

Abstract
Polyketides represent an important class of secondary metabolites that interact with biological targets connected to a variety of disease-associated pathways. Remarkably, nature’s assembly lines, polyketide synthases, manufacture these privileged structures through a combinatorial mixture of just a few structural units. This review highlights the role of these structural elements in shaping a polyketide’s conformational preferences, the use of computer-based molecular modeling and solution NMR studies in the identification of low-energy conformers, and the importance of conformational analogues in probing the bound conformation. In particular, this review covers several examples wherein conformational analysis complements classic structure-activity relationships in the design of biologically active natural product analogues.
1. The role of conformational entropy in molecular recognition
A small molecule’s biological activity is predicated on the interactions it makes with a target receptor’s binding site. Optimum binding requires a complementary spatial relationship of structural features in order to achieve favorable ligand-protein interactions. Synthetically-derived compounds within the medicinal chemistry realm frequently employ rigid structures, which limit the number of rotatable bonds and have become associated with “drug-like” properties.1 In contrast, bioactive natural products often possess significant degrees of conformational flexibility, which may impart potentially beneficial properties for transport, solubility, selectivity, and binding. Nevertheless, apparently flexible molecules of even intermediate complexity will typically adopt preferred conformational profiles. In the case of the polyketide class of natural products, we can see many of the structural features that evolution includes to impart conformation preferences. Some of the hallmarks of this class include sp3-stereogenic centers (methyl and hydroxyl substituents), sp2-hybridized carbons (E- and Z-alkenes and carbonyl groups) and single bonds with restricted rotation (s-sp2-sp2) such as amides, esters, and polyenes. Even when uninvolved in binding, the steric and electronic interactions these functionalities introduce limit the number of potential low-energy conformations while maintaining overall backbone flexibility. As such, these highly substituted molecules preferentially adopt certain conformations, giving us what Hoffmann aptly described as “flexible molecules with a defined shape.”2 As long as the profile contains a conformer population complementary to the active site, the resulting change in free energy upon binding becomes more favorable.
The role of conformation in ligand-protein binding reaches beyond the bound state. Binding affinity involves both enthalpic and entropic contributions. While optimizing a drug candidate ideally involves the improvement of both terms, this is rarely achievable in practice.3 Additionally, improving enthalpic contributions (the direct non-covalent interactions between the ligand and the binding site) can introduce unforeseen complications. For instance, installing functional groups that increase ligand affinity for a target protein through enthalpic means will typically also affect water affinity. If the new groups form suboptimal protein contacts, the resulting effects on solvation and solubility can negatively influence both enthalpic and entropic parameters. As classic structure-activity relationships typically focus on the optimization of target-ligand interactions, they often ignore the potential impact these modifications have on ligand conformational preferences. Therefore, it is critical for one to consider conformational effects when applying a traditional medicinal chemistry approach to polyketide analogue design, evaluating both the structure-activity relationship and the conformation-activity relationship. Recent efforts from several labs have demonstrated the multipurpose nature of structural features within polyketide natural products and several examples are included below.
2. Conformational control elements in polyketides
As researchers discovered in the mid-20th century, conformation plays an important role in a variety of chemical transformations. The rate acceleration for anti-periplanar elimination reactions is one such example. Therefore, it comes as no surprise that many stereochemical control elements also impact polyketide conformation. As the attenuation of conformational freedom represents the ultimate goal in the evolutionary design of these biologically active privileged structures, the most powerful restrictions utilize steric interactions between substituents or modifications of the carbon skeleton itself in the form of unsaturation.
One must take into account general steric minimization when considering a molecule’s preferred conformation, particularly when looking at cyclic molecules. The preference of substituents in six-membered rings to adopt equatorial rather than axial orientations to minimize gauche interactions is a fundamental example of this principle. However, the picture becomes more complicated as rings increase in size, flexibility, and heteroatom substitution. In rings larger than six members, strain arises as bond angles deviate from their idealized values and additional transannular interactions are introduced, which in concert with the appropriate substituents will force specific conformations over others. In large macrocycles with minimized ring strain, local acyclic structural features and subtle interactions between remote substituents will ultimately control the molecule’s conformational preferences (Figure 1).
Figure 1.
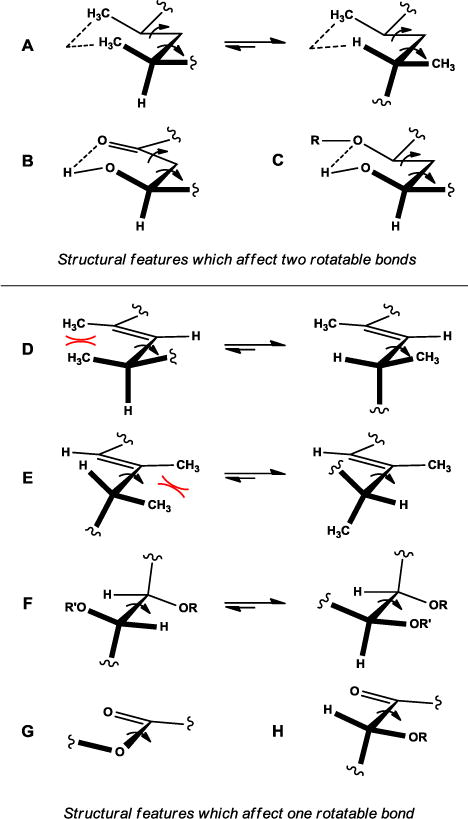
In addition to rings, structural features within polyketides control conformational preferences about individual single bonds or pairs of adjacent single bonds through minimization of steric, electronic, and electrostatic interactions as well as through hydrogen-bonding.
3.1 Conformational mimics of polyketide natural products
Polyketide natural products contain a variety of diverse and demanding functionalities, typically possessing extensive carbon skeletons with multiple stereogenic centers. Since these compounds usually exist in limited natural supply, investigators often rely upon total synthesis to provide sufficient material for therapeutic studies. Subsequently, the design of analogues that simplify or eliminate moieties with no effect on biological activity represents a worthwhile goal. Structural simplification not only shortens a molecule’s synthetic step count but also increases material throughput for clinical investigations. The Wender laboratory has termed this approach function-oriented synthesis,4 wherein biologically-active lead structures are simplified to incorporate only the activity-determining features. From a conformational standpoint, this design strategy involves identifying structural elements that minimally affect either the conformation of regions responsible for protein interactions or the overall ensemble, ultimately creating a simplified structural analogue which retains the activity of the parent natural product.
3.1 Bryostatin
Bryostatin’s recent preclinical success exemplifies the therapeutic importance and potential impact of polyketide research on human health.5 Isolated by Pettit and co-workers from the marine bryozoan Bugula neritina, this diverse collection of polyketides contains over 20 complex natural products.6 The bryostatin family shares many important structural features such as three fully-functionalized tetrahydropyran rings, a unique methoxycarbonyl methylidene group, and several sites of oxygenation (Figure 2). These small molecules elicit a wide array of biological responses such as restoring apoptotic function in cancer cells,7 improving memory in animal models,8 and inducing latent HIV activation.9 Bryostatin’s impressive range of effects has been attributed to its ability to activate protein kinase C (PKC) by binding to the C1-domain.10 Interestingly, bryostatin shares many of these properties with phorbol esters such as PMA 2 despite being structurally dissimilar, even competing with it for binding. However, bryostatin 1 (1) lacks several of PMA’s unwanted biological responses, in particular its tumor-promoting capabilities, and will even antagonize them when dosed simultaneously.
Figure 2.
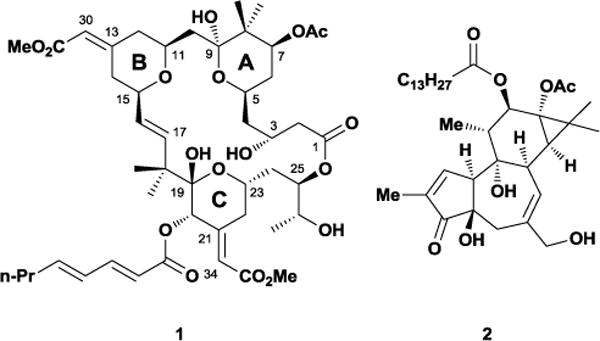
The potent polyketide natural product bryostatin 1 (1) and 12-myristate 13-acetate (PMA) 2.
Unfortunately, bryostatin’s scarce supply has hindered its continued advancement into clinical trials for the treatment of cancer and Alzheimer’s disease. Although synthetic work over the years by numerous laboratories significantly improved the overall step count, these elegant syntheses have produced limited quantities of material.7e Additionally, isolation from Bugula neritina yielded bryostatin in extremely limited amounts (approximately 18 g were isolated from 40,000 L of wet bryozoan),11 making natural extraction methods environmentally costly. Early computational and structure-activity relationship studies revealed that alterations within the C4-C16 region minimally affected bryostatin’s cytotoxicity profile.12 In contrast, deletion or alteration of functionality within the C15-C34 region produced analogues with reduced PKC affinity. Based on these findings, the Wender group hypothesized that exchanging the stereochemically complex C4-C16 region with a tetrahydropyran spacer would yield a simplified scaffold, enabling access to larger quantities of material for further biological studies. To this end, the Wender group designed and synthesized a variety of simplified analogues based around an efficient macrocyclic acetalization reaction (Scheme 1).13
Scheme 1.
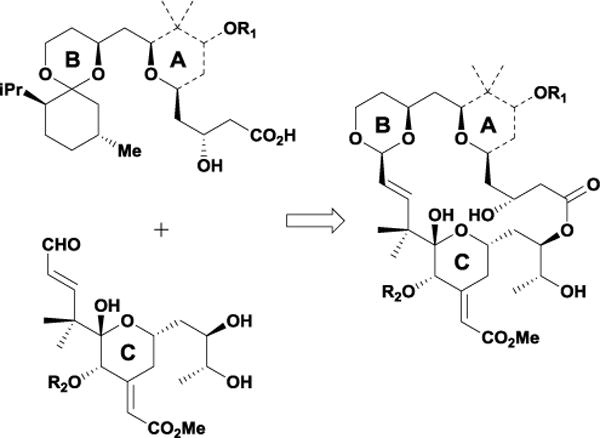
Representative transacetalization macrocyclic ring closure.
From their new pool of analogues, the Wender laboratory selected 3 for detailed NMR analysis as a means of gauging how well its solution structure would match the predicted conformation.14 High-field 1D- and 2D-proton NMR experiments in benzene-d6 yielded spectral data with coupling constants that significantly deviated from rotationally-averaged values, suggesting macrolide 3 exists predominantly as a single conformation at room temperature. While the interproton distances failed to match the computationally-derived conformation calculated previously for bryostatin 10,15 a subsequent constrained gas-phase molecular dynamics simulation found several conformations within 2 kcal/mol of the global minimum. These low-energy conformers satisfied the distance constraints and matched nicely with both reported crystal and solution structures of bryostatin despite the removal of all A- and B-ring substituents and the conversion of the C14 carbon to oxygen. Wender and co-workers attributed this conformation to a transannular hydrogen-bonding network between the C3 and C19 hydroxyl groups and the B-ring acetal oxygen (Figure 3).
Figure 3.
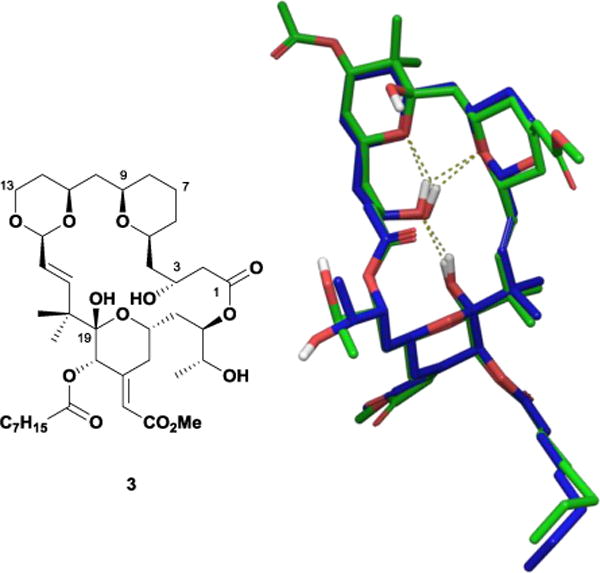
Overlay of acetal 3 (blue) and bryostatin 1 (green) solution conformations showing the internal hydrogen bonding network.
Biological analysis revealed that pyran 3 and several other structurally related analogues successfully bound PKC isozymes in an established bryostatin assay, with the compound exhibiting nanomolar affinity consistent with multiple bryostatins. Furthermore, 3 displayed potent activity against several human cancer cell lines. This data further supported their hypothesis that bryostatin binding requires two functional domains: a recognition domain that interacts with the receptor and a spacing domain that properly orients and constrains the former. The Wender group has since synthesized a multitude of related compounds investigating further modifications to the spacer domain, creating a library of bryostatin analogues which display single-digit nanomolar PKC affinity.7d
Recently, the Wender group began development of second-generation analogues that replace the spacer domain with a salicylate subunit, a commercially available motif that allows the macrocycle to retain key portions of the internal hydrogen-bonding network (Figure 4).16 Computational analysis suggested that salicylate 4 maintained a proper spatial orientation within the recognition domain wherein the C3-ether could make a hydrogen bond with the C19-hemiketal and the aromatic ring would associate with the membrane. Wender and co-workers synthesized 4 in only 23 total steps, employing a highly convergent and step-economical strategy. Biological evaluation showed that this compound bound PKC with low nanomolar potency, approaching that of 1. Given that this analogue required even fewer steps than 3 and reduced the molecule’s structural complexity while retaining activity, the simplified salicylate substructure could be a promising lead scaffold for the development of future compounds.
Figure 4.
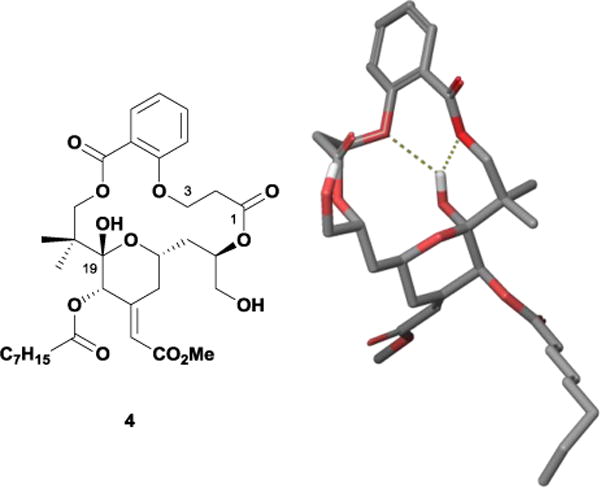
Major solution conformer of salicylate analogue 4.
In 2014, the Keck and Krische laboratories sought an explanation for the interesting biological responses provoked by bryostatin and related structural analogues with regard to the phorbol esters.17 During the process of designing simplified structures, the Keck group discovered that one of their newly-synthesized bryostatin analogues exhibited PMA-like biological responses.18 Subsequent analogues indicated that bryostatin-like activity depended upon the A-ring substituents but not substituents on the B-ring, suggesting that the B-ring could be safely removed altogether. The Keck and Krische laboratories independently synthesized two analogues lacking the B ring, 5 and 7 (Figure 5). Curiously, 5 was the minor product from the final deprotection step, with a ring-expanded analogue 6 representing the bulk of the material. Conformational and computational analysis of 5 and 7 alleged that the compounds retained both the hydrogen-bonding network and solution conformation of bryostatin, although the Keck laboratory also concluded that the ring strain arising from the C15-C17 unsaturated ester of 5 prompted the undesired ring expansion. Interestingly, 5, 6, and 7 all exhibited PMA-like biological responses in U937 histiocytic lymphoma cell assays, suggesting that the B-ring plays a pivotal role in bryostatin-like behavior. Recent work on neristatin 1, which binds to PKC and shares the A/B ring system of 1, has indicated that it also displays bryostatin-like activity.19 The bryostatins demonstrate how structural units can act as both conformational control and protein interaction elements, and how identifying the essential structures for control through conformational analysis can help efficiently design analogue scaffolds. The interesting reports from Keck and Krische suggest that bryostatin’s non-PMA mode of action depends on more than its ability to merely bind PKC, which may require more stringent evaluation of simplified bryostatin analogues moving forward.
Figure 5.
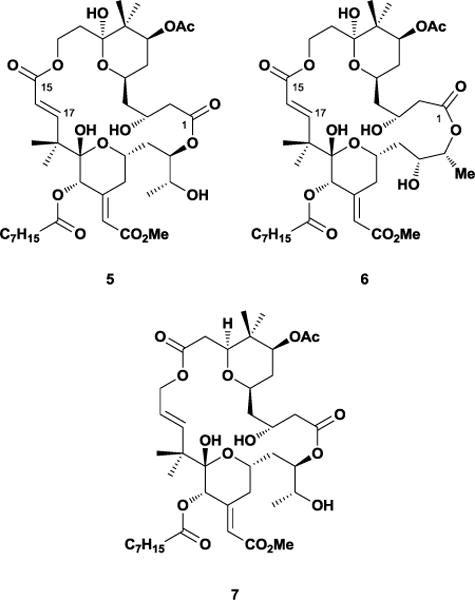
Keck’s analogues 5 and 6 and Krische’s bryostatin analogue 7.
