

Abstract
Background
(−)-Solenopsin A is a piperidine alkaloid that is a component of the venom of the fire ant Solenopsis invicta. Previously, we have demonstrated that solenopsin exhibit anti-angiogenic activity and downregulate phosphoinositol-3 kinase (PI3K) in the p53 deficient renal cell carcinoma cell line 786-O. Solenopsin has structural similarities to ceramide, a major endogenous regulator of cell signaling and cancer therapy induced apoptosis.
Methods
Different analogs of solenopsin were synthesized in order to explore structure-activity relationships. The anti-proliferative effect of solenopsin and analogs was tested on six different cell lines, including three tumor cell lines, two normal cutaneous cell lines, and one immortalized hyperproliferative cell line. FRET-based reporters were used to study the affect of solenopsin and analogs on Akt activity and PDK1 activation and sucrose density gradient fractionation was performed to examine recruitment of PTEN to membrane rafts. Western-blotting was used to evaluate the affect of solenopsin and analogs on the Akt and the MAPK 44/42 pathways in three different tumor cell lines. Measurement of cellular oxygen consumption rate together with autophagy staining was performed to study mitochondrial function. Finally, the affect of solenopsin and analogs on ROS production was investigated.
Results
In this paper we demonstrate that solenopsin analogs with potent anti-proliferative effects can be synthesized from inexpensive dimethylpyridines. To determine whether solenopsin and analogs act as ceramide analogs, we examined the effect of solenopsin and analogs on two stereotypic sites of ceramide activity, namely at lipid rafts and mitochondria. We found that native solenopsin, (−)-solenopsin A, inhibits functional Akt activity and PDK1 activation in lipid rafts in a similar fashion as ceramide. Both cis and trans analogs of solenopsin reduce mitochondrial oxygen consumption, increase reactive oxygen, and kill tumor cells with elevated levels of Akt phosphorylation. However, only solenopsin induces mitophagy, like ceramide.
Conclusions
The requirements for ceramide induced mitophagy and inhibition of Akt activity and PDK1 activation in lipid rafts are under strict stereochemical control. The naturally occurring (−)-solenopsin A mimic some of the functions of ceramide and may be therapeutically useful in the treatment of hyperproliferative and malignant disorders of the skin, even in the presence of elevated levels of Akt.
Electronic supplementary material
The online version of this article (doi:10.1186/s13221-015-0030-2) contains supplementary material, which is available to authorized users.
Keywords: Solenopsin A, Ceramide, Akt, Mitophagy, Reactive oxygen
Background
(−)-Solenopsin A is a piperidine alkaloid that is a component of the venom of the fire ant Solenopsis invicta. Previously, we demonstrated that solenopsin exhibited anti-angiogenic activity and downregulated phosphoinositol-3 kinase (PI3K) in the p53 deficient renal cell carcinoma cell line 786-O [1]. Solenopsin structurally resembles ceramides in chemical structure (Figure 1). Ceramides are fatty acid amides of sphingosine, which play a crucial role in homeostasis of the skin and other organs [2-5]. Defects in the production of ceramides may inhibit physiologic cell death, leading to the persistence of inflammatory and neoplastic cells [5-7]. Large-scale extraction of solenopsin from fire ants is not feasible, and current synthetic routes are multistep routes with low yields. Thus, we devised a simple two-step synthesis of solenopsin analogs starting from inexpensive dimethyl pyridine. We examined the effect of solenopsin and analogs on two stereotypic sites of ceramide activity, lipid rafts and mitochondria [7,8]. We show that native (−)-solenopsin A has ceramide-like activity in lipid rafts and mitochondria. Finally, we show that solenopsin and analogs kill tumor cells regardless of phosphatase and tensin homolog (PTEN) status or Akt activation. Given that loss of PTEN and elevation of Akt are major mechanisms of resistance to chemotherapy [9,10], the use of solenopsin and analogs may be of great utility in treating hyperproliferative skin disorders.
Figure 1.

Compounds studied in this paper and synthetic procedures for preparation of compounds S11-S15. (−)-Solenopsin A is a component of the venom of the fire ant solenopsis invicta. (+)-Solenopsin A is the enantiomer of the naturally occurring solenopsin. The structure of solenopsin resembles the structure of ceramides, which are fatty acid amides of sphingosine that play a crucial role in homeostasis of the skin and other organs. Solenopsin analogs S11-S14 were synthesized by deprotonation of 2,6-dimethylpyridine (S12-S14) or 2,4,6-trimethylpyridine (S11) by n-butyllithiium, followed by addition of alkyl bromides. Analog S15 was synthesized by treating pyridine-2-carboxaldehyde with the Grignard reagent decylmagnesium bromide. The solenopsin analogs (S11-S15) were successfully obtained after hydrogenation of the various 2-alkylpyridines.
Methods
Synthesis
(+)-Solenopsin A and (−)-solenopsin A were synthesized as HCl salts as previously described [1]. Compound S11-S14 were synthesized by deprotonation of 2,6-dimethylpyridine (S12-S14) or 2,4,6-trimethylpyridine (S11) by n-butyllithium, followed by addition of alkyl bromides (Figure 1). S15 was synthesized by treating pyridine-2-carboxaldehyde with the Grignard reagent decylmagnesium bromide (Figure 1). The solenopsin analogs (S11-S15) were successfully obtained after hydrogenation of the various 2-alkylpyridines (Figure 1). Detailed synthetic procedures and characterization data can be found in the Additional file 1.
