

Abstract
Over the last 30 years, Hanwoo has been selectively bred to improve economically important traits. Hanwoo is currently the representative Korean native beef cattle breed, and it is believed that it shared an ancestor with a Chinese breed, Yanbian cattle, until the last century. However, these two breeds have experienced different selection pressures during recent decades. Here, we whole-genome sequenced 10 animals each of Hanwoo and Yanbian cattle (20 total) using the Illumina HiSeq 2000 sequencer. A total of approximately 3.12 and 3.07 billion sequence reads were mapped to the bovine reference sequence assembly (UMD 3.1) at an average of approximately 10.71- and 10.53-fold coverage for Hanwoo and Yanbian cattle, respectively. A total of 17,936,399 single nucleotide polymorphisms (SNPs) were yielded, of which 22.3% were found to be novel. By annotating the SNPs, we further retrieved numerous nonsynonymous SNPs that may be associated with traits of interest in cattle. Furthermore, we performed whole-genome screening to detect signatures of selection throughout the genome. We located several promising selective sweeps that are potentially responsible for economically important traits in cattle; the PPP1R12A gene is an example of a gene that potentially affects intramuscular fat content. These discoveries provide valuable genomic information regarding potential genomic markers that could predict traits of interest for breeding programs of these cattle breeds.
Keywords: Hanwoo, Signature of selection, SNP, Whole-genome sequencing, Yanbian cattle
INTRODUCTION
Over the last 100 years, selective breeding has contributed to significant genetic improvement in economically important traits, including meat, and milk traits in diverse cattle breeds worldwide. Hanwoo (HAN) is an indigenous cattle breed in Korea and it has been intensively bred for beef during over the past 30 years, leading to genetic improvement in production traits (Lee et al., 2014). Yanbian cattle (YAN) are mainly found in the Korean autonomous province of Yanbian as well as northeast provinces in China (Cheng, 1984; Kim et al., 1981). It is presumed that HAN and YAN shared an ancestor until the last century. Although there are a limited number of publications available (Dadi et al., 2014; Lee et al., 2014), the migration routes of HAN and YAN remain to be further clarified. It is generally accepted that HAN may have been introduced to the northeast areas in China along with Koreans who moved into the area around 100 years ago. YAN are used as draft animals as were HAN until the middle of the last century, but YAN have recently attracted attention as a promising beef cattle breed in China.
In cattle, genome-wide genetic variants have been intensively catalogued since the completion of the bovine sequencing project (Elsik et al., 2009). Of the genetic variants, single nucleotide polymorphisms (SNPs) have been the most widely used to identify genes or genomic regions that are responsible for phenotypic variations in cattle (Lu et al., 2013; Utsunomiya et al., 2013). In particular, numerous SNPs have been catalogued using the bovine reference genome assemblies (Liu et al., 2009; Zimin et al., 2009) and whole-genome resequencing (WGS) of multiple cattle breeds (Eck et al., 2009; Kawahara-Miki et al., 2011; Stothard et al., 2011). Furthermore, WGS-derived SNPs were successfully applied in genome-wide association studies and produced higher accuracy predictions of economically important traits such as dairy traits (Daetwyler et al., 2014) as well as detection of signatures of selection throughout the genome (Qanbari et al., 2014). Currently, four Korean native cattle breeds are registered with the Domestic Animal Diversity Information Service through the Food and Agriculture Organization of the United Nations: Hanwoo, Chikso, Heugu, and Jeju Heugu (FAO, 2014). With recent advances in WGS technology, a representative individual per each of the four breeds was recently resequenced and yielded a substantial number of SNPs (Choi et al., 2013a; 2013b; 2014; Lee et al., 2013). However, there are overall few publications available that investigated the native genome resources at the population level.
The objective of this study is two-fold. First, we whole-genome resequenced a total of 10 individuals each of HAN and YAN (20 total) to detect numerous genetic variants that could be used in further breeding programs. To our knowledge, this is the first study to whole-genome resequence YAN. Second, using the SNPs derived from this study, we scanned the whole genome to identify selective sweeps that may potentially have arisen as a result of recent selection of HAN and YAN.
MATERIALS AND METHODS
Library construction and sequencing
We generated whole-genome resequencing data from HAN (N = 10) and YAN (N = 10). The HAN samples were obtained from the Hanwoo Experiment Station, National Institute of Animal Science, Rural Development Administration, Pyeongchang, Korea, and the YAN samples were obtained from Yanbian University in the People’s Republic of China. All the samples resequenced in this study were male individuals. From each animal, 3 μg of genomic DNA was randomly sheared using the Covaris System to generate approximately 90-bp inserts. The fragmented DNA was end-repaired using T4 DNA polymerase and Klenow polymerase, and Illumina paired-end adaptor oligonucleotides were ligated to the sticky ends. We analyzed the ligation mixture by electrophoresis on an agarose gel and then sliced as well as purified 200–250-bp fragments. Clusters of PCR colonies were then sequenced on a HiSeq 2000 sequencing platform (paired-end 90-bp reads) using recommended protocols from the manufacturer.