3.2 Laulimalide
Laulimalide 8, a polyketide isolated from the sponge Cacospongia mycofijiensis, exhibits low nanomolar cytotoxicity against multiple cancers and retains significant activity against paclitaxel-resistant cell lines.20 The potent microtubule-stabilizing agent also binds tubulin at a non-taxane binding site21 and acts synergistically with paclitaxel and other taxane binders.22 In the interests of exploring laulimalide’s therapeutic potential, the Eisai Research Institute synthesized gram-scale quantities of 8 for in vivo biological testing.23 Though laulimalide exhibited a favorable pharmacokinetic profile compared to paclitaxel, it unfortunately displayed poor tumor growth inhibition accompanied by severe toxicity and mortality.24 These severe side effects precluded dosing at higher concentrations. As such, improving laulimalide’s therapeutic usefulness requires analogue development.
Synthetic efforts spurred by laulimalide’s impressive in vitro activity revealed that 8 spontaneously degrades under mildly acidic conditions to yield isolaulimalide 9, a side product that displays significantly reduced cytotoxicity (Figure 6). Early analogue efforts circumvented this undesired degradation pathway primarily through removal/modification of the C16-C17 epoxide, the C2-C3 alkene, or capping the C20 alcohol.25 However, these relatively simple modifications diminished laulimalide’s cytotoxicity profile.26 Recognizing the importance of conformation to the analogue design process, several researchers investigated laulimalide’s conformational preferences to gain a better understanding of the molecule’s pharmacophore.
Figure 6.
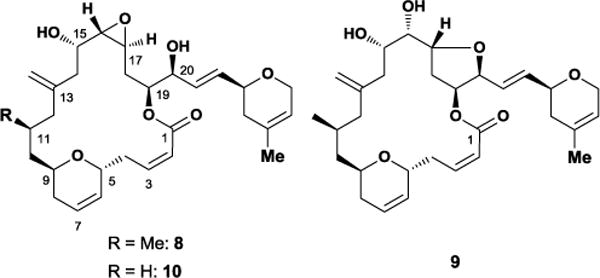
Laulimalide 8, isolaulimalide 9 and 11-desmethyl laulimalide 10.
In 1996, Jefford and co-workers provided the first insights into laulimalide’s conformational preferences using X-ray crystallography.27 While this static picture afforded valuable information on the molecule’s predominant conformation, elucidating laulimalide’s bound conformation required a more rigorous analysis of the macrolide’s solution behavior. Nearly a decade after solving laulimalide’s crystal structure, the Paterson and Snyder groups analyzed the molecule’s solution conformation utilizing two different approaches. The Paterson group used an NMR-constrained conformational search28 while the Snyder group employed the NAMFIS (NMR Analysis of Molecular Flexibility in Solution) method.29
Specifically, Paterson and co-workers used experimental coupling constant values for predicting dihedral angle preferences and NOESY experiments for identifying critical through-space interactions. The Paterson laboratory then generated files containing restrained torsional angles and performed a constrained Monte Carlo conformational search, which provided several low energy conformers within 2 kcal/mol of the global minimum. While this analysis afforded low energy conformers resembling laulimalide’s solid-state conformation, the percent contribution of these solution conformers remained ambiguous. Snyder’s NAMFIS analysis complemented the above-mentioned methodology by changing the mole fraction of each conformer until a sum of square differences between the experimental NMR data and computed variables gave a “best fit” dataset. Interestingly, both groups found that the primary solution conformers bore a great deal of similarity to laulimalide’s solid-state conformation. The solid-state crystal structure and major solution conformation showed that 8 preferred an open and gross-flattened conformation wherein the side chain, the C2–C3/C21–C22 unsaturated moieties, and the exocyclic epoxide all lie in the same plane. The polyketide’s dihydropyran rings also adopted a half-chair conformation and curved slightly under the macrocyclic ring.
Given the difficulty with accessing laulimalide from isolation and total synthesis efforts, the Paterson and Wender groups designed a simplified analogue lacking the C11-Me, 11-desmethyllaulimalide 10 (Figure 6).30 Both laboratories targeted this particular functional group since its inclusion increased the step count in synthetic routes towards 8. High-field NMR experiments and Monte Carlo conformational searches also indicated a high degree of flexibility within the C9-C12 region, which suggested that removing the C11-Me would not significantly impact laulimalide’s overall shape or biological activity (Figure 7). As a result, the Wender and Paterson groups synthesized the simplified analogue 10 utilizing highly convergent strategies. Notably, removing the C11-Me group reduced the Wender group’s synthetic step count and decreased the starting material cost by 22-fold. High-field 1D-NMR experiments showed that 10 retained similar 3JH-H coupling constants throughout the northern and southern regions of the molecule. In particular, the 11-desmethyllaulimalide H14a/b-H15 dipolar couplings nearly matched identically with that of laulimalide 8. However, the 3JH10a-H11a and 3JH11-12a coupling constants deviated slightly from laulimalide’s normal dipolar couplings indicating a minor conformational bias within the C9-C12 region.
Figure 7.
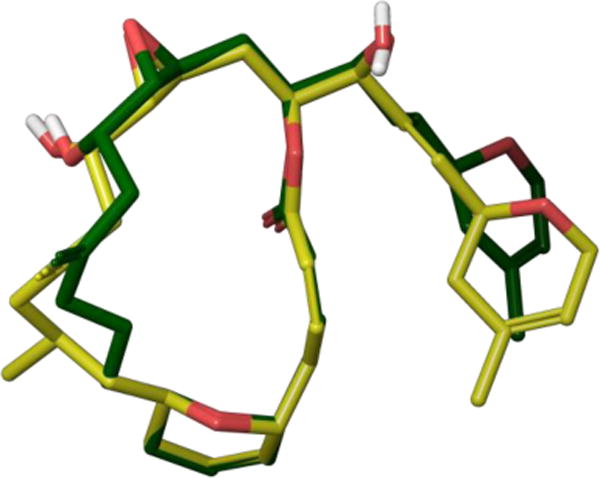
Overlay of 8 (yellow) and 10 (green) lowest-energy solution conformers.
Biological evaluation revealed that 10 possessed nanomolar cytotoxicity against multiple cancer cell lines including cell lines overexpressing the P-glycoprotein pump, though to a lesser degree than 8.31 10 also promoted tubulin polymerization at a critical concentration of only 1.1 ± 0.1 μM. Recently, Prota and co-workers published a co-crystal structure of laulimalide bound to tubulin that conclusively defines the molecule’s binding site on β-tubulin and fully elucidates the bioactive conformation.32 Interestingly, laulimalide’s tubulin bound conformation matches well with both the solid-state and major solution conformations. As shown in Figure 8, a dihydropyran ring flip accounts for the only main difference between the two conformations. Given the activity demonstrated by 10 and the success of molecular modeling in predicting laulimalide’s conformational preferences, additional development in this area should determine whether it is possible to further simplify the structure and retain activity.
Figure 8.
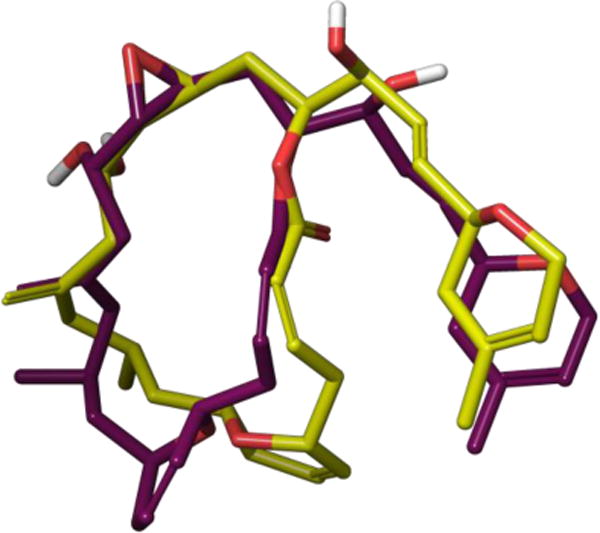
Overlay of laulimalide’s major solution conformation (yellow) with its tubulin-bound conformation (purple).
3.3 Exiguolide
In 2008, the Cossy laboratory recognized the structural similarity between the bryostatins and the natural product exiguolide 11 (Figure 9).33 Interestingly, both 20-membered marine polyketides contain analogous structural motifs such as a bis-tetrahydropyran unit and methoxycarbonyl methylidene group. Ohta and co-workers isolated 11 in 2006 from the sponge Geodia exigua off the coast of Japan and determined its molecular structure via extensive NMR analysis.34 The complex macrolide inhibits the fertilization of sea urchin (Hemicentrotus pulcherrimus) gametes while also displaying potent in vitro anti-cancer activity against multiple human cancer cell lines.35 A recent COMPARE analysis suggests exiguolide’s antiproliferative activity may arise through a distinct mechanism of action compared to other anti-cancer agents such as paclitaxel, vincristine, and doxorubicin.36 Exiguolide’s unknown mechanism of action along with its structural similarity with the bryostatins has prompted the interest of many synthetic chemists. However, the molecule’s extreme scarcity has limited SAR studies, which in turn has hindered further investigation of this promising anti-cancer agent.
Figure 9.
The marine polyketide, (–)-exiguolide.
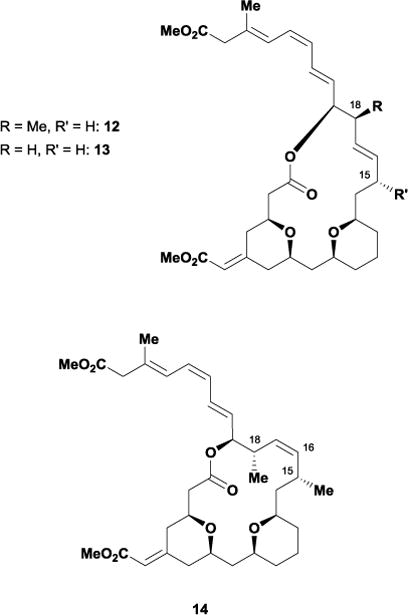
Recently, the Sasaki laboratory complemented existing SAR studies37 by focusing on the relationship between exiguolide’s conformation and the molecule’s cytotoxicity.38 Since previous studies established the importance of the triene side chain and C5-Z-enoate, Sasaki and co-workers investigated how functional groups on exiguolide’s macrocyclic backbone impacted cytotoxicity. In particular, the Sasaki group focused on removing the C15 and C18 allylic methyl groups since these motifs complicated their overall synthetic strategy. Their synthesis employed a stereoselective domino cross-metathesis/intramolecular oxa-conjugate addition for building the methylene bis-tetrahydropyran core.39 Using this advanced intermediate as a divergent starting point, the Sasaki laboratory constructed the 15-desmethyl analogue 12 and 15,18-bis-desmethyl exiguolide 13 using a synthetic strategy similar to the one utilized in their exiguolide total synthesis. The Sasaki group also prepared the corresponding (16,17-Z)-exiguolide 14, as they anticipated this inverted olefin geometry would alter the macrocycle conformation and serve as an excellent control for gauging the relationship between exiguolide’s conformation and its biological activity.
To evaluate the conformational effects of their structural modifications, the Sasaki laboratory performed NMR-based conformational analyses on 11 and each of their newly synthesized analogues. Using 1H-NMR and 2D-NOESY experiments in conjunction with energy-minimized structures generated through MMFF94s calculations, the group concluded that removing the C15 and C18 methyl groups minimally affects the macrocyclic backbone, with 11, 12 and 13 adopting almost identical conformations. Additionally, 14 possesses a significantly altered conformation, affecting both the local C14-C29 domain and bis-tetrahydropyran core (Figure 11).
Figure 11.
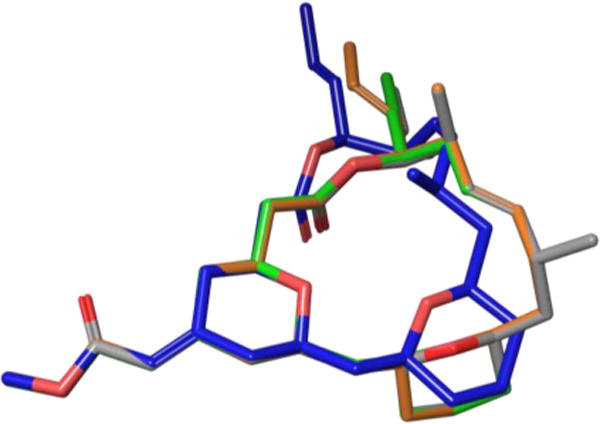
Conformational overlay of 11 (gray), 12 (orange), 13 (green), and 14 (blue).
Biological testing of 14 showed no growth inhibition against A549 and NCI-H460 cancer cell lines, establishing the importance of maintaining exiguolide’s natural conformational profile. While 12 demonstrated comparable activity to the parent natural product (A549 IC50 = 3.14 μM vs. 1.66 μM for 11) 13 exhibited no activity at >100 μM, leading them to suggest that the C18 methyl group plays some role in activity unrelated to conformation. However, as the removal of either the C15 or C18 methyl should significantly alter local conformational preferences through the loss of A1,3-strain, it is possible that the overall conformational preferences of 13 were changed to a greater degree than their preliminary analysis anticipates. More detailed conformational analysis employing some of the techniques outlined in this review and the additional synthesis of an 18-desmethylexiguolide analogue should hopefully clarify the effects of the C15 and C18 methyl groups’ effects on biological activity.
3.4 Discodermolide
In 1990, Gunasekera and Longley isolated discodermolide 15, a linear polyketide natural product, from the marine sponge Discodermia dissoluta (Figure 12).40 While initially reported as an immunosuppressive agent, follow up studies later established 15 as a potent microtubule-stabilizing agent that retains low nanomolar cytotoxicity against paclitaxel-resistant cell lines.41 As expected, this anti-tumor activity spurred numerous synthetic efforts over the years, with a sizeable number of total syntheses reported to date.42 One noteworthy undertaking by Novartis utilized a mixture of the Paterson and Smith routes, eventually producing more than 60 grams of material for use in clinical studies. However, phase I trials were ultimately halted as a result of severe lung toxicities.43
Figure 12.
The microtubule-stabilizing agent, discodermolide 15.
Since certain discodermolide structural features may serve a non-essential role for human chemotherapy, removing these extraneous features would create a more accessible synthetic target and aid the development of more therapeutically benign discodermolide analogues. Several independent laboratories have focused primarily on determining discodermolide’s bioactive conformation for assisting with this analogue design. In 2001, the Smith44 and Snyder45 groups independently conducted extensive conformational studies with 15 using molecular modeling and high-field NMR experiments. Interestingly, these studies showed that discodermolide’s acyclic structure possesses a surprisingly limited conformational profile, with the most favored conformation exhibiting a “hairpin” motif as a result of several key features.
First, the Z-olefins within the polyketide backbone introduce A1,3-strain, stabilizing the C7-C10, C12-C15, and C20-C23 regions of the molecule. The multiple polypropionate substitutions also force discodermolide in conformations that minimize syn-pentane interactions, creating two major turns in the C10-C12 and C16-C20 backbone. Finally, the six-membered lactone ring interconverts between chair, half-chair, and skew-boat conformations, with a hydrogen-bonding interaction between the C7-hydroxyl and the lactone ring oxygen having some moderate influence over the C5-C7 torsional angles (Figure 13). Later transfer-NOE spectroscopy performed in the presence of both unassembled tubulin and assembled microtubules determined that the bound conformation of discodermolide closely matches that of the solution structure, save for some subtle differences in the orientation and shape of the ring.46
Figure 13.
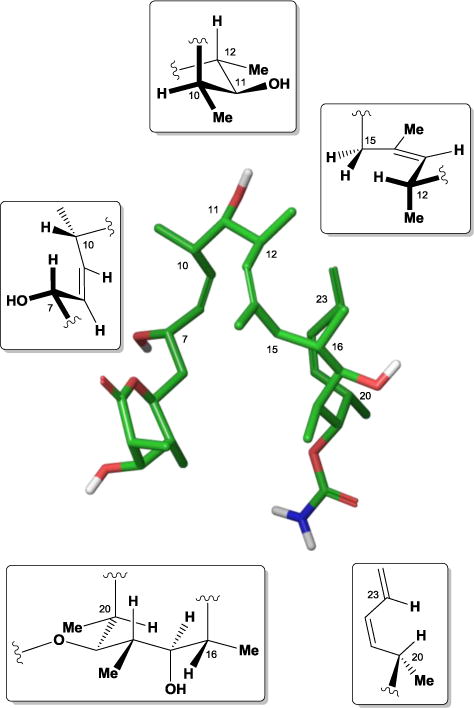
Discodermolide’s major solution conformation and conformational control elements.