Cells and culture conditions
In this study eight different cell lines were used: human A375 melanoma cells, human A2058 melanoma cells, immortalized murine endothelial SVR cells [11-13], primary human melanocytes, primary human keratinocyts, HaCaTs (immortalized human keratinocytes), murine embryonic NIH3T3 fibroblast cells, and human UM-SCC1A squamous carcinoma cells. All cell lines were grown in DMEM with 10% fetal bovine serum, except for primary keratinocytes which were grown in serum free keratinocyte growth media and primary melanocytes that were grown in complete melanocyte growth media
Proliferation studies
A375, A2058, SVR, primary melanocyte, primary keratinocyte, and HaCaT cells were treated with test compounds for 24 hours, followed by cell counting with a Coulter Counter. All compounds were tested in quadruplicates. For A375s, A2058s, SVRs, and primary melanocytes 50,000 cells/well were plated. Due to difficulty growing the cells, primary keratinocytes and HaCaTs were plated at a concentration of 20,000 cells/well, and 15,000 cells/well respectively. All cells were treated with 20 μM of ceramide C2. A375s, A2058s, and SVRs were treated with 10 μM of (−)-solenopsin A, (+)-solenopsin A, or analogs S11-S15, whereas primary melanocytes, primary keratinocytes, and HaCaTs were treated with 20 μM of solenopsin and analogs.
FRET-based reporter construct
The development of the biocensors Lyn-PARE and AktAR has been described previously [14,15]. Briefly, AktAR was generated by a fluorescent protein pair, cerulean and cpVE172, sandwiching a forkhead-associated binding domain (FHA1) and an Akt substrate domain (FOXO) [15]. Lyn-PARE was generated by sandwiching full-length PDK1 between a FRET pair, ECFP (cyan fluorescent protein) and citrine (yellow fluorescent protein) [16], and a motif generated from the Lyn-kinase gene was added to the 5’-end to target the construct to raft microdomains [14].
Cell transfection and imaging
Cell transfection and imaging was conducted as previously described [14,15]. NIH3T3 cells were treated for 1 h with DMSO solutions of ceramide C2 (50 μM), (+)-solenopsin A (10 and 20 μM), (−)-solenopsin A (10 and 20 μM), and analogs S11-S15 (10 μM). A more detailed description of the experimental procedure can be found in the Additional file 1.
Sucrose density gradient fractionation
Cells were treated for 1 h with 20 μM DMSO solutions of (+)-solenopsin A, (−)-solenopsin A, analogs (S12-S15), or 50 μM of ceramide. Lipid raft fractionation was performed with a 5-40% sucrose discontinuous gradient as previously described [17,18]. After ultracentrifugation, thirteen 385 μL fractions were collected, starting from the top of the tube. Equal volumes of each fraction were analyzed by Western blot with rabbit polyclonal antibodies for caveolin-1 and PTEN. A more detailed description of the experimental procedure can be found in the Additional file 1.
Western blot analysis
Cells were grown in T-25 flasks until 80% confluent followed by treatment for 24 h with 10 μM DMSO solutions of (+)-solenopsin A, (−)-solenopsin A, or analogs (S11-S15). Sample aliquots normalized for protein quantities were size fractionated by 10% SDS-PAGE, and the proteins were transferred to a PVDF membrane. The blots were incubated in blocking solution; TBS with 5% (wt/vol) powdered nonfat milk for 1 h at room temperature, followed by incubation over night with rabbit polyclonal p-Akt S473, p-MAPK 44/42, and Β-actin.
Measurement of oxygen consumption rate (OCR)
UM-SCC1A cells were plated 15,000 cells/well in 200 μl DMEM supplemented with 10% FBS and 1% penicillin-streptomycin in each well of a 96-well Seahorse plate and incubated overnight at 37°C with 5% CO2. Cells were treated with 10 μM of compound or DMSO and incubated for 24 hours. OCR was measured as pmoles O2/minute using the Seahorse Biosciences instrument per manufacturer’s instructions. Protein amounts in each well were quantified using the Thermo Scientific Pierce Protein Assay, per manufacturer’s instructions.
Autophagosome staining
UM-SCC1A cells were treated with DMSO (control) or 10 μM of (−)-solenopsin A or analog S14 for 18 h. The cells were stained using a Cyto-ID Autophagy Staining assay from Life Technologies.
Measurement of ROS with dihydroethidium (DHE)
Briefly, A375 and SVR cells were treated for 24 h with 10 μM DMSO solutions of (+)-solenopsin A, (−)-solenopsin A, or analogs (S11-S15). Cells were washed, pelleted, suspended in 10 μM DHE and incubated for 10 min. Thereafter cells were counted using a Becton Dickinson FACScan flow cytometer. Mean values of DHE fluorescence intensity were compared and all samples were repeated in triplicate. A more detailed description of the experimental procedure can be found in the Additional file 1.
Determination of cytotoxicity by MTT assay
MTT reagent was added to the tissue inserts after 72 hours treatment with 10 μM of Solenopsin A and analogs S12 and S14 followed by incubation for 3 hours at 37°C with 5% CO2. Thereafter, MTT was extracted from the tissues and the absorbance was measured at 570 nm. Cell viability was calculated using a spreadsheet provided by MatTek; viability of less than 50% was determined to be irritant and cytotoxic. H&E staining of the tissues from the cultures inserts were also performed; see Additional file 1 for more detailed experimental procedures.