Mapping, SNP calling, and annotation
The sequences were aligned to the bovine reference genome assembly (UMD 3.1) using the Burrows-Wheeler Aligner (BWA, version 0.7.7) with default parameters (Li and Durbin, 2010). SAMtools (version 0.1.19) was used for converting sequence alignment/map (SAM) and binary version of a SAM (BAM) file formats, sorting, and indexing alignments (Li et al., 2009). We used Picard tools (version 1.106) to generate quality matrices for mapping. Duplicate reads were marked with Picard tools (version 1.106) and excluded from downstream analysis. We performed local re-alignment and re-calibration using the Genome Analysis Toolkit (GATK, version 3.1) framework (McKenna et al., 2010). The initial SNP discovery was performed using multi-sample SNP-calling procedure in the GATK package. To reduce the false discovery rate, the filtering steps was conducted by using these criteria: Phred scaled polymorphism probability (QUAL) < 30.0, variant confidence normalized by depth (QD) < 2.0, mapping quality (MQ) < 40.0, strand bias (FS) > 60.0, HaplotypeScore > 13.0, MQRankSum < −12.5, and ReadPos Rank-Sum < −8.0. Detailed description of the terms can be found at the GATK site (https://www.broadinstitute.org/gatk/guide/). All of the filtered SNPs were assigned to 19 functional categories using snpEff version 4.0 (Cingolani et al., 2012) and the Ensembl Bos taurus gene set version 76 (UMD 3.1). In order to utilize the representative gene set of downstream analysis, the “-canon” option in the snpEff program was enabled.
Functional enrichment analysis
We performed a functional enrichment analysis of genes that were found to have population-specific nonsynonymous SNPs (nsSNPs) using the Database for Annotation Visualization and Integrated Discovery (DAVID) tool (Huang da et al., 2009). To construct a network for functional GO network, the Enrichment Map Cytoscape plugin was used to build networks of interrelated terms based on the number of genes shared between gene ontology (GO) terms (Merico et al., 2010). Terms were represented as nodes (circles). Edges linking nodes represented gene sharing, and their thickness represented the degree of gene set overlap.
Selective sweep identification
To detect putative selective sweeps, we performed whole-genome screening to identify genomic regions with excess homozygosity, according to a previously described method (Rubin et al., 2010; 2012). Briefly, all of the SNPs derived from each breed were used to calculate the Z transformation of the pooled heterozygosity (ZHp) in each of the two breeds in this study. The numbers of major and minor allele sequence reads were counted; then, any SNP position whose minor allele frequency was less than 0.05 was removed. Subsequently, we applied the 50% overlapping sliding windows that were 150 kb, where the ZHp was eventually calculated in each of the window. In addition, we removed windows with fewer than 10 SNPs, and we used the Animal QTLdb to retrieve quantitative trait loci (QTL) information and visualize the QTL with putative selective sweeps (Hu et al., 2013).
RESULTS AND DISCUSSION
Sequencing and SNP detection
We extracted genomic DNA from 10 HAN and 10 YAN samples (20 total). A total of ∼7.2 million initial reads were generated by the Illumina HiSeq 2000 sequencer. All of the sequences were aligned against the UMD 3.1 genome assembly using BWA. Potential polymerase chain reaction duplications (6%) were removed from the aligned reads, and the reads were further modified with the Picard toolkit before SNP analysis was performed using GATK. This yielded approximately 6.2 billion reads (626 Giga-bp), covering 98.8% of the reference assembly at an average of 10.6-fold coverage across the region (Table 1). Analysis of the sequence data revealed a total of 7,268,711,330 sequence reads across the reference genome. Of these, 6,192,968,178 reads (85.2%) were finally mapped to the reference genome after recalibration. We identified 17,976,093 SNPs after variant filtration process (Table 2). Of these, 13,936,399 (77.8%) SNPs were previously annotated in dbSNP (version 140). The density of SNPs was also determined to be approximately one per 148 bp, which would be fairly sufficient to locate candidate genomic regions that are associated with various traits of interest in cattle.
Table 1.
