

Abstract
Summary: Network pharmacology-based prediction of multi-targeted drug combinations is becoming a promising strategy to improve anticancer efficacy and safety. We developed a logic-based network algorithm, called Target Inhibition Interaction using Maximization and Minimization Averaging (TIMMA), which predicts the effects of drug combinations based on their binary drug-target interactions and single-drug sensitivity profiles in a given cancer sample. Here, we report the R implementation of the algorithm (TIMMA-R), which is much faster than the original MATLAB code. The major extensions include modeling of multiclass drug-target profiles and network visualization. We also show that the TIMMA-R predictions are robust to the intrinsic noise in the experimental data, thus making it a promising high-throughput tool to prioritize drug combinations in various cancer types for follow-up experimentation or clinical applications.
Availability and implementation: TIMMA-R source code is freely available at http://cran.r-project.org/web/packages/timma/.
Contact: jing.tang@helsinki.fi
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Recently, a network pharmacology paradigm for anticancer drug discovery has been proposed, with an aim of developing multi-targeted drug combinations that consist of distinct chemical agents as a promising strategy to improve treatment efficacy and safety (Al-Lazikani et al., 2012). Most mathematical models for predicting the effects of drug combinations relies on empirical cellular dynamic models, such as those based on signaling pathways or metabolic networks. These types of methods are hardly applicable for predicting cancer-selective drug combinations due to lack of accurate kinetic parameters for each cancer type (Sun et al., 2013). Other methods use the transcriptional responses of drugs, i.e. gene expression profiles before and after drug treatments, to predict drug combinations (Zhao et al., 2014). However, such drug-induced transcriptional phenotypes are not routinely profiled in a typical high-throughput drug screen, and thus provide limited translation potential in the clinical settings.
We have developed a logic-based network pharmacology approach Target Inhibition Interaction using Maximization and Minimization Averaging (TIMMA) for predicting the effects of drug combinations in a given cancer cell sample (Tang et al., 2013). TIMMA combines drug-target interaction networks with single-drug sensitivity profiles, derived either from individual cell line models (Barrettina et al. 2012) or from patient-derived cell samples (Pemovska et al. 2013). The network algorithm starts by searching a set of combinatorial targets that are most predictive of the single-drug sensitivities. A drug combination is then treated as a combination of target inhibitions, the effect of which can be estimated based on the set relationships with the target profiles of the drugs. The outcome of the TIMMA model provides a list of predicted synergy scores for drug combinations, from which a target inhibition network can be inferred.
To enable wider applications of the method, several major limitations need to be overcome. First, the original TIMMA package was written in MATLAB, the accessibility of which in biomedical research community is rather limited compared with the open source R environment. Second, for applying the target set comparison, the drug-target profiles must be binarized, encoding 1 for a true target and 0 for a non-target. A typical drug-target profiling assay, however, often reveals quantitative polypharmacological interactions more complex than what such binary data can capture. Third, the topology of the target inhibition networks was derived manually from the model predictions. The lack of efficient network reconstruction algorithms may become the bottleneck for more straightforward biological interpretations when the network sizes increase. These issues are now addressed in the newly developed R implementation.
2 Implementation
The TIMMA-R workflow starts by preparing two types of input data (Fig. 1). To maximize the prediction power, the first input defines the drugs’ polypharmacological profiles by considering both strong and weak drug-target interactions, so that the effect of a drug combination can be modeled through its (multiple) target interactions. The proteome-wide quantitative drug-target interaction data are available in PubChem, ChEMBL (Gaulton et al., 2012) or CanSAR (Halling-Brown et al., 2012) databases. To minimize the false positive or negative interactions, bioactivity data integration from multiple studies is advised (Tang et al., 2014). The second input data involves drug sensitivity profiles for a given cancer cell line, which can be obtained e.g. from the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al., 2012), the Genomics of Drug Sensitivity in Cancer (GDSC) (Yang et al., 2013) or the Library of Integrated Network-based Cellular Signatures (LINCS) (Vempati et al., 2014) databases. However, the TIMMA-R is not limited to using cell line data only, but can also be applied to patient-derived drug sensitivity profiles (Pemovska et al., 2013). In the following, we briefly report the major updates of the algorithm that will facilitate its usage for high-throughput drug combination discoveries.
Fig. 1.

The TIMMA-R workflow
2.1 Computational speed
With the techniques of vectorization and multi-dimensional matrix computation, the TIMMA-R algorithm has been made more efficient and scalable for large-scale analyzes. For a typical data, consisting of 50 drugs and 500 targets, the average running time on a desktop computer is <50 s, compared with >250 s when using the original MATLAB implementation. Note that the current TIMMA-R aggregates the information from each drug sequentially. A straightforward extension would be thus parallel computing using multiple processors, e.g. one processor for one drug, which can further increase the computational speed.
2.2 Sensitivity analysis
The traditional definition of a drug’s target, originated from the magic bullet paradigm, often considers one or two primary targets that are thought to induce the therapeutic effects (Hopkins, 2008). However, recent proteome-wide bioactivity studies have revealed much more low-affinity, multi-targeted drugs than previously thought (e.g. Davis et al., 2011). An important question for any polypharmacological modeling method is therefore its robustness with respect to experimental uncertainties in drug-target interactions.
We performed a sensitivity analysis using simulated binary drug-target interaction data with 50 drugs and 100 targets, where experimental noise was modeled by flipping either from 0 to 1 (false positive) or vice versa (false negative), for up to 30% of the drug-target interactions. The prediction results between the selected target sets before and after the flipping were compared using the RV coefficient (Lê et al., 2008). The rate of positive interactions in the data were set at 0.3. The simulation was repeated 100 times and summarized in Figure 2. As expected, the RV coefficient decreased as the false interaction rates increase, yet being significantly higher than the random predictions (P < 0.001, paired t-test). These results indicate that TIMMA-R is applicable for those real case studies, where the drug-target interaction data has been either experimentally validated or manually curated.