Results
Structure-activity relationships of solenopsin and analogs
Different analogs of solenopsin were synthesized (Figure 1) in order to explore structure-activity relationships. Three different tumor cell lines, relevant for skin, were used to assess anti-proliferative potency: human A375 melanoma cells, human A2058 melanoma cells, and murine SVR angiosarcoma cells. No significant difference between the naturally occurring compound, (−)-solenopsin A, and its enantiomer (+)-solenopsin A could be seen in any of the tumor cell lines (Figure 2). Both of these compounds have trans geometry. The mixture of the two cis isomers of solenopsin, S12, show weaker anti-proliferative activity than (−)-solenopsin A in all three cell lines (Figure 2), suggesting that the trans isomers are more potent than the corresponding cis isomers. Elongation of the aliphatic side chain with 8 carbons had a negative effect on potency, as analogs S11 and S13 displayed a lower anti-proliferative effect than both solenopsin and S12. Also, the only compound that showed no anti-proliferative effect in any of the cell lines was S11, which in addition to having the longest aliphatic side chain also has an extra methyl group on the piperidine ring. Interestingly, analog S14, which has a 4 carbon longer aliphatic side chain than solenopsin, turned out to be the most potent analog in human melanoma A375 cells and murine angiosarcoma SVR cells (Figure 2a and b). As S14 was significantly more potent than S12 in all three cell lines, it would appear as if a 15-carbon side chain is the optimum length for anti-proliferative activity. S15, which has the same length side chain as solenopsin but lacks the methyl group and has an extra hydroxyl group, also displayed potent anti-proliferative effect in all three cell lines and in human melanoma A2058 cells it was the most potent analog.
Figure 2.
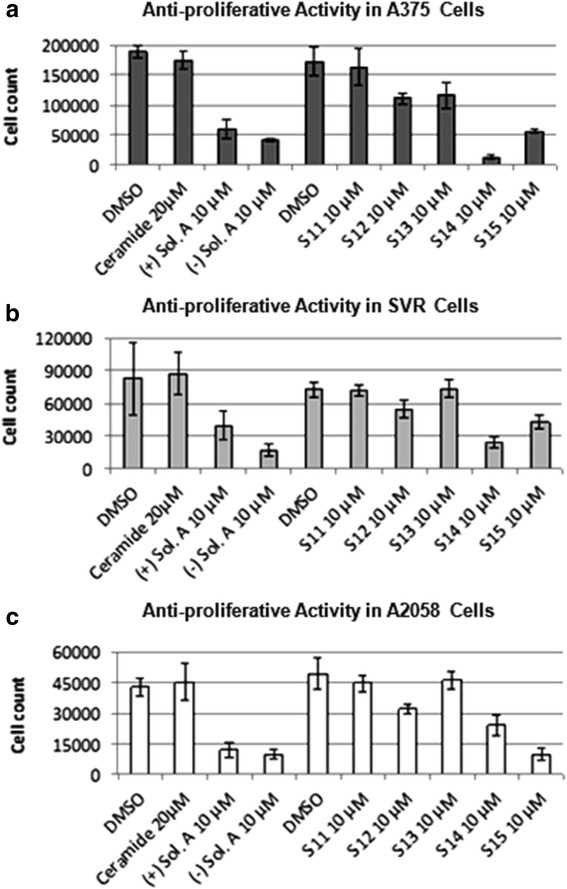
Assessment of anti-proliferative activity for solenopsin and analogs S11-S15. The anti-proliferative effect of (+)-solenopsin A ((+) Sol. A), (−)-solenopsin A ((−) Sol. A), ceramide C2, and solenopsin analogs S11-S15 were evaluated in (a) A375 cells, (b) SVR cells, and (c) A2058 cells. 50,000 cells/well were plated and treated for 24 h with each compound. The first DMSO bar in each chart serves as control for (+)-solenopsin A, (−)-solenopsin A, and ceramide C2. The second DMSO bar is the control for S11-S15. The displayed data are an average of four experiments ± s.d.
The effect of ceramide, solenopsin A, and analogs S11-S15 was also assessed on two normal cutaneous cell lines, namely primary melanocytes and primary keratinocytes (Additional file 1: Figure S1). In addition, the activity of the compounds was assessed in HaCaTs, which are immortalized hyperproliferative human keratinocytes (Additional file 1: Figure S1). The analogs S11 and S13, which were inactive in the tumor cell lines, did not have any activity in these cell lines either. Ceramide only showed activity in primary melanocytes and keratinocytes (Additional file 1: Figure S1), but not in malignant A375s, A2058s, and SVRs, (Figure 2). Interestingly, HaCat cells, which represent premalignant keratinocytes, are resistant to ceramide, supporting our hypothesis that loss of response to ceramide may represent an early event in skin carcinogenesis. Solenopsin A and the analogs S12, S14, and S15 had significant activity in all cell lines, including malignant and primary cell lines (Figure 2 and Additional file 1: Figure S1). Solenopsin A and active analogs were shown to be non-toxic to reconstituted skin equivalents (Additional file 1: Figure S2). Normal keratinization was preserved as assessed by routine histology (data not show).