The formation of this stereochemically-rich lactone ring requires numerous synthetic steps, reducing the practical efficiency of many discodermolide syntheses. Investigations into the necessity of this structurally complex functionality showed that inversion of the C4 and C5 stereocenters did not lead to a loss in potency.47 In addition, structure-activity relationship studies revealed that the 2,3-anhydro derivative also retains nanomolar cytotoxicity.48 As a result, the Smith laboratory hypothesized that the substituents on the ring orient the lactone in a conformation that maximizes binding interactions rather than serve any binding purpose of their own. Thus, they proposed a more conformationally rigid five-membered scaffold as a simplified substitute for the complex six-membered lactone ring. The synthesis of this simplified analogue required only three steps from one of their advanced intermediates and used cheap and commercially available levulinic acid, giving furan 16 in 45% overall yield. The resulting unsubstituted butyrolactone displayed a near 10-fold improvement over 15 against the MCF-7 cancer cell line. The subsequent discovery that the C7-hydroxyl played a non-essential role in promoting cytotoxicity suggested that the entire C1-C7 region could be simplified without penalty.49 Smith and co-workers tested this hypothesis by synthesizing an analogue that replaced the C1-C7 region with a coumarin moiety. Excitingly, the resulting analogue 17 also displayed low nanomolar cytotoxicity against multiple cell lines, despite possessing only 8 of discodermolide’s 13 stereocenters (Figure 14).50 Given this in vitro success, in vivo testing and further development of simplified discodermolide analogues could still yield a promising drug candidate despite the earlier clinical failure of the natural product.
Figure 14.
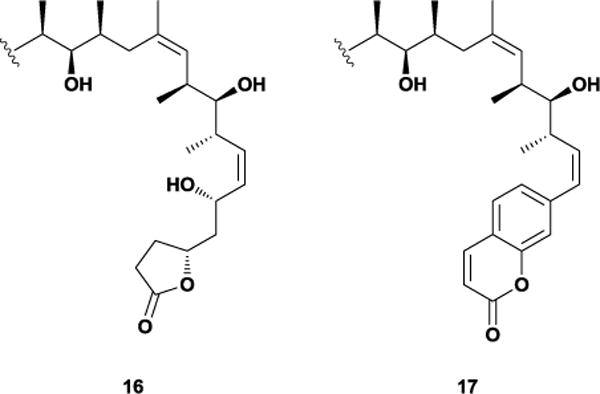
Conformationally-simplified analogues of discodermolide.
3.5 Dictyostatin
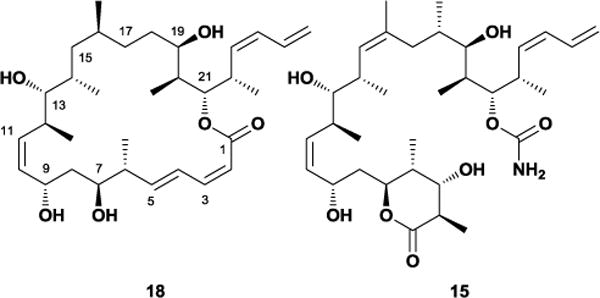
In 1994, Pettit and co-workers isolated dictyostatin 18, a polyketide structurally resembling a cyclic version of discodermolide 15, from a marine sponge collected off the Republic of Maldives (Figure 15).51 Like discodermolide, 18 exhibits potent microtubule-stabilizing activity, retains cytotoxicity against multidrug-resistant cell lines, and binds the taxoid site.52 Given the failure of discodermolide in clinical trials, attention has turned towards dictyostatin as a potential alternative. To date there have been eight published total syntheses of 18,53 with the Paterson54 and Curran55 groups additionally providing a wealth of analogues for testing. Although most of these analogues have exhibited diminished activity, their cumulative biological results have provided a comprehensive picture of dictyostatin’s structure-activity relationships.56 Recent pharmacological studies in mice have shown that dictyostatin crosses the blood-brain barrier to effect prolonged microtubule stabilization in the brain, hinting at its potential to treat neurodegenerative disorders such as Alzheimer’s.57
Figure 15.
Comparison of dictyostatin and discodermolide structures.
While a crystal structure of dictyostatin has yet to be reported, extensive NMR experiments have provided ample data for conformational studies. The low isolation yield of Pettit and co-workers initially led to an incomplete stereochemical assignment, leaving the structure of dictyostatin unclear until its re-isolation in 200352 and subsequent work by Paterson and co-workers in 2004.58 By utilizing both homonuclear (3JH,H) and heteronuclear (2,3JC,H) coupling constants in combination with NOESY experiments they were able to determine the complete relative stereochemistry, which was later confirmed as the absolute configuration through the concurrent synthetic efforts of the Paterson53a and Curran53b groups. The Paterson group further combined this NMR data with Monte Carlo conformational searches to propose a pair of interconverting atropisomers, wherein the lactone adopts either a C1-C2 s-trans or s-cis orientation. Of the two, the lower-energy s-trans structure bears a strong resemblance to the solid state conformation of discodermolide. Later work by Canales et al led to a proposed bioactive conformation of dictyostatin based on TR-NOESY data, which overlays well with the s-trans solution structure save for a major torsional change around the C8-C9 linkage (Figure 16).46b
Figure 16.
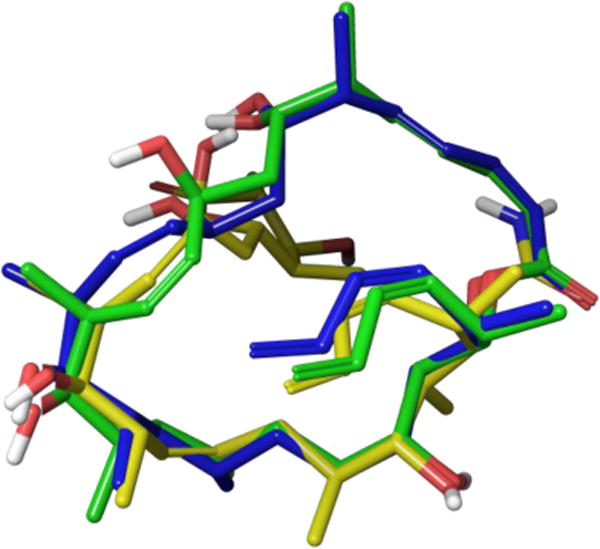
Overlay of s-trans structure (blue) with proposed bioactive conformation (green) and solid state structure of discodermolide (yellow).
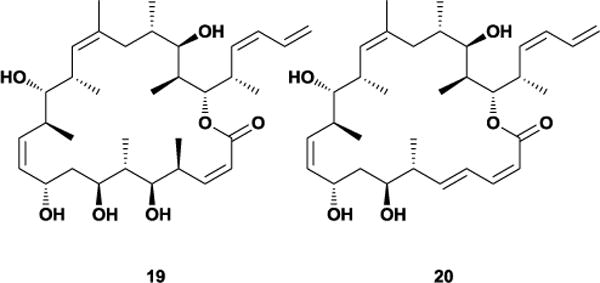
Given the similarities between discodermolide and dictyostatin there has been substantial interest in developing hybrid analogues to explore any shared pharmacophoric elements. Early work by Curran and co-workers pre-dated the elucidation of the true structure of dictyostatin, but still established that simplified macrocyclic analogues could retain modest biological activity.55a Following the stereochemical reassignment of dictyostatin the Paterson group revisited the issue by designing and synthesizing their own hybrid molecule 19, which computationally matched well with the solid-state conformation of discodermolide.59 Initial biological testing revealed that this compound was approximately 10-fold less active than discodermolide, and follow-up studies revealed a further loss in potency against Taxol-resistant cell lines.54e After examination of the NMR-derived bioactive conformations of discodermolide and dictyostatin, Paterson and co-workers further refined their hybrid model to include the C1-C5 dienoate region proposed to be necessary for dictyostatin’s binding affinity.60 The improved hybrid 20 demonstrated nanomolar inhibition against several human cancer cell lines, including Taxol-resistant ones. Interestingly, thermodynamic experiments have found that the acyclic discodermolide’s binding to tubulin is entropically more favorable than the cyclic dictyostatin, suggesting that the latter’s activity may be unrelated to any relative lack of conformational freedom.61
Recently, Snyder and co-workers disputed the conformational picture of dictyostatin as incomplete, arguing that its structure is not rigid enough to support only two conformational families.62 Using NAMFIS to re-evaluate the methanol-d4 NMR data set, they identified sixteen conformations ranging in population from 11.4% to 2.5%; although the previously proposed major solution conformation was not substantially populated, they did find a roughly equal distribution of C1-C2 s-trans and s-cis orientations amongst the set. They also acquired NMR data in DMSO-d6 in order to investigate solvent effects with more quantitative data, ultimately finding fifteen conformations with the s-cis being primarily represented. Among the conformations from the two solvents they found that one of their DMSO structures was a good fit for the TR-NOESY data reported by Canales et al. Subsequent docking of this compound to tubulin uncovered a competitive binding pose that is radically different from the previously reported one, yet compatible with several aspects of known dictyostatin SAR.
While this latest work significantly expands on the conformational profile of dictyostatin, energy calculations on the newly-obtained conformations would have been beneficial in gauging their feasibility. The authors are correct in asserting that force field energies aren’t necessarily accurate but neglected to explore higher ab initio methods to assess relative strain energy, particularly given the prevalence of the higher-energy s-cis form amongst their conformations. Additionally, their binding pose suffers from both the low resolution of the crystal structure used and the inherent inaccuracies of rigid docking to a binding pocket that originally contained a different ligand. Given the significant discrepancies between reported binding poses, additional experimental or crystallographic work is necessary to resolve the issue of dictyostatin’s bound conformation.
3.6 Trienomycin A
The ansamycin natural products are a broad class of polyketides that exhibit potent antibiotic and anti-neoplastic activity, including the well-known rifamycins. In 1985, Umezawa and co-workers isolated the structurally related natural products, trienomycins A-F, from a culture broth of Streptomyces sp. No. 83-16.63 Subsequent biological testing revealed that the trienomycin class displays potent in vitro cytotoxicity against multiple cancer cell lines.64 Of the six known ansamycins, trienomycin A 21 demonstrated the strongest cytotoxicity with an IC50 value of 0.01 μg/mL against L-5178Y murine leukemia and human PLC hepatoma cell lines. The trienomycins primarily consist of a 21-membered macrolactam ring adorned with a non-redox active phenol, an (E,E,E)-triene, four stereocenters, and an N-acylated D-alanine side chain, varying only in the composition of the terminal side chain functionality (Figure 18).
Figure 18.
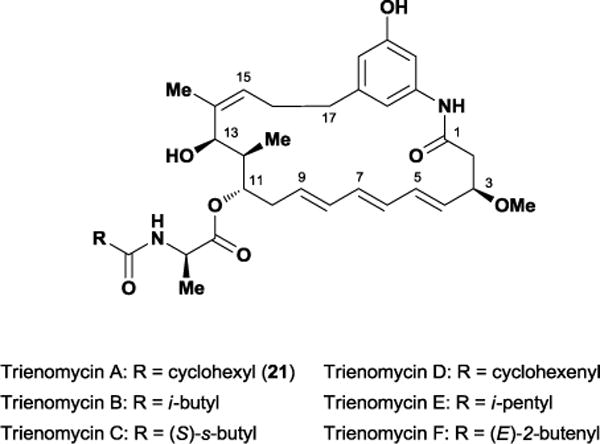
The ansamycin natural products, trienomycins A-F.
Unfortunately, the clinical potential of the trienomycins represents an underexplored avenue as a result of supply issues and minimal structure-activity relationship studies. Funayama and co-workers prepared four semi-synthetic trienomycin A analogues and concluded that the C13-OH, the triene subunit, and the N-acylated side chain remained essential for potent cytotoxicity.65 However, methylation of the free phenol produced an analogue equally active to the parent natural product. These classical SAR studies reveal the difficulty in correlating molecular functionality to biological activity within a polyketide macrocyclic framework.
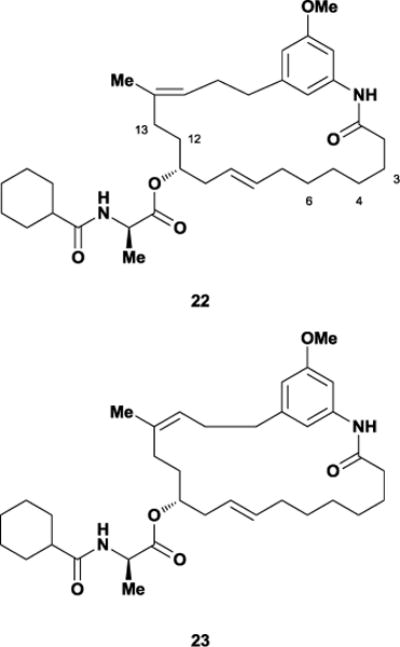
The Blagg laboratory addressed these difficult issues by employing a rational, conformational approach for designing simplified analogues of trienomycin A.66 After generating low-energy conformations of the semi-synthetic analogues with SYBYL,67 Blagg and co-workers showed that while Funayama’s inactive derivatives exhibited significant conformational differences from trienomycin A, the active methyl ether analogue retained the parent geometry. The Blagg group hypothesized that mimicking the conformational preferences of 21 properly orients the critical phenol unit and N-acylated side chain within trienomycin’s binding pocket, thereby promoting tight binding. Interestingly, modeling suggested that removing the C13-OH, C12-Me, and C3-OMe functional groups minimally perturbed the macrolide’s overall conformational preferences. Moreover, additional removal of the C4/C6 alkenes and inversion of the C14-(Z)-alkene geometry produced monoene A 22, which SYBYL calculated would adopt a conformation strongly resembling that of 21 (Figure 19).
Figure 19.
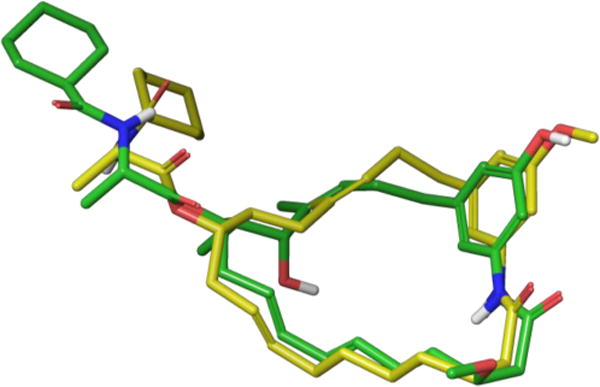
Overlay of trienomycin 21 (green) and monoene A 22 (yellow) structures generated through SYBYL molecular modeling.
The synthesis of 22 required only 13 steps from readily available starting materials. This concise route allowed the multi-gram preparation of not only monoene A but also monoene E 23, which SYBYL calculations indicated would act as a negative control due to its low similarity score. As their model predicted, the conformationally similar 22 displayed potent anti-proliferative activity (0.47 nM, MCF-7 cell line) while the lowest similarity structure 23 displayed no significant activity. Buoyed by these results, the Blagg laboratory embarked on an SAR investigation of the side chain using the new simplified macrocycle as a framework, specifically the effects of altering the amino acid side chain. The resulting analogue work revealed that altering the steric bulk of the α-methyl on the side chain is detrimental to activity, as is lowering the steric bulk of the alkyl amide. Blagg’s approach shows that a simplified analogue of the synthetically-complex trienomycin A could be used for faster determination of SAR and could potentially be used for elucidation of the biological target. However, given the near-certainty that 22 is significantly more flexible than the parent natural product, it is possible that trienomycin’s mode of cytotoxicity may be more dependent on recognition of the side chain rather than the macrocycle.
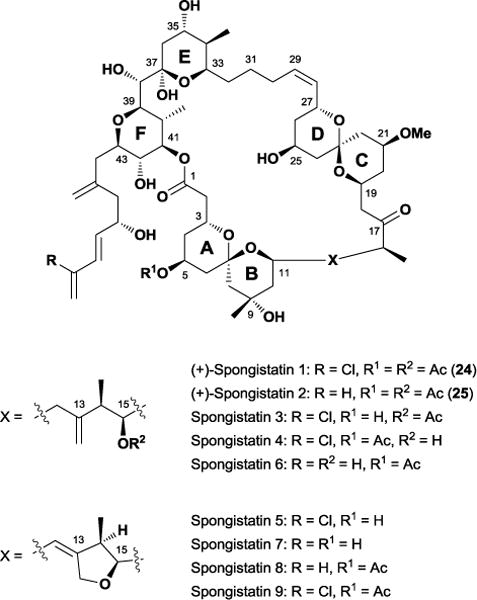
3.7 Spongistatin
The spongistatin class of natural products was first reported in the early 1990s by three separate groups.68 Isolated from an Eastern Indian Ocean sponge of the genus Spongia, members of this family possess potent antineoplastic properties, with spongistatin 1 24 in particular displaying an average IC50 value of 0.12 nM against the NCI panel of 60 human cancer cell lines. The activity observed during cell growth inhibition assays led Hamel to propose that the spongistatins’ mode of action involves tubulin binding, and hypothesized a “polyether” binding site on β-tubulin near the vinca domain to account for its competitive inhibition of maytansine and rhizoxin.69
The complex structure and potent activity of the spongistatins attracted considerable attention from laboratories interested in exploring its potential as a chemotherapeutic agent. Given their scarcity and difficulty to obtain from nature, significant resources have been devoted towards their creation in the laboratory, with several total syntheses of 2470 and 2571 reported to date. Among these, the Smith laboratory took possibly the most ambitious approach in their fourth-generation synthesis of spongistatin 1. Utilizing a multi-component dithiane union tactic, Smith and co-workers assembled multiple, key fragments in a scalable fashion to yield a remarkable 1.009 g of material.72
Having obtained enough material for preclinical development, the Smith group turned towards the task of identifying the moieties necessary for cytotoxicity through SAR studies.73 Given the difficulty in constructing analogues even through their optimized route, the Smith group investigated whether the entire structure of the flexible spongistatin skeleton was necessary for biological activity. Based on the structural features common amongst the natural spongistatins and results from several early analogues,74 the Smith group hypothesized that the spongistatin’s western perimeter represented the main pharmacophoric motif. Furthermore, the Smith laboratory believed that as long as the C37-C39 dihedral angles between the E and F rings maintained a proper orientation, replacing the remainder of the complex molecule with a simplified linker would still produce a molecule with potent activity.