Summary of sequencing results for Hanwoo and Yanbian cattle breeds used in this study.
| Sample Name | No. Sample | Raw_Reads | PCR duplication reads | Mapped reads | Properly paired readsa | A_Coverageb | Ave_Foldc |
|---|---|---|---|---|---|---|---|
| Hanwoo | 10 | 3,625,402,354 | 3,182,976,738 | 3,123,899,715 | 2,989,529,698 | 98.78 | 10.71X |
| Yanbian cattle | 10 | 3,643,308,976 | 3,069,068,463 | 3,069,068,463 | 2,954,705,966 | 98.74 | 10.53X |
|
| |||||||
| Total | 20 | 7,268,711,330 | 6,306,225,775 | 6,192,968,178 | 5,944,235,664 | 98.76 | 10.62X |
Properly Paired reads, “properly paired” means that both ends of the reads were mapped with correct orientation and their fragment sizes were less than 500 bp.
A_Coverage, assembly coverage calculated as the proportion of bases in the genome assembly that were covered by at least one read.
Ave_Fold, average fold that was calculated as the average depth of coverage across the whole genome.
Table 2.
Summary of SNPs identified from Hanwoo and Yanbian cattle in this study.
| Fields | Hanwoo | Yanbian cattle | Total | |
|---|---|---|---|---|
| Sample counts | 10 | 10 | 20 | |
| SNP counta | 13,544,560 | 15,857,687 | 17,926,093 | |
| dbSNP | 11,477,894 | 12,892,393 | 13,936,399 | |
| Ti/Tv ratio | 2.26 | 2.26 | 2.26 | |
| SNP categories | ||||
| Exon | Synonymous variant | 49,684 | 59,232 | 70,079 |
| Non-synonymous variant | 36,457 | 42,595 | 50,827 | |
| Initiator codon variant | 4 | 4 | 5 | |
| Start lost | 34 | 43 | 48 | |
| Stop gained | 399 | 491 | 580 | |
| Stop lost | 16 | 25 | 27 | |
| Stop retained variant | 39 | 43 | 52 | |
| Non coding exon variant | 5,393 | 6,180 | 7,322 | |
| Splice site | Splice region variant | 8,499 | 10,098 | 11,961 |
| Splice acceptor variant | 224 | 269 | 318 | |
| Splice donor variant | 230 | 268 | 331 | |
| Intron | Intron variant | 3,347,348 | 4,047,275 | 4,703,213 |
| Intragenic variant | 40,614 | 49,950 | 56,469 | |
| UTR | 5 prime UTR variant | 4,094 | 4,917 | 5,855 |
| 5prime UTR premature - startcodon gain variant | 674 | 818 | 956 | |
| 3prime UTR variant | 26,280 | 31,680 | 37,323 | |
| Intergenic | Upstreamd gene variant | 531,123 | 637,361 | 742,563 |
| Downstreamd gene variant | 545,133 | 656,190 | 764,392 | |
| Intergenic region | 9,502,323 | 11,284,480 | 13,090,691 | |
| Functional classes | ||||
| Missense | 36,511 | 42,667 | 50,907 | |
| Nonsense | 399 | 491 | 580 | |
| Silent | 49,723 | 59,275 | 70,131 |
SNP count; the overlapped SNP loci between samples were counted as one.
Because the analysis to categorize the SNPs was done non-exclusively, some SNPs were counted at multiple categories.
SNP categories were clustered by six genomic regions: Exon, Splice site, Intron, UTR, Flanking region and Intergenic
Upstream/downstream: 5 Kbp regions that are adjacent to the both ends of a gene were defined as upstream and downstream regions respectively.
The average ratios of homozygous versus heterozygous SNPs are 1:1.8 and 1:2.1 in HAN and YAN, respectively. The higher homozygosity in HAN can be explained by the extensive artificial insemination (AI) used by the systematic selective breeding program for HAN since the 1970s. Alternatively, many genomic regions might still be under genetic drift for YAN. SNP quality was further evaluated by calculating the transition-to-transversion ratio (Ti/Tv) for each SNP, because the Ti/Tv ratio is used as an indicator of potential sequencing errors. These results (HAN: 2.26, YAN: 2.26) support previous observations that were approximately 2.1 and 2.2 in humans and cattle, respectively (Choi et al., 2013b; Fujimoto et al., 2010), indicating that most identified SNPs in this study were reasonably accurate.