Solenopsin inhibits functional Akt activity and PDK1 activation
Ceramides are found in the cell membrane where they act as signaling molecules and play a role in a variety of physiological conditions, such as: differentiation, proliferation, programmed cell death, apoptosis etc. [19]. The underlying mechanisms are complex and the exact means by which ceramides function as signaling molecules are not clear. However, there are a number of studies that show that ceramide inhibit the PI3K/Akt pathway [14]. To determine whether solenopsin A and analogs have similar modes of action as ceramide, we employed a FRET-based Akt activity reporter (AktAR) and a PDK1 activation reporter targeted to membrane rafts (Lyn-PARE) [14,15]. The AktAR construct contains a binding domain (FHA1), a substrate domain (FOXO), and two fluorescent proteins that constitute a FRET pair. This reporter functions as surrogate substrate for Akt and phosphorylation of the substrate leads to a detectable change in FRET [15]. Lyn-PARE contains the full-length PDK1 protein flanked by two fluorescent proteins that constitute a FRET pair. This construct also contains a motif derived from Lyn-kinase, which targets it to membrane rafts. Activation of PDK1 leads to a conformational change and thereby a detectable change in FRET [14]. Previously, these reporters have been used to show that ceramide treatment inhibits PDGF-induced Akt activity and activation of PDK1 in membrane rafts [14]. At 20 μM concentrations both (+)- and (−)-solenopsin A inhibited Akt activity (AktAR) and PDK1 activation (Lyn-PARE) to similar extent as treatment with 50 μM of ceramide (Figure 3). At 10 μM concentrations some inhibition of Akt activity and PDK1 activation could be seen for both (+)- and (−)-solenopsin A. At 10 μM S13 and possibly also S12 displayed some inhibition of Akt activity and PDK1 activation. No significant inhibition could be seen for any of the other solenopsin analogs (Figure 3).
Figure 3.
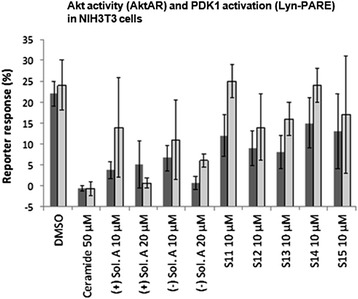
Solenopsin A inhibits Akt activity (AktAR) and PDK1 activation (Lyn-PARE). NIH3T3 cells were transfected with AktAR or Lyn-PARE and serum starved for 24 h, followed by 1 h treatments with DMSO, ceramide C2, (+)-solenopsin A ((+) Sol. A), (−)-solenopsin A ((−) Sol. A) or solenopsin analogs S11-S15. To study Akt activity and PDK1 activation, 50 ng/mL of PDGF was added followed by immediate imaging. FRET ratio of regions of interest at cell cytosol and at cell periphery representing the plasma membrane were used, respectively. Akt activity is measured by AktAR response (dark grey columns) and PDK1 activation by Lyn-PARE response (light grey columns). All the ratios were normalized with the ratio before PDGF addition. Data shown are an average of at least three experiments ± s.d.
Translocation of PTEN to membrane rafts
To examine whether solenopsin recruits PTEN to membrane rafts, as ceramide does, sucrose density gradient fractionation was performed followed by evaluation of the PTEN levels in each fraction. PTEN negatively regulates the PI3K/Akt signaling, which takes place in the lipid raft regions, by converting PIP3 (phosphatidylinostol (3,4,5)-triphosphate) to PIP2 (phosphatidylinositol (4,5)-biphosphate). As most PTEN is usually localized to nonraft regions [14,20,21], studies suggest that ceramide inhibits PI3K/Akt signaling by translocating PTEN from nonraft regions into lipid rafts [15,22,23].
As expected, the amounts of PTEN seem to be higher in lipid raft fractions from cells treated with ceramide (fraction 1–4) (Additional file 1: Figure S3). The compound that appeared to have the largest amount of PTEN in the raft fractions was (−)-solenopsin A (Additional file 1: Figure S3). (+)-solenopsin A and S12-treated cells showed similar amounts of PTEN in the raft fractions as the ceramide treated cells (Additional file 1: Figure S3). The rest of the analogs (S13-S15) appeared to have similar or even lower amounts of PTEN in lipid rafts than the control (Additional file 1: Figure S3). It is worth pointing out that for these experiments the use of a loading control is not possible. Therefore, these results should only be regarded as an indication of the compounds ability to recruit PTEN to membrane rafts. However, the FRET-based analysis was largely consistent with the PTEN localization as demonstrated by sucrose density fractionation.
Solenopsin A and analogs effect on signaling pathways is context dependent
A375 (human melanoma), SVR (murine angiosarcoma), and A2058 (human melanoma) cells treated with solenopsin A and analogs were evaluated by Western-blotting with p-Akt S473, p-MAPK 44/42, and B-actin (Additional file 1: Figure S4 and Additional file 1: Table S1). In A375 human melanoma cells, an up-regulation of p-Akt S473 and p-MAPK 44/42 could be seen for (+)- and (−)-solenopsin A, as well as for analogs S12-S15. In SVR murine angiosarcoma cells on the other hand, p-Akt S473 and p-MAPK 44/42 were down-regulated in all treatment groups, and especially in cells treated with analog S15. The results for the human melanoma A2058 cells were similar to A375 cells but not as pronounced, i.e. there is a slight up-regulation of p-Akt S473 and p-MAPK 44/42 in some of the treatment groups compared to the control. Both human melanoma cell lines (A375 and A2058) have intact p53 and loss of p16ink4a [24], whereas the murine angiosarcoma cell line (SVR) has defective p53 function due to the presence of SV40 large T antigen [11]. This may account for the observed difference in cell signaling between these cell lines.