To facilitate the design of such a linker, the Smith group used molecular modeling as a means of determining spongistatin’s conformational preferences.75 Despite a previously reported solution conformation,76 their calculations and resulting analysis of the macrocyclic torsional angles using an expansion of the Taylor laboratory’s polar map methodology77 indicated a fairly rigid western perimeter. The remainder of the molecule exhibited high flexibility and adopted multiple conformations. To combat the problems they perceived with NMR-constrained molecular dynamics (high-energy conformations) and molecular mechanics (inaccurate energy ordering), Smith and co-workers developed a hybrid software method called Distribution of Solution Conformers (DISCON).78 DISCON uses NMR-derived interproton distances and torsional angles for determining the most populated families from a computationally generated library of conformers, utilizing hierarchical clustering and a genetic algorithm to avoid the overfitting problems that can arise from NMR-based structural optimization in methods like NAMFIS.
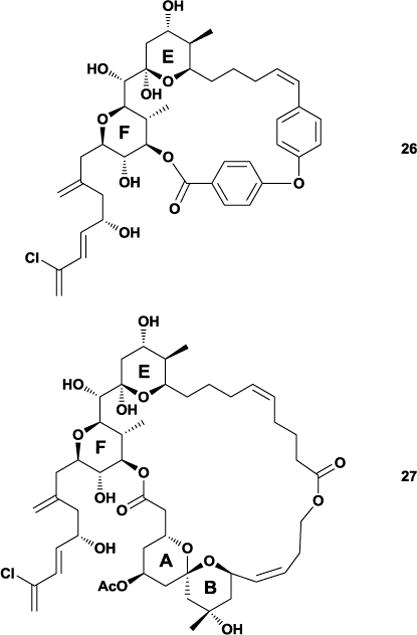
From this DISCON analysis, the Smith laboratory found that 24 existed as an ensemble of four major solution conformations, which when overlaid showed that the western perimeter of the macrocycle maintained a common conformation reflecting an internal hydrogen bonding network in support of their earlier modeling work (Figure 23). To test their linker hypothesis, the Smith group designed an analogue that would possess only the E and F ring systems, ultimately deciding on a biaryl ether tether that would maintain the conformational twist observed during conformational analysis. Synthesis of this molecule (26) ultimately revealed that the strain energy imparted during macrocyclization opened the E ring system during a global deprotection. To combat this issue, the Smith group redesigned their analogue to instead contain a more flexible polymethylene tether. Since previous attempts at replacing the eastern perimeter with a simple tether failed, they also incorporated an internal hydrogen bond acceptor within the molecule as a means of lowering the overall flexibility. Following synthesis of this second-generation analogue (27), DISCON studies found that its major conformations retained the rigidity earlier observed in the ABEF ring system and even imparted a certain degree of restraint on the side chain. Incredibly, biological testing found that 27 displayed nanomolar cytotoxicity against several cancer cell lines and a subsequent cell cycle analysis concluded that the molecule also shared the same microtubule-destabilizing activity as 24.79 This work demonstrates how the pharmacophoric elements of a complex natural product are likely to reside on a conformationally-rigid area, and that replacing flexible regions with simplified tethers is a viable design strategy.
Figure 23.
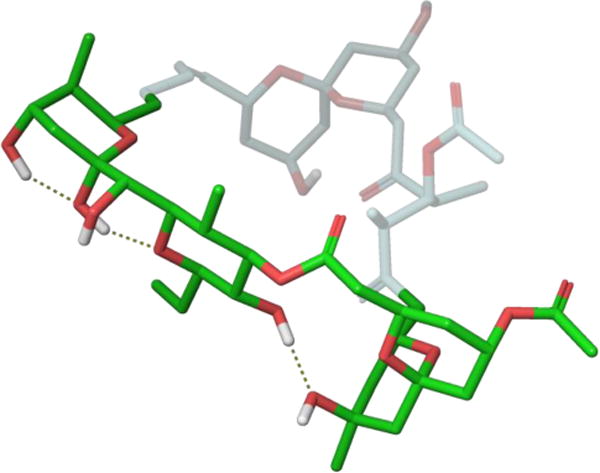
Retained ABEF conformational structure of spongistatin 1 (green).
4. Conformationally-restricted analogues of polyketide natural products
Although flexible polyketides adopt preferred conformational profiles, typically only one of these structures will resemble the ideal bound conformation. As each member of a conformational ensemble has an opportunity for protein binding equivalent to its population in solution, altering their relative populations in solution is likely to affect biological activity. Therefore, analogues that alter a molecule’s conformational preferences to favor either a single conformer or family of conformers could be used as a method for identifying the bioactive conformation in the absence of bound crystallographic or spectroscopic data. Additionally, modifications that preferentially favor the bound structure may lead to various improvements over the unrestricted natural product.
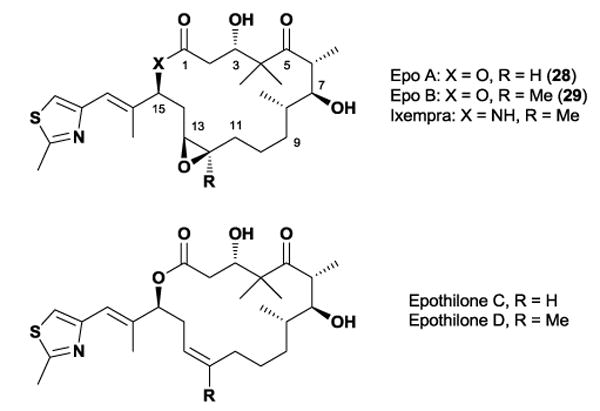
4.1 Epothilone
In 1987, Reichenbach and Höfle isolated the epothilone class of natural products from the soil-dwelling myxobacterium Sorangium cellulosum.80 Nearly a decade later, a group at Merck Research Laboratories discovered that the family not only possessed microtubule-stabilizing properties, but also exhibited similar biological properties as the well-known drug, paclitaxel.81 The epothilones also possessed improved water solubility and retained activity against P-glycoprotein-expressing multiple drug resistant cell lines, including those resistant to paclitaxel.82 The intriguing biological activity, pharmacological properties, and accessibility through fermentation spurred significant interest towards developing the epothilones as an anticancer agent, with several semi-synthetic epothilone-type compounds entering clinical trials and one, an epothilone B lactam analogue, being approved for use in humans as the drug Ixempra (ixabepilone) in 2007.83 In addition to clinical development the epothilones received a staggering amount of attention from the synthetic community, which has been thoroughly covered in reviews elsewhere.84
Early displacement experiments revealed that the epothilones compete for the same tubulin-binding site as paclitaxel, which prompted interest in their three-dimensional structure and potential homology between the two agents. However, epothilone’s unknown relative stereochemistry plagued early efforts to elucidate the molecule’s solution conformational preferences.85 In 1996, Höfle and co-workers used X-ray crystallography as a way of assigning the relative and absolute stereochemistry of epothilone A 28.86 Using a combination of vicinal coupling constants and observed NOEs, they concluded that the solution conformation of 28 was highly similar compared to its solid-state conformation. However, more detailed NMR studies in multiple solvents showed that several conformational families existed in solution. Solution studies conducted by our laboratory showed that the epothilone macrolide core possessed two separate solution conformations in the C1-C8 region, with the major conformation resembling the previously reported solid-state structure.77 A similar analysis of the C11-C15 region demonstrated that flexibility in this region provided additional conformational families, which further complicated the overall picture in solution (Figure 25).87
Figure 25.
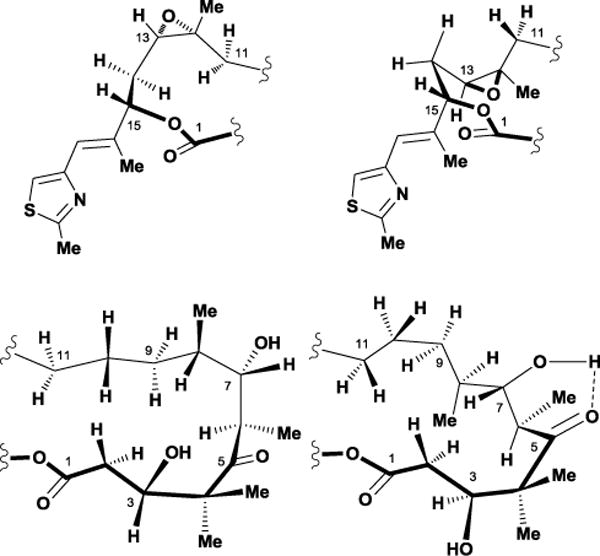
Conformational analysis of the C1-C8 and C11-C15 regions within 29.
In an effort to distinguish between several conformational families in solution, our group investigated multiple structural analogues of the epothilones. We hypothesized that the incorporation of additional A1,3-strain or syn-pentane interactions would alter the relative population of solution conformers, anticipating that the biological activity of the resulting analogues would indicate the bound conformation. To test this theory, we synthesized two diastereoisomeric C14-methyl substituted epothilone analogues, (S)-14-methyl 30 and (R)-14-methyl 31 (Figure 26).87 We postulated that the introduction of a methyl substituent would alter the relative population of the two observed C11-C15 solution conformational families via A1,3-strain. Interestingly, biological testing of both compounds revealed that 30, which resembled the solid-state conformation, retained activity while its C14 epimer 31 lost all cytotoxicity. In a subsequent study, installation of a C14-methoxy group in the same stereochemical orientation resulted in an epothilone D analogue with significantly improved activity over the unmodified compound.88
Figure 26.
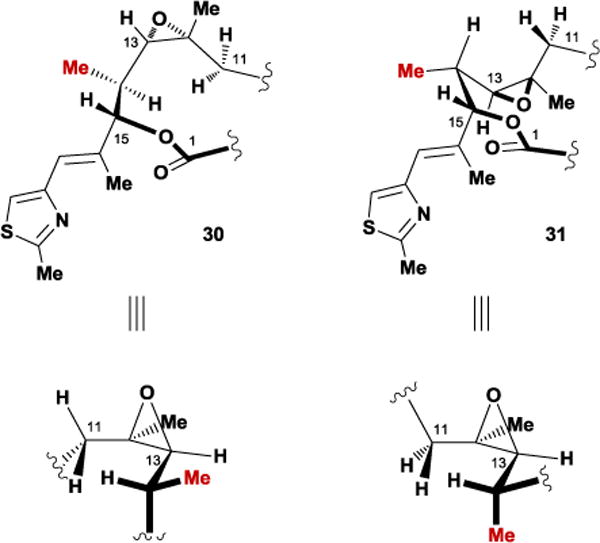
Conformational analysis of the C11-C15 region within the C14-Me epothilone analogues.
The conformational preferences of the C11-C14 region also explained the puzzling results observed by the Danishefsky laboratory in their synthesis of (E)-9,10-dehydro epothilone B 32.89 In the last step of the synthesis, Danishefsky and co-workers found that epoxidation of the (E)-9,10-dehydro epothilone D alkene precursor proceeded with an unusual preference for the α-face, which they rationalized on the change in conformational preferences induced by the 9,10-unsaturation. The Danishefsky group also attributed the molecule’s significant increase in potency to conformation as well, since the unsaturation rigidifies the C9-C10 torsional angle and stabilizes the C25 methyl through minimization of A1,3-strain. The resulting conformation resembles the solid-state and major solution conformations (Figure 27). In addition, analogues probing the conformational families within the C5-C10 region supported a bound conformation mimicking the Höfle solid-state conformation.90 Intriguingly, transfer-NOE and cross-correlated relaxation NMR studies with monomeric tubulin also supported this conclusion.91
Figure 27.
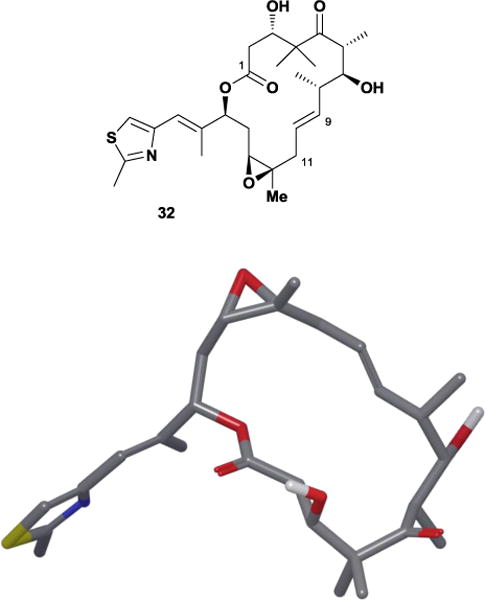
(E)-9,10-dehydroepothilone B 32 and its major solution conformation.
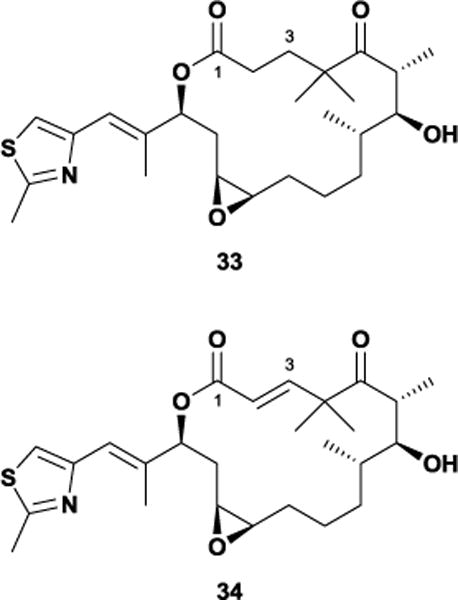
In 2004, Nettles and co-workers published an electron crystallographic structure of epothilone A bound to α,β-tubulin in zinc-stabilized sheets, which reported a ligand conformation that diverged significantly from conformations derived via NMR spectroscopy and the previously mentioned analogue studies.92 One potential reason for this may be that the crystal structure had insufficient resolution to define epothilone’s conformation, forcing the investigators to supplement their work with a NAMFIS-derived structure. Given these contradicting results, Erdélyi and co-workers embarked on a study of epothilone’s conformational preferences in aqueous solution as a way of rectifying the dissimilarity between the reported bound and free structures.93 They also addressed the conformational role of the 3-hydroxy functional group, as two analogues lacking this moiety, C3-deoxy 33 and 2,3-dehydro 34, remained active despite its proposed H-bonding interaction within the tubulin-binding site.94 Their analysis revealed that the previous NMR-derived conformation of unbound epothilone A was a probable component of their aqueous conformational ensemble, and found that the EC-derived conformation was not significantly populated in water. Moreover, Erdélyi et al observed that the C3-deoxy analogues possessed similar conformational preferences and that removal of the C3-hydroxy both with and without the incorporation of a trans double bond between C2 and C3 does not alter the preferred overall macrolide conformation.
Recently, Prota and co-workers resolved the issue of epothilone’s bioactive conformation by obtaining a high-resolution X-ray co-crystal structure of epothilone A bound to α,β-tubulin.95 They accomplished this extraordinary feat through the incorporation of stathmin-like protein RB3 and tubulin tyrosine ligase to the complex. This resulted in a protein-ligand complex with high enough resolution to directly define the conformation of bound epothilone A, which matched well with both the bound and solution structures reported in earlier NMR and analogue studies (Figure 29). The complicated history of the epothilones shows how accurate prediction of a polyketide’s conformational preferences can lead to the prediction of its bioactive structure, which is of critical importance when a well-defined protein co-crystal structure cannot be easily obtained.
Figure 29.
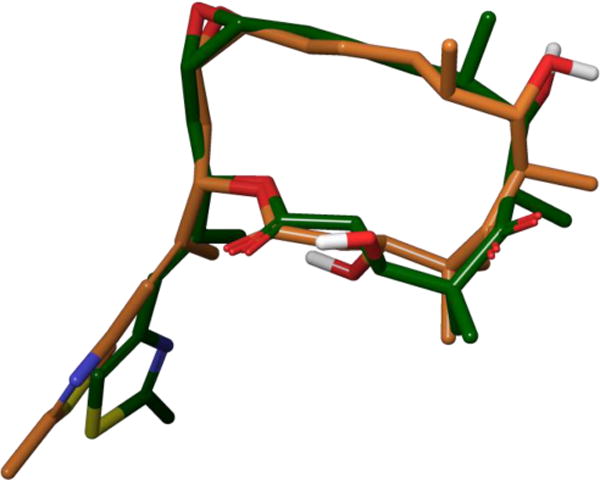
Overlay of the major solution conformation (green) and tubulin-bound conformation (orange) of 28.