SNP annotation and functional enrichment analysis of nsSNPs
All SNPs were annotated according to the functional categories using Ensembl gene annotation and dbSNP databases. Most of the SNPs were detected in intergenic and intron regions (75.9% and 24.3%, respectively), whereas fewer SNPs (0.95%) were identified within genic areas, including exonic, splice sites, and untranslated regions (Fig. 1 and Table 2). The numbers of functionally annotated SNPs were slightly higher than numbers of the detected SNP loci, because a SNP locus may have multiple annotations (Table 2). For example, a SNP locus discovered at the region of overlapped genes has two or more functional annotations. Among the genic SNPs, 951 nsSNPs and 453 nsSNPs were population specific for HAN and YAN, respectively (Supplementary Tables S1 and S2), providing useful resources to be used in genetic analysis of the phenotypic differences in the two cattle populations.
Fig. 1.
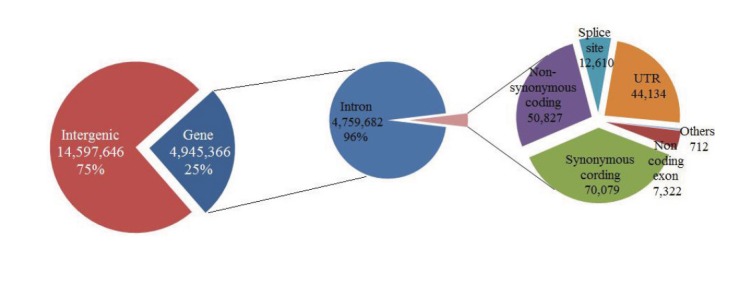
The SNP distribution according to the functional categories
Because these SNPs may be a consequence of selective pressures that are implicated with traits of interest in cattle, we further performed functional enrichment analysis of the genes including the population-specific nsSNP sets. YAN had no significant over-represented GO term from the enrichment analysis, while we observed several GO terms were significantly enriched for the genes where the HAN-specific nsSNPs were identified, which include regulation of response to stimulus (GO:0048583), response to wounding (GO:0009611), wound healing (GO:0042060), and hemostasis (GO:0007599) pairs (Fig. 2). Recent studies have shown signatures of selection or mutations in the genes that potentially affect immune response in cattle (Qanbari et al., 2014; Utsunomiya et al., 2013). Thus, these nsSNP set can be useful genomic resources to further test how these genes are genetically implicated with immune response or infection process in HAN.
Fig. 2.
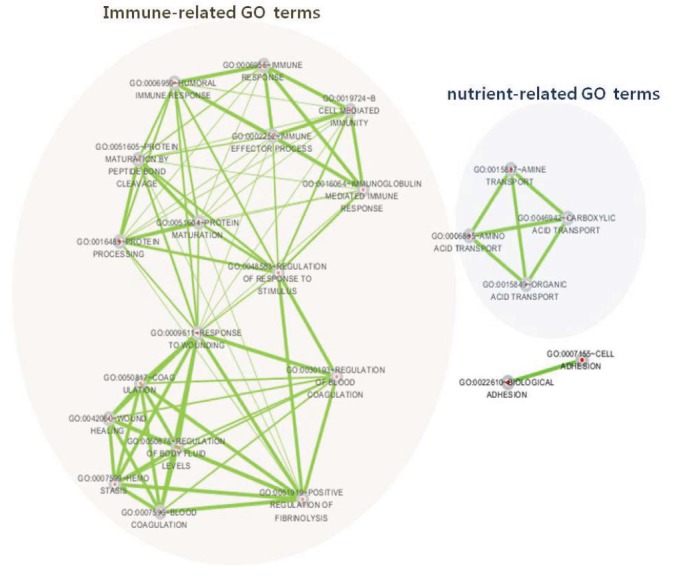
Network of functional terms of nsSNP in Hanwoo. Nodes (circles) are annotated functional terms. Edges connecting nodes represent gene share, being thickness proportional to the number of genes shared between terms (i.e., the degree of gene set overlap).
Additionally, a nutrient-related GO network was constructed between several transporters such as carboxylic acid and amine-organic acid (Fig. 2). Carboxylic acid and amine-organic acid contribute to meat flavor chemistry, as do organic compounds such as hydrocarbons, alcohols, aldehydes, and fatty acids (i.e., palmetic, stearic, or oleic acid) (Wasseriwan, 1979). Muscle is the main source of amino acids and protein. The contents of many different transporters can change the formation of carcass characteristics (Kozova et al., 2009). These transporters (amine, carboxyl acid or organic acid) as called food (muscle) proteins also have some connections between or within muscles during meat (meat muscle) fermentation like as the ion channel. For example, Amino acids are known to affect structural properties in intramuscular connective tissue and toughness of various skeletal muscles (Quang et al., 1989). Carboxylic acid is also involved in flavor-forming pathways by converting amino acid sources into flavor compounds through the transamination route (Liu et al., 2008). Meat tenderness and flavor are affected by the proportion and composition of intramuscular fat (marbling). From these reasons, these transporters can be one of the factors to be implicated with in meat characteristics such as bovine connective tissue components of intramuscular fat muscle.