Solenopsin A and analogs affect mitochondrial function
Although solenopsin A recruits PTEN to lipid rafts (Additional file 1: Figure S3), it does not appear to be enough to dephosphorylate Akt (Additional file 1: Figure S4) in the human melanoma cell lines A375 and A2058 (both are wild type p53) like it did in 786-O renal cell carcinoma cells (mutant p53) [1]. One explanation for the anti-proliferative activity of solenopsin and analogs could be that they, like ceramide, also localize to mitochondria and increase the production of reactive oxygen [25], thereby causing mitophagy and cell death. Cellular oxygen consumption rate (OCR) can be indicative of mitochondrial function. To investigate if solenopsin A and analogs alter mitochondrial function in human head and neck squamous carcinoma cells like ceramide [8], UM-SCC1A cells were treated with 10 μM of solenopsin and analogs, followed by incubation for 24 hours. Cells treated with (+)- and (−)-solenopsin A, S12, S14, and S15 displayed reduced OCR compared to control, while cells treated with S13 have slightly reduced OCR and cells treated with S11 show little change in OCR compared to the control (Figure 4). To verify that mitophagy is the reason for the observed decrease in OCR, a Cyto-ID Autophagy Staining assay was used. The dye stains autophagosomes and co-localizes with LC3-II. UM-SCC1A cells were treated with DMSO (control) and 10 μM of (−)-solenopsin A or analog S14 for 18 h. A significant increase in fluorescence could be seen for (−)-solenopsin A compared to the control, which verifies that (−)-solenopsin A induces autophagy (Additional file 1: Figure S5). Analog S14, on the other hand, does not appear to induce autophagy; instead it looks as if S14 may even decrease basal levels of autophagy (Additional file 1: Figure S5).
Figure 4.

Solenopsin A and analogs reduce oxygen consumption rate. UM-SCC1A cells were plated 15,000/well and treated for 24 h with 10 μM of (+)-solenopsin A ((+) Sol. A), (−)-solenopsin A ((−) Sol. A), and solenopsin analogs S11-S15. Oxygen consumption rate (OCR) was measured as pmoles O2/minute using a Seahorse Biosciences instrument. Data shown are an average of three experiments ± s.d.
Solenopsin A and analogs elevate ROS levels
Ceramide is known to increase ROS production [25-27] and given that Akt phosphorylation was elevated by solenopsin and analog treatment in human melanoma cells, and that elevated phosphorylation of Akt is a common response to superoxide, we examined levels of superoxide. There was a marked increase in superoxide levels, as measured by DHE fluorescence, in both A375 human melanoma and SVR murine angiosarcoma cells treated with the compounds (+)- and (−)-solenopsin A, S12, S14, and S15 ranging from 1.7-2.3 fold compared to vehicle treated cells (Figure 5). Compounds S11, and S13 had no effect on superoxide levels (Figure 5).
Figure 5.
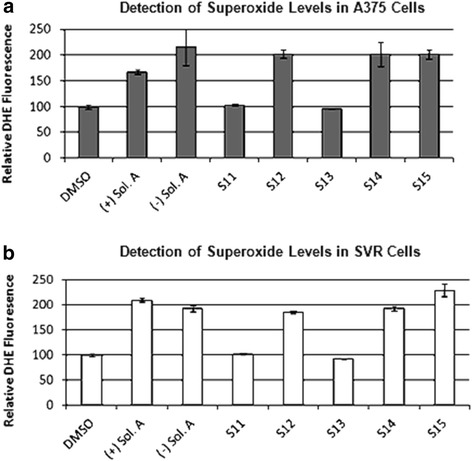
Solenopsin A and analogs increase superoxide levels. Investigation of solenopsin and analogs S11-S14 effect on superoxide levels in (a) A375 cells, and (b) SVR cells. Cells were treated for 24 h with 10 μM of (+)-solenopsin A ((+) Sol. A), (−)-solenopsin A ((−) Sol. A), and solenopsin analogs S11-S15. Cells were trypsined and incubated for 10 min in 10 μM dihydroethedium, followed by analysis with a FACScan flow cytometer. Data shown are an average of three experiments ± s.d.
Discussion
Ceramides play a role in physiologic cell death, as they are involved in removal of undesired cells, and thus limit both inflammation and neoplasia [28,29]. Ceramides have also been implicated in mediating cell death due to chemotherapy and radiation, and inability to generate ceramides is linked to resistance to these treatments [7,28-30]. Thus, restoration of ceramide is a potential anti-angiogenic and anti-inflammatory modality, but is complicated by difficult synthesis, low stability and rapid metabolism. Therefore, analogs that do not suffer from these disadvantages could be therapeutically beneficial. We noted the similarity of solenopsin to ceramide and hypothesized that solenopsin and analogs might act as ceramide-like agonists. We thus evaluated their ceramide-like activity at both the lipid membrane and mitochondria.
A major obstacle to the widespread use of solenopsin is obtaining sufficient quantities for preclinical and clinical studies. Extraction of solenopsin from ants is not feasible, and therefore large scale synthesis is required. All current synthetic methods suffer from reliance on expensive reagents and multiple steps [31-33]. In this paper, we demonstrate that solenopsin analogs can be synthesized by lithiation of inexpensive industrial dimethylpyridines, followed by alkylation of the lithiated pyridines with alkyl halides, which can be varied. Finally, the alkylated pyridine is hydrogenated to give the solenopsin analogs (Figure 1). Each of these steps is amenable to scale up for industrial production [32,34]. In a previous study by our group we showed that solenopsin analogs with shorter aliphatic side chains lacked anti-proliferative activity in murine angiosarcoma SVR cells [1]. Here we demonstrate that solenopsin analogs with 8 carbon longer side chains (S11 and S13) also lack significant anti-proliferative activity in murine SVR angiosarcoma and human melanoma (A375 and A2058) cells. On the other hand, S14, which has a 4 carbon longer side chain, and S15, which has the same length side-chain but contains a hydroxyl group and lacks the methyl group, are equally as potent as (−)-solenopsin A in all three cell lines. Thus, optimal length of the aliphatic side chain appears to be associated with high anti-proliferative activity.