4.2 Geldanamycin
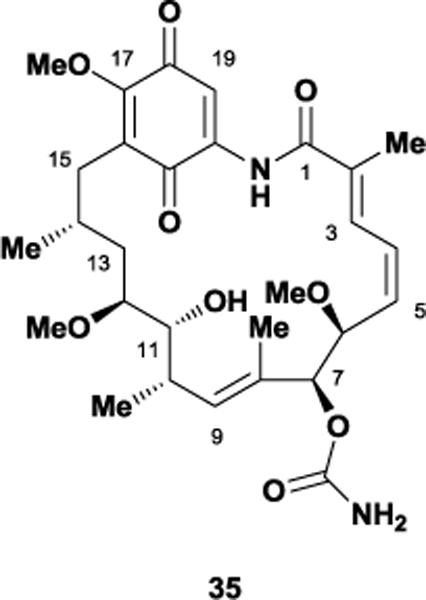
As mentioned previously, the ansamycin natural products have received considerable attention as potential chemotherapeutic agents for the treatment of several diseases. In particular, the benzoquinone ansamycin, geldanamycin 35, exhibits potent anti-cancer activity by binding the ATP site on heat shock protein 90, a molecular chaperone involved in the folding and maturation of proteins upregulated in tumors. In 1970, DeBoer and co-workers isolated 35 from Streptomyces hygroscopicus var. geldanus,96 with its molecular structure being elucidated soon thereafter by the Rinehart laboratory.97 The 21-membered polyketide contains a trisubstituted benzoquinone ring, six stereocenters, and an α,β-unsaturated dienamide. As expected, this structurally daunting macrocyclic framework has challenged numerous synthetic chemists.
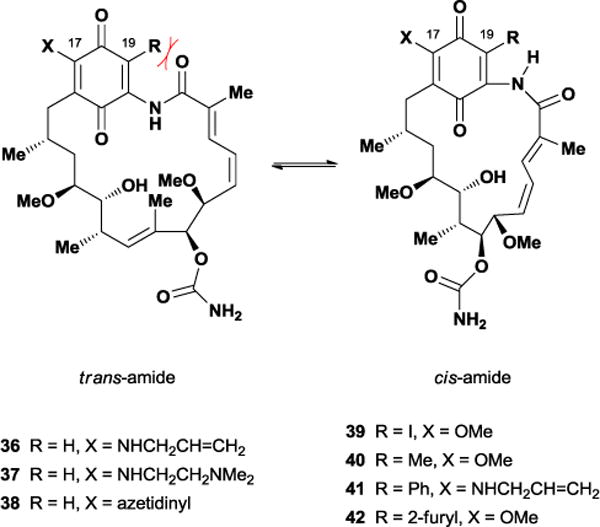
In 2002, Andrus and co-workers accomplished the first total synthesis of 35 utilizing glycolate aldol chemistry and a late stage oxidation for construction of the challenging quinone unit.98 Six years later, the Panek laboratory also completed its total synthesis in only 20 linear steps and 2% overall yield.99 Biosynthetic production would later provide large quantities of geldanamycin and expedite the synthesis of multiple semi-synthetic analogues, with most modifications occurring at the C17 quinone position. Structural alterations at this position included addition-elimination reactions with several nucleophiles such as alkoxides, hydroxide, phenoxide, and amines. The C17 position was targeted as a means of improving the solubility of geldanamycin, however, even the most promising allyl amine derivative 36 still retained significant toxicity. Literature reports suggest the hepatoxicity of geldanamycin arises via the reaction of biological nucleophiles such as glutathione with the C19 quinone.100 Curiously, geldanamycin possesses two condition-dependent conformations; an extended trans-amide solution/solid-state conformation supported by X-ray crystallography101 and NOE experiments,102 and a closed ‘C-clamp’ cis-amide bound conformation arising from protein co-crystallography.103 This discontinuity sparked a number of different studies, which disagreed on either the energetic barrier of the isomerization or its overall necessity for binding and inhibition.104 Moody and co-workers hypothesized that substitution at geldanamycin’s C19 position could potentially kill two birds with one stone, blocking glutathione’s nucleophilic addition while also forcing the unbound macrocycle into a cis-amide conformation. In doing so, they believed they could attenuate geldanamycin’s toxicity issues while also investigating the conformational requirements of its binding.105
Starting from commercially available geldanamycin, Moody and co-workers prepared a wide variety of C19-substituted analogues. As modification of this site through nucleophilic addition possessed limited scope, the Moody group maximized analogue diversity by employing Stille coupling reactions with 19-iodogeldanamycin 39, which was readily obtained in a single step from 35. After successfully synthesizing 19-methylgeldanamycin 40 in 86% yield, the Moody laboratory attached several electron-rich and electron-deficient aromatic groups to the C19 position in good to excellent yields. Since 17-substituted derivatives such as 36 and 37 showed earlier clinical promise,106 the Moody group also synthesized multiple analogues of their 19-substituted geldanamycin derivatives.
Interestingly, the 19-substituted analogues displayed higher polarity than geldanamycin. Moody and co-workers undertook conformational NMR studies as a means of examining whether observed polarity resulted from the hypothesized trans- to cis-amide isomerization. While 2D NMR work on geldanamycin and the 17-amino derivative 38 suggested a preference for the trans-amide conformation, the 19-substituted compounds revealed substantial 1H and 13C chemical shift differences that implied a major change in the chemical environment throughout the macrocycle. Quantitative NOE studies performed on 19-phenyl-AAG 41 strongly suggested that the cis-amide conformation formed predominantly in solution. The resulting ‘C-clamp’ form seen in protein crystallography studies moves the hydrophobic surface area into the heart of the structure while exposing hydrophilic moieties to the solvent resulting in the observed increase in polarity. X-ray crystallography studies on 19-substituted analogue 42 confirmed these results, with the resulting crystal structure overlaying well with that of protein-bound geldanamycin (Figure 32).
Figure 32.
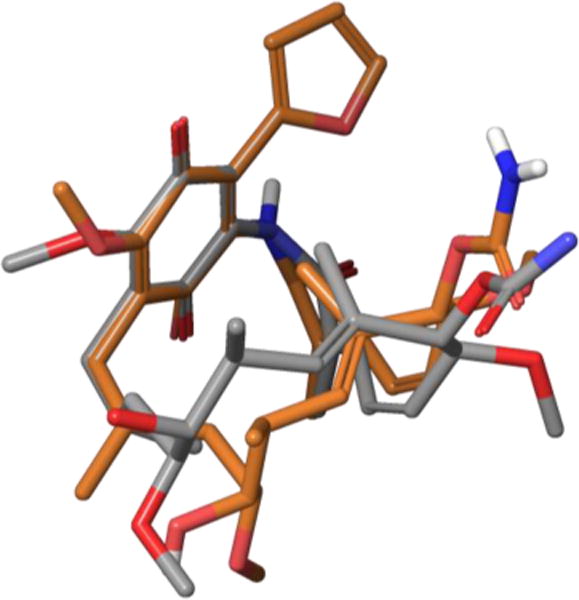
Overlay of solid-state 42 (orange) with protein-bound 35 (gray).
Using N-acetylcysteine methyl ester as a model for glutathione attack, Moody and co-workers determined that while geldanamycin and 17-amino derivatives will react under basic conditions, 19-methyl and 19-phenyl analogues do not. To determine the biological implications of blocking nucleophilic attack at C19, the Moody group tested several 19-substituted derivatives using normal endothelial and epithelial cells. The resulting data showed that these analogues exhibit less toxicity than their parent compounds (35-37) in all tested cases. Protein crystallography studies confirmed that the 19-substituted compounds bind in the ATP site on Hsp90, although the added steric bulk alters the quinone ring’s bound position thus penalizing the molecule’s binding affinity. This work reiterates how simple alterations to a polyketide’s structure can have profound implications to its conformational preferences.
5. Polyketide Candidates for Conformation-Activity Relationship Studies
In addition to the studies outlined above, several biologically-active polyketides have received considerable attention with regards to conformational analysis. However, analogues designed to take advantage of their conformational preferences have yet to be reported, making them prime candidates for additional research in this area.
5.1. Peloruside A
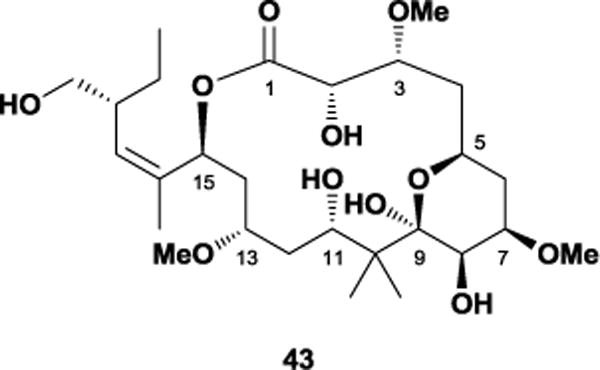
In 2000, West and co-workers isolated the polyketide, peloruside A 43, from the marine sponge Mycale hentscheli off the coast of New Zealand.107 This unique polyketide exhibited potent cytotoxicity with a mode of action similar to that of paclitaxel.108 Additionally, 43 possessed comparable activity to paclitaxel in both normal and P-glycoprotein multi-drug resistant cell lines.109 Interestingly, incubation studies found that unlike paclitaxel, excess laulimalide displaced peloruside, indicating that 43 shares a non-taxol binding site with the former. A subsequent study revealed that it also acts synergistically with paclitaxel and epothilone, which enhances overall antimitotic action.110 As the initial isolation yielded only 3 mg from 170 g of wet sponge, the Page laboratory attempted culturing the parent sponge on a large scale. Although successful in growing specimens, low yields and reproducibility proved that aquaculture was an unreliable source.111
Because of peloruside’s activity profile, complex structure, and limited availability, it has been an attractive target for synthetic groups. As the NMR solution studies in the isolation paper provided only the relative stereochemistry, peloruside’s initial total synthesis by De Brabander and co-workers in 2003 established the molecule’s absolute stereochemistry.112 In 2005, our laboratory completed the first total synthesis of natural (+)-peloruside A,113 with an additional four total syntheses reported to date.114 Although a number of analogues have been synthesized and evaluated for biological activity, the severe loss of activity demonstrated by peloruside congeners and analogues thus far has shed little light on the overall SAR picture.115
In 2006, Jiménez-Barbero and co-workers contributed to the puzzle by investigating peloruside’s tubulin binding site along with both the free and bound conformations.116 For the solution conformation, they utilized conformational searches and molecular dynamics simulations to generate a conformational library, which they compared with data generated through extensive NMR experiments in D2O. Their calculations indicated that peloruside A exists in two conformational families in solution, which differ mainly in the relative orientation of the C9-C15 region (Figure 34). To determine the bioactive conformation, Jiménez-Barbero and co-workers employed similar transfer-NOE experiments used by Carlomagno and Griesinger in their study of the epothilones.91 They concluded that the TR-NOESY cross-peaks strongly agreed with the earlier observed major solution conformation B, thus identifying it as the bound structure. Finally, the group used the above NMR-derived conformation in conjunction with docking studies as a means of proposing a binding site for peloruside on α-tubulin.
Figure 34.
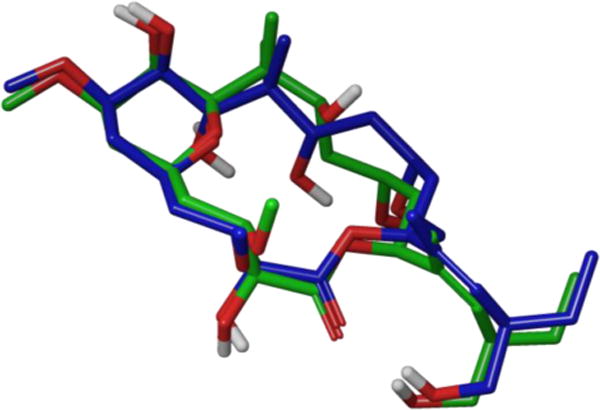
Overlay of peloruside major solution conformers A (blue) and B (green) in solution.
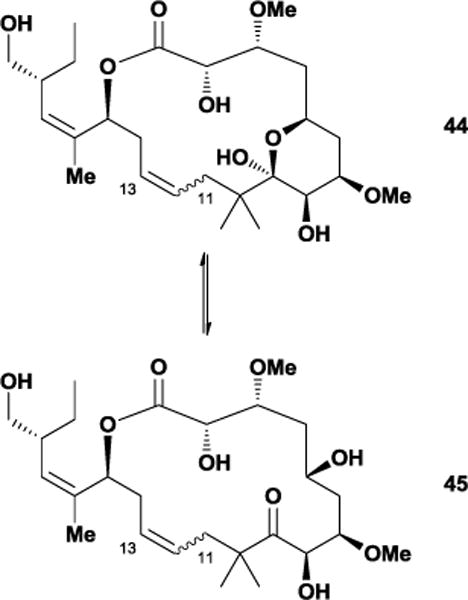
The relatively small number of conformations observed by Jiménez-Barbero and co-workers in their molecular dynamics simulations raised several questions within our laboratory. In particular, we believed that the lack of rigidifying features should not bias conformational preferences to such a degree. Since the previous study utilized only the MM3 force field, our group used additional force fields in Monte Carlo conformational searches as a way of expanding peloruside’s potential conformational space. After employing the MM2, MM3, and OPLS-2001 force fields in generating conformational libraries, we used our polar map methodology77 to confirm that the rigidity conferred by the lactone ester (s-trans), C2,C3-diol monoether (gauche effect), and C5-C9 dihydropyran (chair conformation) produced a relatively small number of conformational families.117 Our group also found that the C9-C15 region exhibited the previously-observed flexibility. We envisioned that replacement of the C11 and C13 stereogenic centers with a set of olefinic Z and E isomers could potentially offer additional insight into the tubulin-bound conformation of peloruside A by altering the relative populations of macrocyclic conformations.
Unfortunately, the completed analogues were highly unstable due to equilibration of the closed dihydropyran 44 to the isomeric open keto-alcohol 45, which degraded within days.118
The location of the peloruside/laulimalide-binding site on tubulin was also a matter of some debate. While Jiménez-Barbero and co-workers proposed an α-tubulin site based on their docking studies, experiments performed by Schriemer and co-workers identified a unique site on β-tubulin. They based this prediction on hydrogen/deuterium exchange mass spectrometry experiments, which tubulin mutation studies further supported in 2011.119 Both the bound conformation and the binding site were unequivocally confirmed earlier this year with the publication of a tubulin-peloruside A co-crystal structure.32 Prota and co-workers found that peloruside binds to the same β-tubulin site as laulimalide. Interestingly, its bound conformation is highly similar to the solution structures found earlier, differing only in the previously established flexible C9-C15 region (Figure 36). Given the scarcity of natural peloruside A and the activity loss demonstrated by its analogues and congeners, there would be considerable value in designing bioactive conformationally-simplified structures to facilitate further biological studies.
Figure 36.
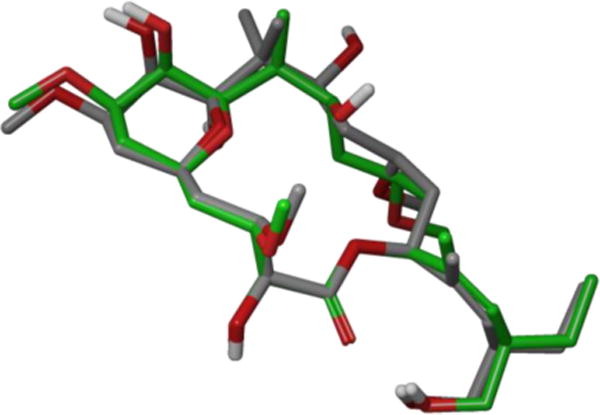
Peloruside’s tubulin bound conformation (gray) overlaid with proposed Díaz bound conformation B (green).
5.2. Zampanolide and Dactylolide
In 1996, Tanaka and co-workers isolated the natural product (−)-zampanolide 46 from the marine sponge Fasciospongia rimosa off the coast of Okinawa, Japan.120 Over a decade later, Field and co-workers also isolated the same molecule from a Tongan marine sponge, Cacospongia mycofijiensis, collected near ‘Eua Tonga.121 Biological evaluation performed by Field and co-workers revealed that zampanolide possessed low nanomolar cytotoxicity against multiple cancer cell lines and exhibited potent microtubule-stabilizing activity. Similar to other compounds discussed in this review, 46 showed no sensitivity to ovarian cancer cells overexpressing the P-glycoprotein multiple drug resistance pump. Intriguingly, Riccio and co-workers isolated a structurally similar polyketide, (+)-dactylolide 47, from the marine sponge Dactylospongia sp. off the coast of the Vanuatu islands.122 Although significantly less cytotoxic than zampanolide, studies by the Field laboratory established that 47 targets microtubules and promotes the in vitro polymerization of tubulin in a similar fashion as zampanolide. Moreover, Field and co-workers also discovered that both zampanolide and dactylolide promote microtubule polymerization by irreversibly alkylating β-tubulin via a unique covalent reaction between their α,β-unsaturated enone and the β-tubulin residue H229.123
Though zampanolide and dactylolide exhibit remarkable biological activities, low isolation yields and minimal quantities of material obtained from total synthesis have hindered further biological studies. As a result, both our group and the Díaz laboratory embarked on separate investigations of dactylolide’s solution conformational preferences as a way of gaining insight into the bioactive conformation of dactylolide and zampanolide’s macrolide core.123,124 Our laboratory envisioned that a detailed understanding of the molecule’s solution conformational preferences and bioactive conformation would greatly assist with future analogue design efforts.
