku1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Goodnow CC. Immunology: DNA drives autoimmunity. Nature. 2002;416:595–598. doi: 10.1038/416595a. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang Y, Sharma N, Tarasova NI, Timofeeva OA, Winkler-Pickett RT, Tanigawa S, Perantoni AO. STAT1 activation regulates proliferation and differentiation of renal progenitors. Cell. Signal. 2010;22:1717–1726. doi: 10.1016/j.cellsig.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Krishnan K, Greenlund AC, Gupta S, Lim JT, Schreiber RD, Schindler CW, Krolewski JJ. Phosphorylated interferon-alpha receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. EMBO J. 1996;15:1064–1074. [PMC free article] [PubMed] [Google Scholar]
- Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y, Cao X. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat. Immunol. 2010;11:487–494. doi: 10.1038/ni.1876. [DOI] [PubMed] [Google Scholar]
- Yin T, Shen R, Feng GS, Yang YC. Molecular characterization of specific interactions between SHP-2 phosphatase and JAK tyrosine kinases. J. Biol. Chem. 1997;272:1032–1037. doi: 10.1074/jbc.272.2.1032. [DOI] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.