One of the modes of ceramide’s activity is downregulation of Akt, a serine-threonine kinase that plays a central role in protecting tumor cells from apoptosis [35,36]. Akt, which originally was discovered as a viral oncogene, has transforming activity in multiple cell types. PTEN is a tumor suppressor that inhibits Akt activation in lipid rafts. We used A375 human melanoma cells, which express wild type PTEN [37], to assess the effect of solenopsin A on PTEN localization. Indeed, solenopsin A did appear to cause relocalization of PTEN to lipid rafts (Additional file 1: Figure S3). FRET-based reporters were used to show that solenopsin A inhibits Akt activity (AktAR) and PDK1 activation (Lyn-PARE) in lipid rafts to the same extent as ceramide in NIH3T3 murine embryonic fibroblast cells (Figure 3). Unexpectedly, we found that solenopsin A increases Akt phosphorylation in cells with wild type p53 (A375 and A2058), while it decreases Akt activation in cells with defective p53 function (SVR and 786-O) (Additional file 1: Figure S4), thus demonstrating a context dependent effect on tumor cells [1,38]. Taken together it appears as if relocalization of PTEN to lipid rafts is insufficient to block Akt activation, at least in cells with wild type p53. In addition, solenopsin A is highly effective in killing A375 human melanoma cells, despite no reduction of Akt phosphorylation. This implies that solenopsin’s killing of tumor cells with wild type PTEN may not depend exclusively on Akt deactivation, but may involve additional events, such as mitochondrial induced cell death. Indeed, investigation of solenopsin and analogs affect on OCR-levels revealed a similar trend as the proliferation assays. Solenopsin A and the analogs S12, S14 and S15 all markedly decreased the OCR levels (Figure 4) in UM-SCC1A cells. Interestingly, autophagy staining showed that (−)-solenopsin A (trans geometry) but not S14 (cis geometry) induced mitophagy (Additional file 1: Figure S5), indicating strict stereochemical requirements for induction of mitophagy. Thus, the trans geometry is not essential for anti-proliferative activity, although it appears to be required for ceramide-like mitophagy activity. Further, our study of superoxide levels displayed a similar pattern as the proliferation and OCR assays, i.e. solenopsin A, S12, S14 and S15 substantially increased superoxide, whereas S11 and S13 showed no effect (Figure 5). Figure 6 is a suggested model, based on our results, which show the effect of ceramide and solenopsin on cell function. The FDA requires all new topical and transdermal products to be evaluated for skin irritation and sensitization [39]. A 3D cell culture of human skin keratinocytes has been evaluated by European Center for the Validation of Alternative Methods as an alternative to the rabbit draize test, to avoid animal usage [40]. In our study, solenopsin and analogs S12, S14 were found to be non-irritant (Additional file 1: Figure S2). Thus, solenopsin and analogs have the potential for further development for topical treatment of both tumors and inflammatory disorders that are deficient in ceramide.
Figure 6.
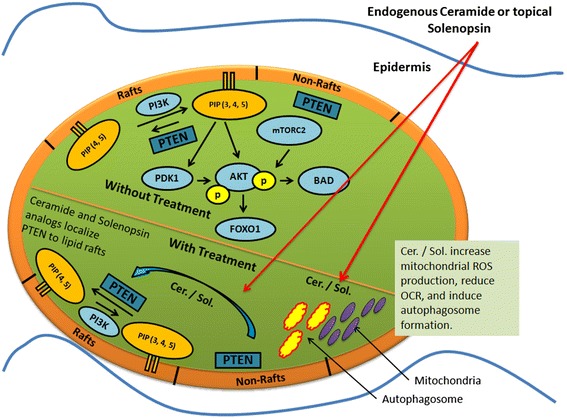
A model for the effect of Ceramide and Solenopsin on cell function. Ceramide (Cer.) and solenopsin (Sol.) derivatives inhibit the AKT pathway by translocating PTEN from non-raft regions into the rafts. Ceramide and solenopsin also increase mitochondrial ROS production, reduce oxygen consumption (OCR), and induces autophagosome formation.
Conclusions
In this work we show that the naturally occurring (−)-solenopsin A exhibit ceramide-like biological activity in human melanoma cell lines and could therefore be useful in treating hyperproliferative skin disorders. First, we demonstrate that native (−)-solenopsin A mimics canonical functions of ceramide on cells, namely inhibition of Akt activity and PDK1 activation in lipid rafts as well as induction of mitophagy (Figure 6). Differences in cis-trans geometry and chain length abrogate these functions. Second, the response to solenopsin differs in cells of various genetic backgrounds, with no effect or elevation of Akt phosphorylation in cells with wild type p53 (A375, A2058), while decreasing Akt phosphorylation in cells with defective p53 (SVR, 786-O). Solenopsin and analogs are also effective in killing cells with elevated Akt phosphorylation, which is an adverse prognostic factor in most cancers. Based upon the novel hypothesis that restoration of ceramide-like signaling may be beneficial in the treatment of neoplastic and hyperproliferative skin disorders, our assessment of solenopsin and analogs on normal cutaneous cells and premalignant cells demonstrate the potential of solenopsin and analogs to treat hyperproliferative disorders when compared to ceramide.