Functional impact of fixed SNPs in HAN
We also identified fixed SNPs in each breed from the SNP annotation results. The fixed SNPs were defined as those where all specimens had the alternate allele type. We observed 1,173 and 1,058 fixed SNPs in HAN and YAN, respectively. Among these fixed SNPs, we only considered the proportion of the fixed SNPs that were present in at least 50% of each population because of missing (or unknown) genotype information in individuals. According to the criteria, 339 and 84 fixed SNPs were detected in HAN and YAN, respectively. These fixed SNPs might represent variants during selection in the population and may be useful information for further investigating breed identification. Interestingly, some fixed SNPs in HAN (28 of 104 coding SNPs, 27%) were located in fertility-related genes such as ubiquitously transcribed tetratricopeptide repeat containing Y-linked (UTY); prostate transmembrane protein, androgen induced 1 (PMEPA1); sex-determining region Y (SRY); and eukaryotic translation initiation factor 1A, Y-linked (EIF1AY). SRY is used as a candidate marker for sperm quality and fertility in bulls (Mishra et al., 2013), and EIF1AY and UTY are also known to play key roles in spermatogenesis and bull fertility (Liu et al., 2009). Since the national breeding program started in the 1970s (Lee et al., 2014), proven bulls have been intensively selected based on production traits through progeny tests in HAN (Kim et al., 1981). The use of AI in HAN has been used to obtain high selection intensity with a more rapid advance in genetic selection, which is similar to that in the Holstein industry (Norman et al., 2003). In Korea, classical semen parameters (i.e., viability, motility, and abnormal shapes) are normally checked before AI. Moreover, bull fertility is significantly correlated with sperm shape (r = 0.85, p < 0.05) (Ostermeier et al., 2001). Thus, HAN fertility might be indirectly selected for based on selection for meat quality traits during artificial selection. This might also affect genomic regions that are associated with the reproduction traits in HAN, which deserve to be validated in further studies, using multiple individuals as well as diverse cattle breeds. Because there are a limited number of publications available for genetic dynamic of fertility traits in Hanwoo, these findings would grant a useful opportunity to perform further association studies between fertility-related genes and reproductive fitness in HAN.
Selective sweep analysis
Using all of the WGS-derived SNPs from this study, we scanned the whole genome to detect regions with high degrees of fixation, which are indicative of signature of selection, in each breed. We applied 50% overlapping sliding windows that were 150 kb along all 29 autosomes; however, 30 windows with fewer than 10 SNPs were excluded. A total of 33,482 windows were used to calculate the Z transformations of the pooled heterozygosity (ZHp) in each window. The ZHp scores ranged from −7.95 to 1.54 and from −10.96 to 1.61, with median values of 0.24 and 0.22 for HAN and YAN, respectively (Fig. 3, Supplementary Tables S3 and S4). Because extremely low ZHp scores indicate putative selective sweeps because of excess homozygosity, we focused on the ZHp score in the extreme lower end of the distribution. We observed 195 and 173 windows with ZHp values of less than or equal to −4 in HAN and YAN, respectively, whereas 73 and 68 windows showed ZHp scores less than −5 that corresponded to approximately the lowest 0.2% for HAN and YAN, respectively.
Fig. 3.
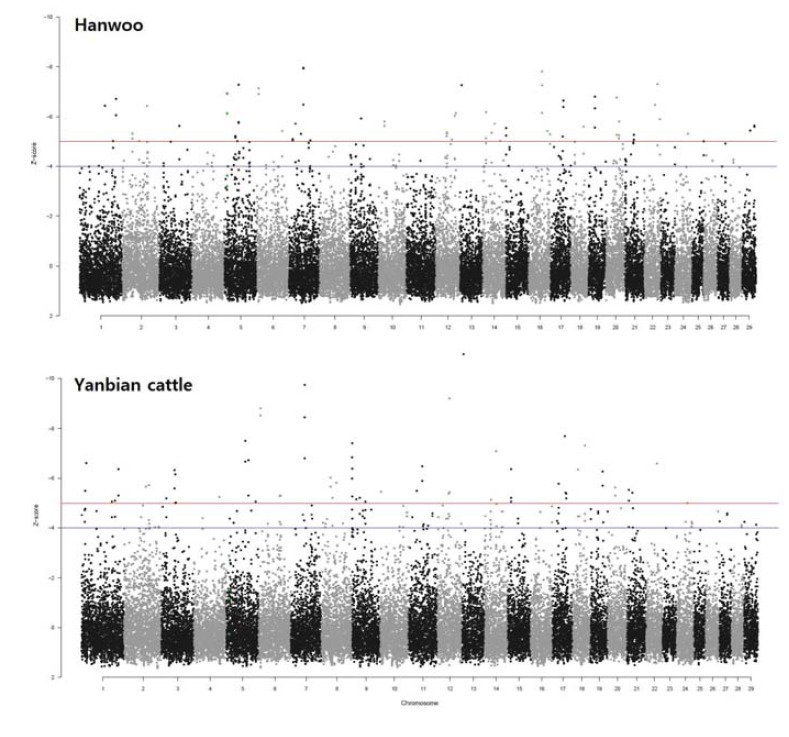
Distribution of ZHp scores across all 29 Bos taurus autosomes for HAN and YAN. The upper and lower plots indicate the ZHp score distribution of Hanwoo and Yanbian cattle, respectively. Even numbered chromosomes are presented in gray and odd numbered chromosomes are black. The blue and red horizontal lines indicate ZHp thresholds of −4 and −5 that could be strong candidates of selective sweeps in this study.