Fig. 2.
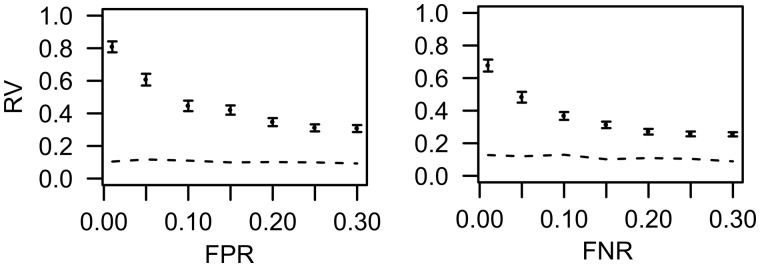
Robustness of TIMMA-R predictions using simulated drug-target interactions with a varying degree of false negative or false positives. y-axis: average RV coefficient, where error bars indicate standard error of the means; x-axis: false positive rate or false negative rate; Dotted trace, random prediction
2.3 Modeling of multi-class drug-target data
Compared with the conventional drug-target binarization, a more realistic drug-target modeling classifies the targets of a given drug into several classes in terms of their binding affinities (Tyner et al., 2013). TIMMA-R modeling of such multi-class data are straightforward, as the set relationships still hold between two drugs, except that the number of possible scenarios for a target combination goes from to where is the number of targets and is the number of interactions classes. Even though the target combination space increases, the prediction accuracy of TIMMA-R under the categorical setting stays at the same level as using the binary data (Supplementary Material). On the other hand, we found that introducing more drug-target classes may not always lead to better prediction accuracy. This may be due to the multi-classification scheme of Tyner et al. (2013), where weak interactions, such as Kd or IC50 values close to 10 , were considered as one of the active classes. Such a classification might be sub-optimal for characterizing the response of patient-derived samples given that the majority of the drugs’ in vivo or ex vivo efficacy is expected to be elicited via their targets with nanomolar potency. Given that the drug-target data are already sparse in the binary case, we do not recommend over-interpreting the drug-target interactions with more than three classes (see Supplementary Material for more detailed discussion).
2.4 Network reconstruction
The TIMMA-R predictions can be formulated as a complete truth table, based on which the minimized Boolean expression is determined by the enhanced Quine-McCluskey algorithm using the Qualitative Comparative Analysis (QCA) package in R (Duşa, 2010). The minimized Boolean expression is a union of target combination scenarios leading to the same (maximal) response. For network visualization, we utilized a two-terminal graph, with series and parallel components, to represent the predicted response of cancer survival signaling pathways (see Supplementary Material). With the network reconstruction algorithm, TIMMA-R is able to produce illustrative visualizations that facilitate the interpretation of the drug combination predictions. The nested network format enables network visualization and analysis in Cytoscape.
5 Conclusion
The TIMMA-R package will facilitate effective integration of drug-target and drug sensitivity profiles which are becoming increasingly available for the anticancer research. The package does not only offer an efficient prioritization tool for selecting the most promising drug combinations for further experimental testing, but also produces data-driven hypotheses for their target interactions. Such network-centric investigation of the underlying mechanisms of action in the given cellular context will greatly improve our knowledge about the functional cross-talks between parallel cancer signaling pathways to be utilized in anticancer treatment applications.
Funding
Academy of Finland (Grants 272437, 269862, 279163); the Biocentrum Helsinki and the Cancer Society of Finland.
Conflict of Interest: none declared.
Supplementary Material
References
- Al-Lazikani B., et al. (2012) Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol., 30, 679–692. [DOI] [PubMed] [Google Scholar]
- Barretina J., et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.I., et al. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol., 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Duşa A. (2010) A mathematical approach to the boolean minimization problem. Qual. Quant., 44, 99–113. [Google Scholar]
- Gaulton A., et al. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res., 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halling-Brown M.D., et al. (2012) canSAR: an integrated cancer public translational research and drug discovery resource. Nucleic Acids Res., 40, D947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A.L. (2008) Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol., 4, 682–690. [DOI] [PubMed] [Google Scholar]
- Lê S., et al. (2008) FactoMineR: an R package for multivariate analysis. J. Stat. Softw., 25, 1–18. [Google Scholar]
- Pemovska T., et al. (2013) Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov., 3, 1416–1429. [DOI] [PubMed] [Google Scholar]
- Sun X., et al. (2013) High-throughput methods for combinatorial drug discovery. Sci. Transl. Med., 5, 205rv1. [DOI] [PubMed] [Google Scholar]
- Tang J., et al. (2013) Target inhibition networks: predicting selective combinations of druggable targets to block cancer survival pathways. PLoS Comput. Biol., 9, e1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J., et al. (2014) Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model., 54, 735–743. [DOI] [PubMed] [Google Scholar]
- Tyner J.W., et al. (2013) Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res., 73, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vempati U.D., et al. (2014) Metadata standard and data exchange specifications to describe, model, and integrate complex and diverse high-throughput screening data from the library of integrated network-based cellular signatures (LINCS). J. Biomol. Screen., 19, 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., et al. (2013) Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res., 41, D955–D961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., et al. (2014) Predicting cooperative drug effects through the quantitative cellular profiling of response to individual drugs. CPT Pharmacomet. Syst. Pharmacol., 3, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.