In 2012, the Díaz laboratory used two complementary NMR spectroscopy based methods, STD (saturation transfer difference)125 and transfer-NOE spectroscopy, to determine dactylolide’s bioactive conformation while bound to α,β-tubulin heterodimers and fully formed microtubules. In order to accurately determine dactylolide’s bioactive conformation, Díaz and co-workers explored the molecule’s available conformational space employing Monte Carlo conformational searches. Of these conformations, they selected the lowest-energy conformer that best matched their experimental TR-NOESY data (Figure 38).
Figure 38.
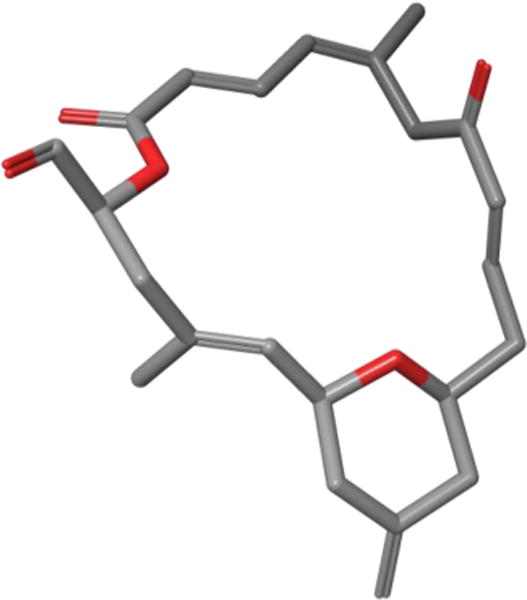
TR-NOESY derived bioactive conformation of dactylolide while bound to tubulin.
Since dactylolide undergoes fast exchange with tubulin, the conformation reported by Díaz and co-workers represents only one of several conformational families instead of an individual structure. In 2013, our laboratory thoroughly explored dactylolide’s solution conformational behavior utilizing a combination of Monte Carlo conformational searches, coupling constants, long-range NOE’s, and Smith’s DISCON software.124 A conformational library was generated using the combined results from three separate force fields, then deconvoluted using DISCON’s clustering analysis. The resulting ensemble consisted of three interconverting conformational families that matched the NMR data more than any single structure (Figure 39). As noted in our polar coordinate map analysis, the minimization of A1,3-strain controlled most of the backbone torsional angles within the macrolactone core. Interestingly, the two major families, conformers A and B, showed similar conformational profiles except for the orientation of the northern fragment and α,β-unsaturated enone. In contrast, the minor conformational family C somewhat resembled the other two families, except the C6–C7 bond rotates 180° giving the macrolide core a twisted shape.
Figure 39.
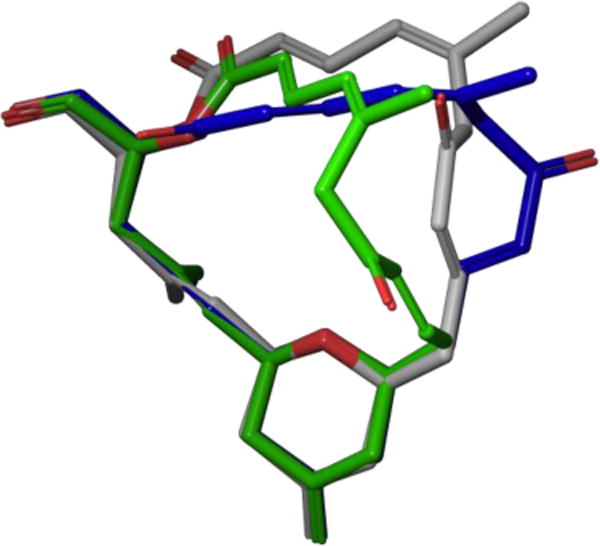
DISCON-derived solution conformations, major conformers A (blue) and B (gray) and minor conformer C (green).
Direct comparison of zampanolide and dactylolide’s coupling constant values shows a remarkable similarity between the conformational preferences of the two molecules. Thus, one may assume zampanolide’s side chain minimally affects the macrolactone conformation. In 2013, Prota and co-workers reported the high-resolution crystal structure of 46 covalently bound to tubulin, providing the bound conformation.95 As shown in Figure 40, the only major difference between zampanolide’s tubulin bound conformation and solution conformation appears in the enone region of the molecule. Our solution conformational analysis suggests this flexible C9-C10 enone freely rotates on a plane perpendicular to the pyran ring, with a preference for torsional angles of 0°, 120°, and −120°. The preference for these particular torsions results from the minimization of A1,3-strain through exclusively eclipsed conformations. However, upon closer examination of the C9 stereochemistry in the tubulin bound complex, we noticed the C7-C9 enone must adopt the s-cis orientation represented in solution conformer C prior to conjugate addition.
Figure 40.
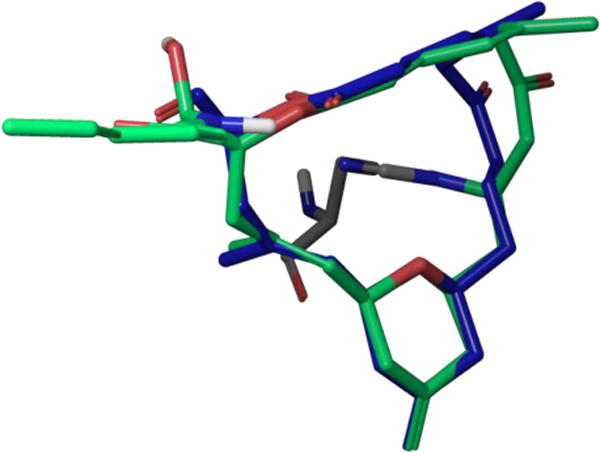
Overlay of zampanolide co-crystal structure (green) bound to tubulin H229 (gray) with solution conformer A (blue).
Since zampanolide makes few interactions with the Taxol binding site, our laboratory believes that the design of potent, bioactive zampanolide analogues should revolve around structural alterations that minimally affect the molecule’s native bioactive conformation. For instance, the Altmann laboratory showed that a C13-desmethylene dactylolide analogue 48 displays roughly equipotent antiproliferative activity compared with (−)-dactylolide. However, further excising the entire tetrahydropyran subunit from 48 afforded ether 49, which possessed reduced antiproliferative activity by over 20-fold.126 One possible reason for this observed loss of cytotoxicity may be the removal of the C15 stereogenic center, which results in the subsequent loss of rigidifying A1,3-strain. Thus, removing the tetrahydropyran motif incorporates unnecessary degrees of flexibility within the macrolactone backbone, ultimately altering the molecule’s overall shape and potentially its tubulin binding mode. Since introduction of the zampanolide side chain to 49 restores much of this lost activity, this suggests that while conformation may not be of paramount importance to tubulin binding it does play a role. Further work is necessary to determine the overall conformational requirements of zampanolide’s biological activity.
6. Conclusions
Polyketide natural products possess a wide range of structural features that play a role in defining their conformational preferences through fundamental steric, electronic and electrostatic interactions. These evolutionarily driven features confer the overall structure with defined conformer populations while retaining backbone flexibility, which likely provides energetic assistance during protein binding and improved physical properties such as solubility and membrane transport. Computer-based molecular modeling can provide an accurate representation of the conformational preferences of even the most complex polyketide structures. The resulting library of potential conformers can be ordered energetically through the use of solution NMR-derived coupling constant and NOE data. As is clear from the case studies covered within this review, protein-bound conformations determined by X-ray crystallographic analysis often show significant similarities to low-energy conformers populated in solution.
7. Future outlook
From one perspective, conformational mimics can be designed to maintain native conformer populations with simplified structural features. This design strategy can address supply issues for clinically bound polyketides in service of function-oriented synthesis principles. Alternatively, the addition of structural features that attenuate conformational preferences can provide insight into the bound conformation when coupled with biological activity studies. In either case, it is clear that investigators cannot disregard the conformational requirements of a polyketide’s biological activity in their analogue design programs.
Supplementary Material
Figure 10.
Exiguolide analogues, 15-desmethyl 12, 15,18-bis-desmethyl 13, and (16,17-Z)-exiguolide 14.
Figure 17.
Dictyostatin-discodermolide hybrid molecules.
Figure 20.
Monoene A 22 and E 23 analogues.
Figure 21.
The spongistatin class of natural products.
Figure 22.
Simplified spongistatin analogues designed by the Smith lab.
Figure 24.
The epothilone class of natural products.
Figure 28.
C3-deoxyepothilone 33 and 2,3-dehydroepothilone 34.
Figure 30.
The ansamycin natural product, geldanamycin.
Figure 31.
Geldanamycin analogues and trans- to cis-amide equilibrium.
Figure 33.
Peloruside A.
Figure 35.
Spontaneous equilibration of pyran lactol 44 to open form 45.
Figure 37.
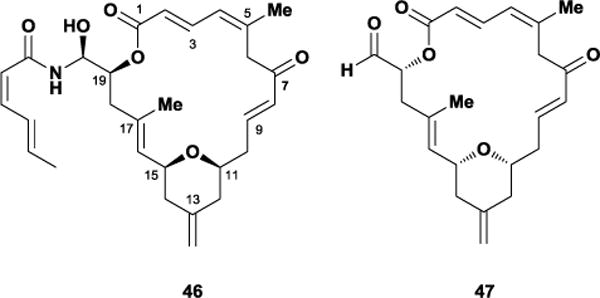
Zampanolide 46 and dactylolide 47.
Figure 41.
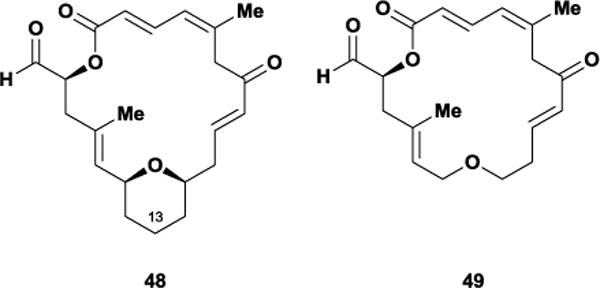
Altmann’s dactylolide analogues, C13-desmethylene 48 and ether 49.
Acknowledgments
This manuscript was inspired by the seminal contributions of Reinhard W. Hoffman. Research on conformation-activity relationships in polyketide natural products in the author’s laboratory have been supported by the National Institutes of Health and the National Science Foundation.
Footnotes
Footnotes should appear here. These might include comments relevant to but not central to the matter under discussion, limited experimental and spectral data, and crystallographic data.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
References
- 1.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann RW. Angew Chem Int Ed Engl. 1992;31:1124–1134. [Google Scholar]
- 3.Chodera JD, Mobley DL. Annu Rev Biophys. 2013;42:121–142. doi: 10.1146/annurev-biophys-083012-130318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 5.For information on the bryostatins in clinical trials, see: http://clinicaltrials.gov
- 6.(a) Pettit GR, Day JF, Hartwell JL, Wood HB. Nature. 1970;227:962–963. doi: 10.1038/227962a0. [DOI] [PubMed] [Google Scholar]; (b) Pettit GR, Herald CL, Doubek DL, Herald DL, Arnold E, Clardy J. J Am Chem Soc. 1982;104:6846–6848. [Google Scholar]
- 7.For reviews covering the biological properties of the bryostatins, see:; (a) Mutter R, Wills M. Bioorg Med Chem. 2000;8:1841–1860. doi: 10.1016/s0968-0896(00)00150-4. [DOI] [PubMed] [Google Scholar]; (b) Hale KJ, Hummersone MG, Manaviazar S, Frigerio M. Nat Prod Rep. 2002;19:413–453. doi: 10.1039/b009211h. [DOI] [PubMed] [Google Scholar]; (c) Kortmansky J, Schwartz GK. Cancer Invest. 2003;21:924–936. doi: 10.1081/cnv-120025095. [DOI] [PubMed] [Google Scholar]; (d) Wender PA, Baryza JL, Hilinski MK, Horan JC, Kan C, Verma VA. In: Drug Discovery Research: New Frontiers in the Post-Genomic Era. Huang Z, editor. Wiley-VCH; Hoboken, NJ: 2007. pp. 127–162. [Google Scholar]; (e) Hale KJ, Manaviazar S. Chem─Asian J. 2010;5:704–754. doi: 10.1002/asia.200900634. [DOI] [PubMed] [Google Scholar]; (f) Wender PA, Loy BA, Schrier AJ. Isr J Chem. 2011;51:453–472. doi: 10.1002/ijch.201100020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yu L, Krische MJ. In: Total Synthesis: At the Frontier of Organic Chemistry. Li JJ, Corey EJ, editors. Springer; Heidelberg, Germany: 2013. pp. 103–130. [Google Scholar]
- 8.(a) Alkon DL, Sun MK, Nelson TJ. Trends Pharmacol Sci. 2007;28:51–60. doi: 10.1016/j.tips.2006.12.002. [DOI] [PubMed] [Google Scholar]; (b) Sun MK, Alkon DL. Eur J Pharmacol. 2008;584:328–337. doi: 10.1016/j.ejphar.2008.02.014. [DOI] [PubMed] [Google Scholar]; (c) Wang D, DS Darwis, BG Schreurs, DL Alkon. Behav Pharmacol. 2008;19:245–256. doi: 10.1097/FBP.0b013e3282feb0d2. [DOI] [PubMed] [Google Scholar]; (d) Sun MK, Alkon DL. CNS Drug Rev. 2006;12:1–8. doi: 10.1111/j.1527-3458.2006.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kuzirian AM, Epstein HT, Gagliardi CJ, Nelson TJ, Sakakibara M, Taylor C, Scioletti AB, Alkon DL. Biol Bull. 2006;210:201–214. doi: 10.2307/4134558. [DOI] [PubMed] [Google Scholar]; (f) Sun MK, Alkon DL. Eur J Pharmacol. 2005;512:43–51. doi: 10.1016/j.ejphar.2005.02.028. [DOI] [PubMed] [Google Scholar]; (g) Scioletti AB, Kuzirian AM, Epstein HT, Nelson TJ, Alkon DL. Biol Bull. 2004;207:159. doi: 10.1086/BBLv207n2p159. [DOI] [PubMed] [Google Scholar]; (h) Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van Der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozokowski AP, VanLeuven F, Alkon DL. Proc Natl Acad Sci USA. 2004;101:11141–11146. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Khan TK, Nelson TJ, Verma VA, Wender PA, Alkon DL. Neurobiol Dis. 2009;34:332–339. doi: 10.1016/j.nbd.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. Nat Med. 2014;20:425–429. doi: 10.1038/nm.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mehla R, Mehla-Bivalkar S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, Chauhan A. PLoS One. 2010;5:e11160. doi: 10.1371/journal.pone.0011160. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Perez M, de Vinuesa AG, Sanchez-Duffhues G, Marquez N, Bellido ML, Munoz-Fernandez MA, Moreno S, Castor TP, Calzado MA, Munoz E. Curr HIV Res. 2010;8:418–429. doi: 10.2174/157016210793499312. [DOI] [PubMed] [Google Scholar]; (d) Sanchez-Duffhues G, Vo MQ, Perez M, Calzado MA, Moreno S, Appendino G, Munoz E. Curr Drug Targets. 2011;12:348–356. doi: 10.2174/138945011794815266. [DOI] [PubMed] [Google Scholar]
- 10.(a) Berkow RL, Kraft AS. Biochem Biophys Res Commun. 1985;131:1109–1116. doi: 10.1016/0006-291x(85)90205-0. [DOI] [PubMed] [Google Scholar]; (b) De Vries DJ, Herald CL, Pettit GR, Blumberg PM. Biochem Pharmacol. 1988;37:4069–4073. doi: 10.1016/0006-2952(88)90097-4. [DOI] [PubMed] [Google Scholar]
- 11.Schaufelberger DE, Koleck MP, Beutler JA, Vatakis AM, Alvarado AB, Andrews P, Marzo LV, Muschik GM, Roach J, Ross JT, Lebherz WB, Reeves MP, Eberwein RM, Rodgers LL, Testerman RP, Snader KM, Forenza S. J Nat Prod. 1991;54:1265–1270. doi: 10.1021/np50077a004. [DOI] [PubMed] [Google Scholar]
- 12.(a) Wender PA, Cribbs CM, Koehler KF, Sharkey NA, Herald CL, Kamano Y, Pettit GR, Blumberg PM. Proc Natl Acad Sci USA. 1988;85:7197–7201. doi: 10.1073/pnas.85.19.7197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pettit GR, Sengupta D, Blumberg PM, Lewin NE, Schmidt JM, Kraft AS. Anti-Cancer Drug Des. 1992;7:101–113. [PubMed] [Google Scholar]; (c) Pettit GR. J Nat Prod. 1996;59:812–821. doi: 10.1021/np9604386. [DOI] [PubMed] [Google Scholar]
- 13.Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Shiozaki M. J Am Chem Soc. 1998;120:4534–4535. [Google Scholar]
- 14.Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Siedenbiedel C, Pettit GR. Proc Natl Acad Sci USA. 1998;95:6624–6629. doi: 10.1073/pnas.95.12.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamano Y, Zhang HP, Morita H, Itokawa H, Shirota O, Pettit GR, Herald DL, Herald CL. Tetrahedron. 1996;52:2369–2376. [Google Scholar]