Among the putative selective sweeps, we located several loci that potentially contribute to phenotypic differences between HAN and YAN. For example, a putative selective sweep was observed in a locus with an extremely low ZHp score of −6.93 in HAN (window #396 in chromosome 5), which included a gene that encodes protein phosphatase 1 regulatory subunit 12A (PPP1R12A). Alternatively, the ZHp score in the same window was distinctively lower for YAN (ZHp: −2.88) (Supplementary Table S4). Furthermore, we observed a cluster with a higher degree of homozygosity in the adjacent windows including the PPP1R12A gene in HAN (Fig. 4 and Supplementary Table S3); alternatively, windows surrounding the adjacent windows did not show such distinct difference of the ZHp values between HAN and YAN. This phenomenon is a typical pattern for inferring selective sweep, indicating that this genomic region or the gene may be implicated with selection of HAN during recent decades.
Fig. 4.
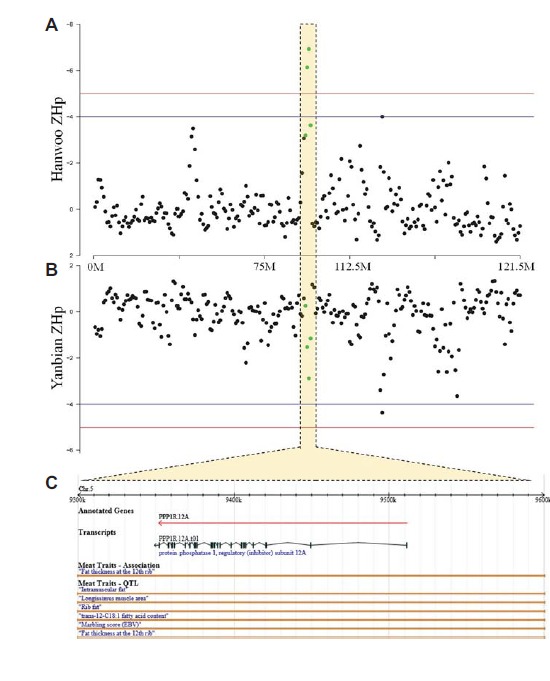
A putative selective sweep at the PPP1R12A locus in Hanwoo. (A) and (B) present the Z score of the pooled heterozygosity (ZHp) plot for Hanwoo and Yanbian cattle, respectively. The red and blue horizontal lines in these plots indicate ZHp scores of −5 and −4, respectively. Each circle-point represents ZHp score in each window used in this study. The yellow-highlighted marks the vicinity of the sweep including the PPP1R12A gene. ZHp scores in the interval were indicated as a green color. (C) The annotated gene structure of the PPP1R12A gene as well as underlying QTLs for the genomic region as visualized using the Animal QTLdb.
PPP1R12A, also called myosin phosphatase target subunit 1 (MYPT1), is known to be involved in diverse cellular processes (Ito et al., 2004; Matsumura and Hartshorne, 2008), including insulin signaling regulation in skeletal muscle (Zhang et al., 2014). There are limited available publications that have investigated this gene in cattle. Sun et al. (2013) reported that PPP1R12A was one of the highly expressed genes in pig intramuscular adipose tissue, which indicates that the gene may be associated with intramuscular fat content (marbling). In the Korean beef industry, intramuscular fat content is the most important palatability factor and has been selected for in the HAN selective breeding program. Consequently, selective breeding led to the genetic improvement in this economically important trait (Lee et al., 2014). Genetic improvement by the selection may have left a genomic footprint, which would be shown as an extreme ZHp value. Interestingly, the gene was found to overlap with QTL for marbling in cattle (McClure et al., 2010; Nalaila et al., 2012; Peters et al., 2012). Although concluding detailed functions of genes in the selective sweeps is beyond the scope of this study, the findings of this research provide useful genomic information for further studies to better understand genetic mechanisms that underlie economically important traits in cattle.