Supporting data
The data set supporting the results of this article is included within the article and its additional file: Supplementary Material Solenopsin.
Additional file
Supplementary Material Solenopsin.
Acknowledgements
This work was funded by Dr. Arbiser's NIH R01 AR047901, the Margolis Foundation, Rabinowitch-Davis Foundations and Dr. Zhang's NIH R01 DK073368.
Abbreviations
- DHE
Dihydroethidium
- FOXO
Forkhead box protein
- OCR
Oxygen consumption rate
- PI3K
Phosphoinositol-3 kinase
- PIP2
Phosphatidylinositol (4,5)-biphosphate
- PIP3
Phosphatidylinostol (3,4,5)-triphosphate
- PTEN
Phosphatase and tensin homolog
Footnotes
Competing interest
Jack Arbiser and J Philip Bowen are coinventors on US Patent 8,168,657. The authors declare that they have no competing interests.
Authors’ contributions
IK designed and performed the majority of the experiments, interpreted the results, and wrote most of the manuscript. XZ designed and performed the FRET-based reporter experiments, interpreted results, and edited the manuscript. RT designed and performed the OCR and autophagy staining experiments and interpreted results. ATS did the last synthesis step (hydrogenation) of the solenopsin analogs and characterized the final products. MYB did the proliferation experiments with the non-tumor cell lines, interpreted results, and wrote part of the manuscript. PB designed and performed the 3D skin model experiments and interpreted results. AKB provided intellectual input and directed the 3D skin model experiments. JPB contributed with (+)- and (−)-solenopisn A and provided intellectual input. GQ, SLF, MDB, and KSP synthesized and characterized the natural product (−)-solenopsin A and the enantiomer (+)-solenopsin A. XL designed and helped perform the sucrose density fractionation experiments. GC provided intellectual input and directed the sucrose density fractionation experiments. BO provided intellectual input and directed the OCR and autophagy staining experiments. JZ provided intellectual input, directed the FRET-based reporter experiments, and edited the manuscript. EBW provided intellectual input, direction in the synthesis design, and edited the manuscript. RSA designed and performed the ROS experiments and interpreted results. JLA formulated the research ideas, provided intellectual input, directed the research designs, provided resources, interpreted results, and wrote major parts of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Isabella Karlsson, Email: anna.isabella.r.karlsson@emory.edu.
Xin Zhou, Email: xzhou30@jhmi.edu.
Raquela Thomas, Email: thomasrj@musc.edu.
Allorie T Smith, Email: atsmith@uu.edu.
Michael Y Bonner, Email: mybonne@emory.edu.
Pooja Bakshi, Email: bakshi@live.mercer.edu.
Ajay K Banga, Email: banga_ak@mercer.edu.
J Phillip Bowen, Email: bowen_jp@mercer.edu.
Ghassan Qabaja, Email: g_qabaja@uncg.edu.
Shavon L Ford, Email: shavon.ford@catalent.com.
Matthew D Ballard, Email: mdballar@uncg.edu.
Kimberly S Petersen, Email: kspeters@uncg.edu.
Xuechen Li, Email: xuechen.li@emory.edu.
Guangping Chen, Email: gchen3@emory.edu.
Besim Ogretmen, Email: ogretmen@musc.edu.
Jin Zhang, Email: zhang32@jhmi.edu.
E Blake Watkins, Email: bwatkins@uu.edu.
Rebecca S Arnold, Email: rsarnol@emory.edu.
Jack L Arbiser, Email: jarbise@emory.edu.
References
- 1.Arbiser JL, Kau T, Konar M, Narra K, Ramchandran R, Summers SA, et al. Solenopsin, the alkaloidal component of the fire ant (Solenopsis invicta), is a naturally occurring inhibitor of phosphatidylinositol-3-kinase signaling and angiogenesis. Blood. 2007;109:560–565. doi: 10.1182/blood-2006-06-029934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rabionet M, Gorgas K, Sandhoff R. Ceramide synthesis in the epidermis. Biochim Biophys Acta. 1841;2014:422–434. doi: 10.1016/j.bbalip.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Mencarelli C, Martinez-Martinez P. Ceramide function in the brain: when a slight tilt is enough. Cell Mol Life Sci. 2013;70:181–203. doi: 10.1007/s00018-012-1038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Barros M, Coant N, Truman JP. Snider AJ. Sphingolipids in colon cancer. Biochim Biophys Acta: Hannun YA; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuya H, Shimizu Y, Kawamori T. Sphingolipids in cancer. Cancer Metastasis Rev. 2011;30:567–576. doi: 10.1007/s10555-011-9304-1. [DOI] [PubMed] [Google Scholar]
- 6.Gomez-Munoz A, Gangoiti P, Arana L, Ouro A, Rivera IG, Ordonez M, et al. New insights on the role of ceramide 1-phosphate in inflammation. Biochim Biophys Acta. 1831;2013:1060–1066. doi: 10.1016/j.bbalip.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- 8.Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 10.Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1:707–717. [PubMed] [Google Scholar]
- 11.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, et al. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci U S A. 1997;94:861–866. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arbiser JL, Klauber N, Rohan R, van Leeuwen R, Huang MT, Fisher C, et al. Curcumin is an in vivo inhibitor of angiogenesis. Mol Med. 1998;4:376–383. [PMC free article] [PubMed] [Google Scholar]