- 16.Wender PA, Nakagawa Y, Near KE, Staveness D. Org Lett. 2014;16:5136–5139. doi: 10.1021/ol502491f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Kraft MB, Poudel YB, Kedei N, Lewin NE, Peach ML, Blumberg PM, Keck GE. J Am Chem Soc. 2014;136:13202–13208. doi: 10.1021/ja5078188. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Andrews IP, Ketcham JM, Blumberg PM, Kedei N, Lewin NE, Peach ML, Krische MJ. J Am Chem Soc. 2014;136:13029–13216. doi: 10.1021/ja507825s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Kedei N, Telek A, Czap A, Lubart ES, Czifra G, Yang D, Chen J, Morrison T, Goldsmith PK, Lim L, Mannan P, Garfield SH, Kraft MB, Li W, Keck GE, Blumberg PM. Biochem Pharmacol. 2011;81:1296–1308. doi: 10.1016/j.bcp.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kedei N, Telek A, Michalowski AM, Kraft MB, Li W, Keck GE, Blumberg PM. Biochem Pharmacol. 2013;85:313–324. doi: 10.1016/j.bcp.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kedei N, Kraft MB, Keck GE, Herald CL, Melody N, Pettit GR, Blumberg PM. J Nat Prod Article ASAP. doi: 10.1021/acs.jnatprod.5b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Cancer Res. 1999;59:653–660. [PubMed] [Google Scholar]
- 21.Pryor DE, O’Brate A, Bilcer G, Díaz JF, Wang Y, Kabaki H, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E. Biochemistry. 2002;41:9109–9115. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 22.Gapud EJ, Bai R, Ghosh AK, Hamel E. Mol Pharmacol. 2004;66:113–121. doi: 10.1124/mol.66.1.113. [DOI] [PubMed] [Google Scholar]
- 23.BM Gallagher, Jr, Fang FG, Johannes CW, Pesant M, Tremblay MR, Zhao H, Akasaka K, Li XY, Liu J, Littlefield BA. Bioorg Med Chem Lett. 2004;14:575–579. doi: 10.1016/j.bmcl.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Towle MJ, Cheng H, Saxton P, Reardon C, Wu J, Murphy EA, Kuznetsov G, Johannes CW, Tremblay MR, Zhao H, Pesant M, Fang FG, Vermeulen MW, Gallagher BM, Jr, Littlefield BA. Anticancer Research. 2007;27:1509–1518. [PubMed] [Google Scholar]
- 25.(a) Wender PA, Hegde SG, Hubbard RD, Zhang L. J Am Chem Soc. 2002;124:4956–4957. doi: 10.1021/ja0258428. [DOI] [PubMed] [Google Scholar]; (b) Wender PA, Hedge SG, Hubbard RD, Zhang L, Mooberry SL. Org Lett. 2003;5:3507–3509. doi: 10.1021/ol035339f. [DOI] [PubMed] [Google Scholar]
- 26.Mooberry SL, Randall-Hlubek DA, Leal RM, Hegde SG, Hubbard RD, Zhang L, Wender PA. Proc Natl Acad Sci. 2004;101:8803–8808. doi: 10.1073/pnas.0402759101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jefford CW, Gernardinelli G, Tanaka J, Higa T. Tetrahedron Lett. 1996;37:159–162. [Google Scholar]
- 28.Paterson I, Menche D, Britton R, Håkansson AE, Silva-Martinez MÁ. Tetrahedron Lett. 2005;46:3677–3682. [Google Scholar]
- 29.Thepchatri P, Cicero DO, Monteagudo E, Ghosh AK, Cornett B, Weeks ER, Snyder JP. J Am Chem Soc. 2005;127:12838–12846. doi: 10.1021/ja042890e. [DOI] [PubMed] [Google Scholar]
- 30.(a) Paterson I, Bergmann H, Menche D, Berkessel A. Org Lett. 2004;6:1293–1295. doi: 10.1021/ol049791q. [DOI] [PubMed] [Google Scholar]; (b) Wender PA, Hilinski MK, Soldermann N, Mooberry SL. Org Lett. 8:1507–1510. doi: 10.1021/ol060233g. [DOI] [PubMed] [Google Scholar]
- 31.Paterson I, Menche D, Håkansson AE, Longstaff A, Wong D, Barasoain I, Buey RM, Díaz JF. Bioorg Med Chem Lett. 2005;15:2243–2247. doi: 10.1016/j.bmcl.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 32.Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann KH, Miller JH, Díaz JF, Steinmetz MO. Angew Chem Int Ed. 2014;53:1621–1625. doi: 10.1002/anie.201307749. [DOI] [PubMed] [Google Scholar]
- 33.Cossy J. Comptes Rendus Chimie. 2008;11:1477–1482. [Google Scholar]
- 34.Ohta S, Uy MM, Yanai M, Ohta E, Hirata T, Ikegami S. Tetrahedron Lett. 2006;47:1957–1960. [Google Scholar]
- 35.Fuwa H, Sukuzi T, Kubo H, Yamori T, Sasaki M. Chem –Eur J. 2011;17:2678–2688. doi: 10.1002/chem.201003135. [DOI] [PubMed] [Google Scholar]
- 36.Kong D, Yamori T. Bioorg Med Chem. 2012;20:1947–1951. doi: 10.1016/j.bmc.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Crane EA, Zabawa TP, Farmer RL, Scheidt KA. Angew Chem Int Ed. 2011;50:9112–9115. doi: 10.1002/anie.201102790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuwa H, Mizunuma K, Sasaki M, Suzuki T, Kubo H. Org Biomol Chem. 2013;11:3442–3450. doi: 10.1039/c3ob40131f. [DOI] [PubMed] [Google Scholar]
- 39.Fuwa H, Sasaki M. Org Lett. 2010;12:584–587. doi: 10.1021/ol902778y. [DOI] [PubMed] [Google Scholar]
- 40.(a) Gunasekera SP, Gunasekera M, Longley RE, Shulte K. J Org Chem. 1990;55:4912–4912. [Google Scholar]; Additions and corrections; (b) J Org Chem. 1991;56:1346. [Google Scholar]
- 41.Kowalski RJ, Giannakakou P, Gunasekera SP, Longley RE, Day BW, Hamel E. Mol Pharmacol. 1997;52:613–622. [PubMed] [Google Scholar]
- 42.For an overview of total syntheses of discodermolide, see:; Smith AB, III, Freeze BS. Tetrahedron. 2008;64:261–298. doi: 10.1016/j.tet.2007.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mita A, Lockhart AC, Chen TL, Bochinski K, Curtright J, Cooper W, Hammond L, Rothenberg M, Rowinsky E, Sharma S. J Clin Oncol ASCO Annu Meet Proc. 2004;14S:2025. [Google Scholar]
- 44.Smith AB, III, LaMarche MJ, Falcone-Hindley M. Org Lett. 2001;3:695–698. doi: 10.1021/ol006967p. [DOI] [PubMed] [Google Scholar]
- 45.Monteagudo E, Cicero DO, Cornett B, Myles DC, Snyder JP. J Am Chem Soc. 2001;123:6929–6930. doi: 10.1021/ja015569u. [DOI] [PubMed] [Google Scholar]
- 46.(a) Sánchez-Pedregal VM, Kubicek K, Meiler J, Lyothier I, Paterson I, Carlomagno T. Angew Chem Int Ed. 2006;45:7388–7394. doi: 10.1002/anie.200602793. [DOI] [PubMed] [Google Scholar]; (b) Canales A, Matesanz R, Gardner NM, Andreu JM, Paterson I, Díaz JF, Jiménez-Barbero J. Chem Eur J. 2008;14:7557–7569. doi: 10.1002/chem.200800039. [DOI] [PubMed] [Google Scholar]
- 47.Shaw SJ, Sundermann KF, Burlingame MA, Myles DC, Freeze BS, Xian M, Brouard I, Smith AB., III J Am Chem Soc. 2005;127:6532–6533. doi: 10.1021/ja051185i. [DOI] [PubMed] [Google Scholar]
- 48.Martello LA, LaMarche MJ, He L, Beauchamp TJ, Smith AB, III, Horwitz SB. Chem Biol. 2001;8:843–855. doi: 10.1016/s1074-5521(01)00055-2. [DOI] [PubMed] [Google Scholar]
- 49.Smith AB, III, Xian M. Org Lett. 2005;7:5229–5232. doi: 10.1021/ol052070m. [DOI] [PubMed] [Google Scholar]
- 50.Shaw SJ, Menzella HG, Myles DC, Xian M, Smith AB., III Org Biomol Chem. 2007;5:2753–2755. doi: 10.1039/b708884c. [DOI] [PubMed] [Google Scholar]
- 51.Pettit GR, Cichacz ZA, Gao F, Boyd MR, Schmidt JM. J Chem Soc Chem Commun. 1994:1111–1112. [Google Scholar]
- 52.Isbrucker RA, Cummins J, Pomponi SA, Longley RE, Wright AE. Biochem Pharmacol. 2003;66:75–82. doi: 10.1016/s0006-2952(03)00192-8. [DOI] [PubMed] [Google Scholar]
- 53.(a) Paterson I, Britton R, Delgado O, Meyer A, Poullennec KG. Angew Chem Int Ed. 2004;43:4629–4633. doi: 10.1002/anie.200460589. [DOI] [PubMed] [Google Scholar]; (b) Shin Y, Fournier JH, Fukui Y, Brückner AM, Curran DP. Angew Chem Int Ed. 2004;43:4634–4637. doi: 10.1002/anie.200460593. [DOI] [PubMed] [Google Scholar]; (c) O’Neil GW, Phillips AJ. J Am Chem Soc. 2006;128:5340–5341. doi: 10.1021/ja0609708. [DOI] [PubMed] [Google Scholar]; (d) Ramachandran PV, Srivastava A, Hazra D. Org Lett. 2007;9:157–160. doi: 10.1021/ol062737k. [DOI] [PubMed] [Google Scholar]; (e) Paterson I, Britton R, Delgado O, Gardner NM, Meyer A, Naylor GJ, Poullennec KG. Tetrahedron. 2010;66:6534–6545. [Google Scholar]; (f) Zhu W, Jiménez M, Jung WH, Camarco DP, Balachandran R, Vogt A, Day BW, Curran DP. J Am Chem Soc. 2010;132:9175–9187. doi: 10.1021/ja103537u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ho S, Bucher C, Leighton JL. Angew Chem Int Ed. 2013;52:6757–6761. doi: 10.1002/anie.201302565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wünch S, Breit B. Chem Eur J. 2015;21:2358–2363. doi: 10.1002/chem.201406252. [DOI] [PubMed] [Google Scholar]
- 54.(a) Paterson I, Gardner NM, Poullennec KG, Wright AE. Bioorg Med Chem Lett. 2007;17:2443–2447. doi: 10.1016/j.bmcl.2007.02.031. [DOI] [PubMed] [Google Scholar]; (b) Paterson I, Gardner NM, Poullennec KG, Wright AE. J Nat Prod. 2008;71:364–369. doi: 10.1021/np070547s. [DOI] [PubMed] [Google Scholar]; (c) Paterson I, Gardner NM, Guzmán E, Wright AE. Bioorg Med Chem. 2009;17:2282–2289. doi: 10.1016/j.bmc.2008.10.084. [DOI] [PubMed] [Google Scholar]; (d) Paterson I, Gardner NM, Guzmán E, Wright AE. Bioorg Med Chem Lett. 2008;18:6268–6272. doi: 10.1016/j.bmcl.2008.09.109. [DOI] [PubMed] [Google Scholar]; (e) Paterson I, Gardner NM, Naylor GJ. Pure Appl Chem. 2009;81:169–180. [Google Scholar]; (f) Paterson I, Naylor GJ, Fujita T, Guzmán E, Wright AE. Chem Commun. 2010;46:261. doi: 10.1039/b921237j. [DOI] [PubMed] [Google Scholar]; (h) Paterson I, Naylor GJ, Gardner NM, Guzmán E, Wright AE. Chem Asian J. 2011;6:459–473. doi: 10.1002/asia.201000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.(a) Shin Y, Choy N, Balachandran R, Madiraju C, Day BW, Curran DP. Org Lett. 2002;4:4443–4446. doi: 10.1021/ol026942l. [DOI] [PubMed] [Google Scholar]; (b) Fukui Y, Brückner AM, Shin Y, Balachandran R, Day BW, Curran DP. Org Lett. 2006;8:301–304. doi: 10.1021/ol0526827. [DOI] [PubMed] [Google Scholar]; (c) Shin Y, Fournier JH, Brückner A, Madiraju C, Balachandran R, Raccor BS, Edler MC, Hamel E, Sikorski RP, Vogt A. Tetrahedron. 2007;63:8537–8562. doi: 10.1016/j.tet.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jung WH, Harrison C, Shin Y, Fournier JH, Balachandran R, Raccor BS, Sikorski RP, Vogt A, Curran DP, Day BW. J Med Chem. 2007;50:2951–2966. doi: 10.1021/jm061385k. [DOI] [PubMed] [Google Scholar]; (e) Raccor BS, Vogt A, Sikorski RP, Madiraju C, Balachandran R, Montgomery K, Shin Y, Fukui Y, Jung WH, Curran DP. Mol Pharmacol. 2007;73:718–726. doi: 10.1124/mol.107.042598. [DOI] [PubMed] [Google Scholar]; (f) Eiseman JL, Bai L, Jung WH, Moura-Letts G, Day BW, Curran DP. J Med Chem. 2008;51:6650–6653. doi: 10.1021/jm800979v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Vollmer LL, Jimenez M, Camarco DP, Zhu W, Daghestani HN, Balachandran R, Reese CE, Lazo JS, Hukriede NA, Curran DP. Mol Cancer Ther. 2011;10:994–1006. doi: 10.1158/1535-7163.MCT-10-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zanato C, Pignataro L, Ambrosi A, Hao Z, Trigili C, Díaz JF, Barasoain I, Gennari C. Eur J Org Chem. 2011:2643–2661. [Google Scholar]
- 57.Brunden KR, Gardner NM, James MJ, Yao Y, Trojanowski JQ, Lee VMY, Paterson I, Ballatore C, Smith AB. ACS Med Chem Lett. 2013;4:886–889. doi: 10.1021/ml400233e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paterson I, Britton R, Delgado O, Wright AE. Chem Commun. 2004:632–633. doi: 10.1039/b316390c. [DOI] [PubMed] [Google Scholar]
- 59.Paterson I, Gardner NM. Chem Commun. 2007:49–51. doi: 10.1039/b615122a. [DOI] [PubMed] [Google Scholar]
- 60.Paterson I, Naylor GJ, Wright AE. Chem Commun. 2008:4628–4630. doi: 10.1039/b811575c. [DOI] [PubMed] [Google Scholar]
- 61.Buey RM, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I, Mooberry S, Andreu JM, Díaz JF. Chem Biol. 2005;12:1269–1279. doi: 10.1016/j.chembiol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 62.Jogalekar AS, Damodaran K, Kriel FH, Jung WH, Alcaraz AA, Zhong S, Curran DP, Snyder JP. J Am Chem Soc. 2011;133:2427–2436. doi: 10.1021/ja1023817. [DOI] [PubMed] [Google Scholar]
- 63.Funayama S, Okada K, Komiyama K, Umezawa I. J Antibiot. 1985;38:1107–1109. doi: 10.7164/antibiotics.38.1107. [DOI] [PubMed] [Google Scholar]
- 64.Funayama S, Okada K, Iwasaki K, Komiyama K, Umezawa I. J Antibiot. 1985;38:1677–1683. doi: 10.7164/antibiotics.38.1677. [DOI] [PubMed] [Google Scholar]
- 65.Funayama S, Anraku Y, Mita A, Yang ZB, Shibata K, Komiyama K, Umezawa I, Omura S. J Antibiot. 1988;41:1223–1230. doi: 10.7164/antibiotics.41.1223. [DOI] [PubMed] [Google Scholar]
- 66.Brandt GEL, Blagg BSJ. ACS Med Chem Lett. 2011;2:735–740. doi: 10.1021/ml200108y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jain AN. J Med Chem. 2004;47:947–961. doi: 10.1021/jm030520f. [DOI] [PubMed] [Google Scholar]
- 68.(a) Pettit GR, Chicacz ZA, Gao F, Herald CL, Boyd MR, Schmidt JM, Hooper JNA. J Org Chem. 1993;58:1302–1304. [Google Scholar]; (b) Fusetani N, Shinoda K, Matsunaga S. J Am Chem Soc. 1993;115:3977–3981. [Google Scholar]; (c) Kobayashi M, Aoki S, Sakai H, Kawazoe K, Kihara N, Sasaki T, Kitagawa I. Tetrahedron Lett. 1993;34:2795–2798. [Google Scholar]
- 69.(a) Bai R, Pettit GR, Hamel E. J Biol Chem. 1990;265:17141–17149. [PubMed] [Google Scholar]; (b) Bai R, Cichacz ZA, Herald CL, Pettit GR, Hamel E. Mol Pharmacol. 1993;44:757–766. [PubMed] [Google Scholar]; (c) Bai R, Taylor GF, Cichacz ZA, Herald CL, Kepler JA, Pettit GR, Hamel E. Biochemistry. 1995;34:9714–9721. doi: 10.1021/bi00030a009. [DOI] [PubMed] [Google Scholar]