Acknowledgments
This work was supported by Agenda (PJ01040601 and PJ906956) of the National Institute of Animal Science, Rural Development Administration (RDA), Korea; and the 2014 Postdoctoral Fellowship Program of the Rural Development Administration, Korea.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Cheng P. Livestock breeds of China. FAO Animal Production and Health Paper 46. 1984.
- Choi JW, Chung WH, Lee KT, Lee JW, Jung KS, Cho Y, Kim N, Kim TH. Whole genome resequencing of Heugu (Korean Black Cattle) for the genome-wide SNP discovery. Korean J. Food Sci. An. 2013a;33:715–722. [Google Scholar]
- Choi JW, Liao X, Park S, Jeon HJ, Chung WH, Stothard P, Park YS, Lee JK, Lee KT, Kim SH, et al. Massively parallel sequencing of Chikso (Korean brindle cattle) to discover genome-wide SNPs and InDels. Mol. Cells. 2013b;36:203–211. doi: 10.1007/s10059-013-2347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Liao X, Stothard P, Chung WH, Jeon HJ, Miller SP, Choi SY, Lee JK, Yang B, Lee KT, et al. Whole-genome analyses of Korean native and Holstein cattle breeds by massively parallel sequencing. PLoS One. 2014;9:e101127. doi: 10.1371/journal.pone.0101127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadi H, Lee SH, Lee SS, Park C, Kim KS. Inter- and intra-population genetic divergence of East Asian cattle populations: focusing on Korean cattle. Genes Genom. 2014;36:261–265. [Google Scholar]
- Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brondum RF, Liao X, Djari A, Rodriguez SC, Grohs C, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014;46:858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- Eck SH, Benet-Pages A, Flisikowski K, Meitinger T, Fries R, Strom TM. Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol. 2009;10:R82. doi: 10.1186/gb-2009-10-8-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsik CG, Tellam RL, Worley KC, Gibbs RA, Muzny DM, Weinstock GM, Adelson DL, Eichler EE, et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science. 2009;324:522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Agriculture Organization (FAO) Domestic Animal Diversity Information Service (DAD-IS) 2014 http://dad.fao.org/, Accessed September 5, 2014.
- Fujimoto A, Nakagawa H, Hosono N, Nakano K, Abe T, Boroevich KA, Nagasaki M, Yamaguchi R, Shibuya T, Kubo M, et al. Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing. Nat. Genet. 2010;42:931–936. doi: 10.1038/ng.691. [DOI] [PubMed] [Google Scholar]
- Hu ZL, Park CA, Wu XL, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41:D871–879. doi: 10.1093/nar/gks1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: structure, regulation and function. Mol. Cell. Biochem. 2004;259:197–209. doi: 10.1023/b:mcbi.0000021373.14288.00. [DOI] [PubMed] [Google Scholar]
- Kawahara-Miki R, Tsuda K, Shiwa Y, Arai-Kichise Y, Matsumoto T, Kanesaki Y, Oda S, Ebihara S, Yajima S, Yoshikawa H, et al. Whole-genome resequencing shows numerous genes with nonsynonymous SNPs in the Japanese native cattle Kuchinoshima-Ushi. BMC Genomics. 2011;12:103. doi: 10.1186/1471-2164-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Kim YH, Cho WH, Bok KK, Kim BJ, Kang CM. Yanbian Cattle. Yanbian People Publishing House; 1981. pp. 1–5. [Google Scholar]
- Kozova M, Kalac P, Pelikanova T. Changes in the content of biologically active polyamines during beef loin storage and cooking. Meat Sci. 2009;81:607–611. doi: 10.1016/j.meatsci.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Lee KT, Chung WH, Lee SY, Choi JW, Kim J, Lim D, Lee S, Jang GW, Kim B, Choy YH, et al. Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genomics. 2013;14:519. doi: 10.1186/1471-2164-14-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Park BH, Sharma A, Dang CG, Lee SS, Choi TJ, Choy YH, Kim HC, Jeon KJ, Kim SD, et al. Hanwoo cattle: origin, domestication, breeding strategies and genomic selection. J. Anim. Sci. Technol. 2014;56 doi: 10.1186/2055-0391-56-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing, S The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Nauta A, Francke C, Siezen RJ. Comparative genomics of enzymes in flavor-forming pathways from amino acids in lactic acid bacteria. Appl. Environ. Microbiol. 2008;74:4590–4600. doi: 10.1128/AEM.00150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Qin X, Song XZ, Jiang H, Shen Y, Durbin KJ, Lien S, Kent MP, Sodeland M, Ren Y, et al. Bos taurus genome assembly. BMC Genomics. 2009;10:180. doi: 10.1186/1471-2164-10-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Miller S, Sargolzaei M, Kelly M, Vander Voort G, Caldwell T, Wang Z, Plastow G, Moore S. Genome-wide association analyses for growth and feed efficiency traits in beef cattle. J. Anim. Sci. 2013;91:3612–3633. doi: 10.2527/jas.2012-5716. [DOI] [PubMed] [Google Scholar]