- 13.Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501–35507. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 14.Gao X, Lowry PR, Zhou X, Depry C, Wei Z, Wong GW, et al. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc Natl Acad Sci U S A. 2011;108:14509–14514. doi: 10.1073/pnas.1019386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, et al. The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 17.Huang H, Feng X, Zhuang J, Frohlich O, Klein JD, Cai H, et al. Internalization of UT-A1 urea transporter is dynamin dependent and mediated by both caveolae- and clathrin-coated pit pathways. Am J Physiol Renal Physiol. 2010;299:F1389–1395. doi: 10.1152/ajprenal.00718.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen G, Howe AG, Xu G, Frohlich O, Klein JD, Sands JM. Mature N-linked glycans facilitate UT-A1 urea transporter lipid raft compartmentalization. FASEB J. 2011;25:4531–4539. doi: 10.1096/fj.11-185991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castro BM, Prieto M, Silva LC. Ceramide: a simple sphingolipid with unique biophysical properties. Prog Lipid Res. 2014;54:53–67. doi: 10.1016/j.plipres.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Caselli A, Mazzinghi B, Camici G, Manao G, Ramponi G. Some protein tyrosine phosphatases target in part to lipid rafts and interact with caveolin-1. Biochem Biophys Res Commun. 2002;296:692–697. doi: 10.1016/S0006-291X(02)00928-2. [DOI] [PubMed] [Google Scholar]
- 21.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/MCB.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goswami R, Singh D, Phillips G, Kilkus J, Dawson G. Ceramide regulation of the tumor suppressor phosphatase PTEN in rafts isolated from neurotumor cell lines. J Neurosci Res. 2005;81:541–550. doi: 10.1002/jnr.20550. [DOI] [PubMed] [Google Scholar]
- 23.Hajduch E, Turban S, Le Liepvre X, Le Lay S, Lipina C, Dimopoulos N, et al. Targeting of PKCzeta and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem J. 2008;410:369–379. doi: 10.1042/BJ20070936. [DOI] [PubMed] [Google Scholar]
- 24.Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60:1800–1804. [PubMed] [Google Scholar]
- 25.Kogot-Levin A, Saada A. Ceramide and the mitochondrial respiratory chain. Biochimie. 2014;100:88–94. doi: 10.1016/j.biochi.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 26.Lecour S, Van der Merwe E, Opie LH, Sack MN. Ceramide attenuates hypoxic cell death via reactive oxygen species signaling. J Cardiovasc Pharmacol. 2006;47:158–163. doi: 10.1097/01.fjc.0000198520.28674.41. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Junk P, Huwiler A, Burkhardt C, Wallerath T, Pfeilschifter J, et al. Dual effect of ceramide on human endothelial cells: induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation. 2002;106:2250–2256. doi: 10.1161/01.CIR.0000035650.05921.50. [DOI] [PubMed] [Google Scholar]
- 28.Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 29.Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene. 2003;22:5897–5906. doi: 10.1038/sj.onc.1206702. [DOI] [PubMed] [Google Scholar]
- 30.Beckham TH, Cheng JC, Marrison ST, Norris JS, Liu X. Interdiction of sphingolipid metabolism to improve standard cancer therapies. Adv Cancer Res. 2013;117:1–36. doi: 10.1016/B978-0-12-394274-6.00001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reding MT, Buchwald SL. Short Enantioselective Total Syntheses of the Piperidine Alkaloids (S)-Coniine and (2R,6R)-trans-Solenopsin A via Catalytic Asymmetric Imine Hydrosilylation. J Org Chem. 1998;63:6344–6347. doi: 10.1021/jo980808q. [DOI] [PubMed] [Google Scholar]
- 32.Wilkinson TJ, Stehle NW, Beak P. Enantioselective syntheses of 2-alkyl- and 2,6-dialkylpiperidine alkaloids: preparations of the hydrochlorides of (−)-coniine, (−)-solenopsin A, and (−)-dihydropinidine. Org Lett. 2000;2:155–158. doi: 10.1021/ol9912534. [DOI] [PubMed] [Google Scholar]
- 33.Amat M, Llor N, Hidalgo J, Escolano C, Bosch J. Enantioselective synthesis of piperidine, indolizidine, and quinolizidine alkaloids from a phenylglycinol-derived delta-lactam. J Org Chem. 2003;68:1919–1928. doi: 10.1021/jo0266083. [DOI] [PubMed] [Google Scholar]
- 34.Macconnell JG, Blum MS, Fales HM. Alkaloid from fire ant venom: identification and synthesis. Science. 1970;168:840–841. doi: 10.1126/science.168.3933.840. [DOI] [PubMed] [Google Scholar]
- 35.Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 36.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, et al. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 37.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fried L, Arbiser JL. The reactive oxygen-driven tumor: relevance to melanoma. Pigment Cell Melanoma Res. 2008;21:117–122. doi: 10.1111/j.1755-148X.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 39.Tan CH, Rasool S, Johnston GA. Contact dermatitis: allergic and irritant. Clin Dermatol. 2014;32:116–124. doi: 10.1016/j.clindermatol.2013.05.033. [DOI] [PubMed] [Google Scholar]
- 40.Kandarova H, Klausner M, Kubilus J, Ayehunie S, Hayden P, Kaluzhny Y, Letasiova S, Sheasgreen J. Update on validation status and industry utilization of normal human 3D (NHU-3D) animal alternative models. In: MatTek Corporation A, MA, USA; MatTek In Vitro Life Science Laboratories, Bratislava, Slovakia, editor. Presented at 8th World Congress on Alternative and Animal Use; Montreal, Canada. 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Solenopsin.