- 70.(a) Guo J, Duffy KJ, Stevens KL, Dalko PI, Roth RM, Hayward MM, Kishi Y. Angew Chem Int Ed. 1998;37:187–192. [Google Scholar]; (b) Hayward MM, Roth RM, Duffy KJ, Dalko PI, Stevens KL, Guo J, Kishi Y. Angew Chem Int d. 1998;37:192–196. [Google Scholar]; (c) Paterson I, Wallace DJ, Oballa RM. Tetrahedron Lett. 1998;39:8545–8548. [Google Scholar]; (d) Paterson I, Chen DYK, Coster MJ, Acena JL, Bach J, Gibson KR, Keown LE, Oballa RM, Trieselmann T, Wallace DJ, Hodgson AP, Norcross RD. Angew Chem Int Ed. 2001;40:4055–4060. [PubMed] [Google Scholar]; (e) Smith AB, III, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew Chem Int Ed. 2001;40:191–195. doi: 10.1002/1521-3773(20010105)40:1<191::AID-ANIE191>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]; (f) Smith AB, III, Zhu W, Shirakami S, Sfouggatakis C, Doughty VA, Bennett CS, Sakamoto Y. Org Lett. 2003;5:761–764. doi: 10.1021/ol034037a. [DOI] [PubMed] [Google Scholar]; (g) Ball M, Gaunt MJ, Hook DF, Jessiman AS, Kawahara S, Orsini P, Scolaro A, Talbot AC, Tanner HR, Yamanoi S, Ley SV. Angew Chem Int Ed. 2005;44:5433–5438. doi: 10.1002/anie.200502008. [DOI] [PubMed] [Google Scholar]; (h) Smith AB, III, Tomioka T, Risatti CA, Sperry JB, Sfouggatakis C. Org Lett. 2008;10:4359–4362. doi: 10.1021/ol801792k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.(a) Evans DA, Trotter BW, Cote B, Coleman PJ, Dias LC, Tyler AN. Angew Chem Int Ed. 1998;36:2744–2747. [Google Scholar]; (b) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem Int Ed. 2001;40:196–199. [PubMed] [Google Scholar]; (c) Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J Am Chem Soc. 2002;124:5661–5663. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]; (d) Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J Am Chem Soc. 2003;125:12844–12849. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]; (e) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Boldi AM, Murase N, Moser WH, Brook CS. Tetrahedron. 2009;65:6470–6488. doi: 10.1016/j.tet.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith AB, III, Sfouggatakis C, Risatti CA, Sperry JB, Zhu W, Doughty VA, Tomioka T, Gotchev DB, Bennett CS, Sakamoto S, Atasoylu O, Shirakami S, Bauer D, Takeuchi M, Koyanagi J, Sakamoto Y. Tetrahedron. 2009;65:6489–6509. doi: 10.1016/j.tet.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith AB, III, Risatti CA, Atasoylu O, Bennett CS, Liu J, Cheng H, TenDyke K, Xu Q. J Am Chem Soc. 2011;133:14042–14053. doi: 10.1021/ja2046167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.(a) Paterson I, Acena JL, Bach J, Chen DYK, Coster MJ. Chem Commun. 2003;4:462–463. [PubMed] [Google Scholar]; (b) Wagner CE, Wang Q, Melamed A, Fairchild CR, Wild R, Heathcock CH. Tetrahedron. 2008;64:124–136. [Google Scholar]; (c) Smith AB, III, Lin Q. Bioorg Med Chem Lett. 1998;8:567–568. doi: 10.1016/s0960-894x(98)00050-x. [DOI] [PubMed] [Google Scholar]; (d) Smith AB, III, Corbett RM, Pettit GR, Chapuis JC, Schmidt JM, Hamel E, Jung MK. Bioorg Med Chem Lett. 2002;12:2039–2042. doi: 10.1016/s0960-894x(02)00305-0. [DOI] [PubMed] [Google Scholar]
- 75.Atasoylu O, Furst G, Risatti CA, Smith AB., III Org Lett. 2010;12:1788–1791. doi: 10.1021/ol100417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aoki S, Nemoto N, Kobayashi Y, Kobayashi M, Kitagawa I. Tetrahedron. 2001;57:2289–2292. [Google Scholar]
- 77.Taylor RE, Zajicek J. J Org Chem. 1999;64:7224–7228. [Google Scholar]
- 78.DISCON software and documentation is available for download at http://discon.sourceforge.net/
- 79.Smith AB, III, Risatti CA, Atasoylu O, Bennett CS, TenDyke K, Xu Q. Org Lett. 2010;12:1792–1795. doi: 10.1021/ol100418n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gerth K, Bedorf N, Höfle G, Irschik H, Reichenbach H. J Antibiot. 1996;49:560–563. doi: 10.7164/antibiotics.49.560. [DOI] [PubMed] [Google Scholar]
- 81.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 82.(a) Wolff A, Technau A, Bradner G. Int J Oncol. 1997;11:123–126. doi: 10.3892/ijo.11.1.123. [DOI] [PubMed] [Google Scholar]; (b) Giannakakou P, Sackett DL, Yang YK, Zhan Z, Butters JT, Fojo T, Poruchynsky MS. J Biol Chem. 1997;272:17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 83.Hunt JT. Mol Cancer Ther. 2009;8:275–281. doi: 10.1158/1535-7163.MCT-08-0999. [DOI] [PubMed] [Google Scholar]
- 84.For a comprehensive review of the epothilones, see:; Altmann KH, Höfle G, Müller R, Mulzer J, Prantz K. In: The Epothilones: An Outstanding Family of Anti-Tumor Agents. Kinghorn AD, Falk H, Kobayashi J, editors. Springer; Heiderlberg, Germany: 2009. [Google Scholar]
- 85.Victory SF, Vander Velde DG, Jalluri RK, Grunewald GL, Georg GI. Bioorg Med Chem Lett. 1996;6:893–898. [Google Scholar]
- 86.Höfle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Angew Chem Int Ed Engl. 1996;35:1567–1569. [Google Scholar]
- 87.Taylor RE, Chen Y, Beatty A, Myles DC, Zhou Y. J Am Chem Soc. 2003;125:26–27. doi: 10.1021/ja028196l. [DOI] [PubMed] [Google Scholar]
- 88.Frein JD, Taylor RE, Sackett DL. Org Lett. 2009;11:3186–3189. doi: 10.1021/ol900971r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yoshimura F, Rivkin A, Gabarda AE, Chou TC, Dong H, Sukenick G, Morel FF, Taylor RE, Danishefsky SJ. Angew Chem Int Ed. 2003;42:2518–2521. doi: 10.1002/anie.200351407. [DOI] [PubMed] [Google Scholar]
- 90.Taylor RE, Chen Y, Galvin GM, Pabba PK. Org Biomol Chem. 2004;2:127–132. doi: 10.1039/b312213c. [DOI] [PubMed] [Google Scholar]
- 91.Carlomagno T, Blommers MJJ, Meiler J, Hanhke W, Schupp T, Petersen F, Schinzer D, Altmann KH, Griesinger C. Angew Chem Int Ed. 2003;42:2511–2515. doi: 10.1002/anie.200351276. [DOI] [PubMed] [Google Scholar]
- 92.Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 93.Erdélyi M, Pfeiffer B, Hausenstein K, Fohrer J, Gertsch J, Altmann KH, Carlomagno T. J Med Chem. 2008;51:1469–1473. doi: 10.1021/jm7013452. [DOI] [PubMed] [Google Scholar]
- 94.(a) Cachoux F, Isarno T, Wartmann M, Altmann KH. Angew Chem Int Ed. 2005;44:7469–7463. doi: 10.1002/anie.200501760. [DOI] [PubMed] [Google Scholar]; (b) Regueiro-Ren A, Leavitt K, Kim SH, Höfle G, Kiffe M, Gougoutas JZ, Dimarco JD, Lee FYF, Fairchild CR, Long BH, Vite GD. Org Lett. 2002;4:3815–3818. doi: 10.1021/ol026589j. [DOI] [PubMed] [Google Scholar]
- 95.Prota AE, Bargsten K, Zurwerra D, Field JJ, Díaz JF, Altmann KH, Steinmetz MO. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 96.DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. J Antibiot. 1970;23:442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- 97.Rinehart KL, Jr, Sasaki K, Slomp G, Grostic MF, Olson EC. J Am Chem Soc. 1970;92:7591–7593. doi: 10.1021/ja00729a018. [DOI] [PubMed] [Google Scholar]
- 98.Andrus MB, Meredith EL, Simmons BL, Soma Sekhar BBV, Hicken EJ. Org Lett. 2002;4:3549–3552. doi: 10.1021/ol0267432. [DOI] [PubMed] [Google Scholar]
- 99.Qin HL, Panek JS. Org Lett. 2008;10:2477–2479. doi: 10.1021/ol800749w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.(a) Cysyk RL, Parker RJ, Barchi JJ, Jr, Steeg PS, Hartman NR, Strong JM. Chem Res Toxicol. 2006;19:376–381. doi: 10.1021/tx050237e. [DOI] [PubMed] [Google Scholar]; (b) Lang W, Caldwell GW, Li J, Leo GC, Jones WJ, Masucci JA. Drug Metab Dispos. 2007;35:21–29. doi: 10.1124/dmd.106.009639. [DOI] [PubMed] [Google Scholar]; (c) Guo W, Reigan P, Siegel D, Ross D. Drug Metab Dispos. 2008;36:2050–2057. doi: 10.1124/dmd.108.022004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rinehart KL, Shield LS. Prog Chem Org Nat Prod. 1976;33:231–307. doi: 10.1007/978-3-7091-3262-3_3. [DOI] [PubMed] [Google Scholar]
- 102.Schnur RC, Corman ML. J Org Chem. 1994;59:2581–2584. [Google Scholar]
- 103.(a) Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]; (b) Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]; (c) Jez JM, Chen JCH, Rastelli G, Stroud RM, Santi DV. Chem Biol. 2003;10:361–368. doi: 10.1016/s1074-5521(03)00075-9. [DOI] [PubMed] [Google Scholar]
- 104.(a) Lee YS, Marcu MG, Neckers L. Chem Biol. 2004;11:991–998. doi: 10.1016/j.chembiol.2004.05.010. [DOI] [PubMed] [Google Scholar]; (b) Thepchatri P, Eliseo T, Cicero DO, Myles D, Snyder JP. J Am Chem Soc. 2007;129:3127–3134. doi: 10.1021/ja064863p. [DOI] [PubMed] [Google Scholar]; (c) Onuoha SC, Mukund SR, Coulstock ET, Sengerovà B, Shaw J, McLaughlin SH, Jackson SE. J Mol Biol. 2007;372:287–297. doi: 10.1016/j.jmb.2007.06.065. [DOI] [PubMed] [Google Scholar]; (d) Reigan P, Siegel D, Guo W, Ross D. Mol Pharmacol. 2011;79:823–832. doi: 10.1124/mol.110.070086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kitson RRA, Chang CH, Xiong R, Williams HEL, Davis AL, Lewis W, Dehn DL, Siegel D, Roe SM, Prodromou C, Ross D, Moody CJ. Nature Chemistry. 2013;5:307–314. doi: 10.1038/nchem.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, D’Andrea G, Dickler M, Moynahan ME, Sugerman S, Ma W, Patil S, Norton L, Hannah AL, Hudis C. Clin Cancer Res. 2011;17:5132–5139. doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 107.West LM, Northcote PT, Batershill CN. J Org Chem. 2000;65:445–449. doi: 10.1021/jo991296y. [DOI] [PubMed] [Google Scholar]
- 108.Hood KA, West LM, Rouwe B, Northcote PT, Berridge MV, Wakefield SJ, Miller JH. Cancer Res. 2002;62:3356–3360. [PubMed] [Google Scholar]
- 109.Gaitanos TN, Buey RM, Díaz JF, Northcote PT, Teesdale-Spittle P, Andreu JM, Miller JH. Cancer Res. 2004;64:5063–5067. doi: 10.1158/0008-5472.CAN-04-0771. [DOI] [PubMed] [Google Scholar]
- 110.Wilmes A, Bargh K, Kelly C, Northcote PT, Miller JH. Mol Pharm. 2007;4:269–280. doi: 10.1021/mp060101p. [DOI] [PubMed] [Google Scholar]
- 111.Page MJ, Northcote PT, Webb VL, Mackey S, Handley SJ. Aquaculture. 2005;250:256–269. [Google Scholar]
- 112.Liao X, Wu Y, De Brabander JK. Angew Chem Int Ed. 2003;42:1648. doi: 10.1002/anie.200351145. [DOI] [PubMed] [Google Scholar]
- 113.(a) Jin M, Taylor RE. Org Lett. 2005;7:1303–1305. doi: 10.1021/ol050070g. [DOI] [PubMed] [Google Scholar]; (b) Taylor RE, Jin M. Org Lett. 2003;5:4959–4961. doi: 10.1021/ol0358814. [DOI] [PubMed] [Google Scholar]
- 114.(a) Ghosh AK, Xu X, Kim JH, Xu CX. Org Lett. 2008;10:1001–1004. doi: 10.1021/ol703091b. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Welch DS, Speed AWS, Moniz AG, Reichelt A, Ho S. J Am Chem Soc. 2009;131:3840–3841. doi: 10.1021/ja900020a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McGowan MA, Stevenson CP, Schiffler MA, Jacobsen EN. Angew Chem Int Ed. 2010;49:6147–6150. doi: 10.1002/anie.201002177. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hoye TR, Jeon J, Kopel LC, Ryba TD, Tennakoon MA, Wang YN. Angew Chem Int Ed. 2010;49:6151–6155. doi: 10.1002/anie.201002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.(a) Pera B, Razzak M, Trigili C, Pineda O, Canales A, Buey RM, Jiménez-Barbero J, Northcote PT, Paterson I, Barasoain I, Díaz JF. ChemBioChem. 2010;11:1669–1678. doi: 10.1002/cbic.201000294. [DOI] [PubMed] [Google Scholar]; (b) Singh AJ, Razzak M, Teesdale-Spittle P, Gaitanos TN, Wilmes A, Paterson I, Goodman JM, Miller JH, Northcote PT. Org Biomol Chem. 2011;9:4456–4466. doi: 10.1039/c0ob01127d. [DOI] [PubMed] [Google Scholar]; (c) Zimmermann N, Pinard P, Carboni B, Gosselin P, Gaulon-Nourry C, Dujardin G, Collet S, Lebreton J, Mathé-Allainmat M. Eur J Org Chem. 2013:2303–2315. [Google Scholar]; (d) Wullschleger CW, Gertsch J, Altmann KH. Org Lett. 2010;12:1120–1123. doi: 10.1021/ol100123p. [DOI] [PubMed] [Google Scholar]; (e) Wullschleger CW, Gertsch J, Altmann KH. Chem Eur J. 2013;19:13105–13111. doi: 10.1002/chem.201301796. [DOI] [PubMed] [Google Scholar]
- 116.Jiménez-Barbero J, Canales A, Northcote PT, Buey RM, Andreu JM, Díaz JF. J Am Chem Soc. 2006;128:8757–8765. doi: 10.1021/ja0580237. [DOI] [PubMed] [Google Scholar]
- 117.Nicholson CP., PhD . thesis. University of Notre Dame; 2010. [Google Scholar]
- 118.Zhao Z, Taylor RE. Org Lett. 2012;14:669–671. doi: 10.1021/ol203268t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.(a) Huzil JT, Chik JK, Slysz GW, Freedman H, Tuszynski J, Taylor RE, Sackett DL, Schriemer DC. J Mol Biol. 2008;378:1016–1030. doi: 10.1016/j.jmb.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Begaye A, Trostel S, Zhao Z, Taylor RE, Schriemer DC, Sackett DL. Cell Cycle. 2011;10:3387–3396. doi: 10.4161/cc.10.19.17706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tanaka J, Higa T. Tetrahedron Lett. 1996;37:5535–5538. [Google Scholar]
- 121.Field JJ, Singh AJ, Kanakkanthara A, Halafihi T, Northcote PT, Miller JH. J Med Chem. 2009;52:7328–7332. doi: 10.1021/jm901249g. [DOI] [PubMed] [Google Scholar]
- 122.Cutignano A, Bruno I, Bifulco G, Casapullo A, Cécile D, Gomez-Paloma L, Riccio R. Eur J Org Chem. 2001:775–778. [Google Scholar]
- 123.Field JJ, Pera B, Calvo E, Canales A, Zurwerra D, Trigili C, Rodríguez-Salarichs J, Matesanz R, Kanakkanthara A, Wakefield SJ, Singh AJ, Jiménez-Barbero J, Northcote PT, Miller JH, López JA, Hamel E, Barasoain I, Altmann KH, Díaz JF. Chem Biol. 2012;19:686–698. doi: 10.1016/j.chembiol.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Larsen EM, Wilson MR, Taylor RE. Org Lett. 2013;15:5246–5249. doi: 10.1021/ol402462h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mayer M, Mayer B. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 126.Zurwerra D, Glaus F, Betschart L, Schuster J, Gertsch J, Ganci W, Altmann KH. Chem Eur J. 2012;18:16868–16883. doi: 10.1002/chem.201202553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.