- Matsumura F, Hartshorne DJ. Myosin phosphatase target subunit: Many roles in cell function. Biochem. Biophys. Res. Commun. 2008;369:149–156. doi: 10.1016/j.bbrc.2007.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, McKay SD, Gregg SJ, Chapple RH, Northcutt SL, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim. Genet. 2010;41:597–607. doi: 10.1111/j.1365-2052.2010.02063.x. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra C, Palai TK, Sarangi LN, Prusty BR, Maharana BR. Candidate gene markers for sperm quality and fertility in bulls. Veterinary World. 2013;6:905–910. [Google Scholar]
- Nalaila SM, Stothard P, Moore SS, Li C, Wang Z. Whole-genome QTL scan for ultrasound and carcass merit traits in beef cattle using Bayesian shrinkage method. J. Anim. Breed. Genet. 2012;129:107–119. doi: 10.1111/j.1439-0388.2011.00954.x. [DOI] [PubMed] [Google Scholar]
- Norman H, Powell R, Wright J, Sattler C. Timeliness and effectiveness of progeny testing through artificial insemination. J. Dairy Sci. 2003;86:1513–1525. doi: 10.3168/jds.S0022-0302(03)73737-0. [DOI] [PubMed] [Google Scholar]
- Ostermeier GC, Sargeant GA, Yandell BS, Evenson DP, Parrish JJ. Relationship of bull fertility to sperm nuclear shape. J. Androl. 2001;22:595–603. [PubMed] [Google Scholar]
- Peters SO, Kizilkaya K, Garrick DJ, Fernando RL, Reecy JM, Weaber RL, Silver GA, Thomas MG. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J. Anim. Sci. 2012;90:3398–3409. doi: 10.2527/jas.2012-4507. [DOI] [PubMed] [Google Scholar]
- Qanbari S, Pausch H, Jansen S, Somel M, Strom TM, Fries R, Nielsen R, Simianer H. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 2014;10:e1004148. doi: 10.1371/journal.pgen.1004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang N, Zarkadas CG. Comparison of the amino acid composition and connective tissue protein contents of selected bovine skeletal muscles. J. Agric. Food Chem. 1989;37:1279–1286. [Google Scholar]
- Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- Rubin CJ, Megens HJ, Martinez Barrio A, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, et al. Strong signatures of selection in the domestic pig genome. Proc. Nati. Acad. Sci. USA. 2012;109:19529–19536. doi: 10.1073/pnas.1217149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X, Moore SS. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics. 2011;12:559. doi: 10.1186/1471-2164-12-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WX, Wang HH, Jiang BC, Zhao YY, Xie ZR, Xiong K, Chen J. Global comparison of gene expression between subcutaneous and intramuscular adipose tissue of mature Erhualian pig. Genet. Mol. Res. 2013;12:5085–5101. doi: 10.4238/2013.October.29.3. [DOI] [PubMed] [Google Scholar]
- Utsunomiya YT, O’Brien AMP, Sonstegard TS, Van Tassell CP, do Carmo AS, Meszaros G, Soelkner J, Garcia JF. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS One. 2013;8:e64280. doi: 10.1371/journal.pone.0064280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasseriwan A. Symposium on meat flavor chemial basis for meat flavor: A Review. J. Food Sci. 1979;44:6–11. [Google Scholar]
- Zhang X, Ma D, Caruso M, Lewis M, Qi Y, Yi Z. Quantitative phosphoproteomics reveals novel phosphorylation events in insulin signaling regulated by protein phosphatase 1 regulatory subunit 12A. J. Proteomics. 2014;109:63–75. doi: 10.1016/j.jprot.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, Hanrahan F, Pertea G, Van Tassell CP, Sonstegard TS, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10:R42. doi: 10.1186/gb-2009-10-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.