

Abstract
Extensive data suggest that estradiol contributes to the development of breast cancer by acting as a mitogen and exerting direct genotoxic effects after enzymatic conversion to 4-hydroxyestradiol (4-OHE2) via cytochrome P450 1B1 (CYP1B1). The mammary gland, ovary, and uterus all express CYP1B1. Overexpression of this enzyme has been associated with an increased risk of breast cancer and blockade might reduce this carcinogenic effect. For this reason, we conducted systematic in vitro and in vivo studies of a CYP1B1 inhibitor, TMS (2,3′,4,5′-tetramethoxystilbene). We found that TMS blocked the enzymatic conversion of radiolabeled estradiol to both 2-hydroxyestradiol (2-OHE2) and 4-OHE2, but did not inhibit Cyp1b1 message formation. In vivo studies using mass spectrometry showed that TMS inhibited formation of 2-OHE2 and 4-OHE2 and the resulting estrogen-DNA adducts. To examine its biologic actions in vivo, we investigated whether TMS could block the hyperplastic changes that occur in the developing breast of aromatase-transfected mice. We found that TMS induced a significant reduction of ductal structures in mice less than 6 months in age. In older mice, no reduction in breast morphology occurred. These latter studies uncovered unexpected estrogen agonistic actions of TMS at high doses, including a paradoxical stimulation of breast ductal structures and the endometrium. These studies suggest that the enzyme inhibitory properties of TMS, as well as the effects on developing breast, could implicate a role for TMS in breast cancer prevention, but only in low doses and on developing breast.
Keywords: 2,3′,4,5′-Tetramethoxystilbene (TMS); Mouse mammary gland; Ductal branching; Aromatase-transgenie mice; Cytochrome P450 1B1
Introduction
Epidemiologic studies suggest that estrogen contributes to the development of breast cancer. Early menarche, late menopause, high breast density, and increased plasma estrogen levels are all associated with an increased risk of breast cancer. A widely accepted mechanistic hypothesis suggests that estrogens directly activate the transcription of genes involved in cellular proliferation, enhancing mitosis and accelerating the rate of mutations, which lead to cancer. However, the enhanced proliferation induced by estrogen may not explain all of its carcinogenic effects. In certain model systems, the estrogen receptor antagonist fulvestrant fails to block estrogen-induced neoplastic transformation [1].
Several investigators have suggested that metabolism of estrogens may influence the carcinogenic process through the formation of genotoxic metabolites. One pathway involves the conversion of estradiol (E2) to 4-hydroxyestradiol (4-OHE2) via the cytochrome P450 enzyme CYP1B1 [2–4]. 4-OHE2 is then further metabolized to the catecholestrogen-3,4-quinone, resulting in depurinating DNA adducts, error-prone DNA repair, and formation of mutations [5]. Human tissues that are regulated by hormones, such as the mammary gland, ovary, uterus, and prostate, all express cytochrome P450 1B1 (CYP1B1) [6–9].
Based on these considerations, researchers have sought to identify CYP1B1 inhibitors in an effort to find agents that would prove useful in preventing cancer modulated by this enzyme. Past investigations examined the stilbene family as promising lead compounds. The Guengerich laboratory has shown that 2,4,3′,5′-tetramethoxystilbene (TMS) is the most potent and selective CYP1B1 inhibitor in vitro [10]. TMS was unexpectedly found to exert additional biologic effects on breast cancer cell lines, inducing apoptosis through mitochondrial pathways [11]. This study involves a series of experiments to examine further the direct and indirect effects of TMS on CYP1B1 enzymatic activity and transcriptional regulation. In addition, we determined whether TMS alters ductal and lobular morphology in the developing and developed breast. We found that TMS blocked the enzymatic conversion of E2 to both 2-OHE2 and 4-OHE2, but does not alter Cyp1b1 message. In mice less than 6 months of age, TMS reduced ductal and lobular formation in the developing breast. These effects suggest that TMS or a more potent and selective inhibitor might be useful for the prevention of breast cancer.
Materials and methods
Reagents and chemicals
TMS was synthesized as described previously [12]. HistoClear was from National Diagnostics (Atlanta, GA) and Taq DNA polymerase from Invitrogen (Carlsbad, CA). All other reagents were from Sigma-Aldrich (St. Louis, MO).
Measurement of CYP1B1 and CYP1A1 activities in breast cancer cells
LTED [13] or MCF-7 cells were pretreated with TCDD for 48 h to induce CYP1B1 and CYP1A1 activities sufficiently to allow demonstration of inhibition. Cells were incubated at 37°C with 1.5 ml serum-free medium containing [6,7-3H]E2 (1 μCi/ml), TCDD (10 nM), the catechol-o-methyltransferase inhibitor OR486 (10 μM), and 4-OHE2 or 2-OHE2 (5 μM) for 6 h. After incubation, the medium was recovered and treated with β-glucuronidase and sulfatase for hydrolysis of the metabolite conjugates [14]. Catecholestrogens were isolated [14] and separated by thin layer chromatography in a solvent containing chloroform, cyclohexane, and glacial acetic acid (2:2:1). Radioactivity of steroid spots was counted in a Beckman LS6500 scintillation counter. The conversion of radiolabeled E2 to the 4-OHE2 and 2-OHE2 metabolites, respectively, was used to assess CYP1B1 and CYP1A1 activity.
Aromatase-transfected mice
The previously described, MMTV-aromatase over-expressing transgenic mice were a kind gift from Dr. Raj Tekmal [15]. All animal experiments were conducted under Federal and Institutional guidelines and approved by the University of Virginia Animal Care and Use Committee.
TMS and letrozole treatments
TMS was administered at three different doses: low dose, 30 mg via two silastic implants of 15 mg each; mid-dose, 60 mg via two silastic implants of 30 mg each, which were placed subcutaneously into the back of mice. The 30 mg implants delivered 77 microgram/kg/day and the 60 mg implants delivered 144 μg/kg/day (see Supplemental Fig. 1). High dose, subcutaneous injections at 50 mg/kg daily. Letrozole was administered by gavage at 1 mg/kg/mouse/day.
In vivo experiments
Ovariectomy plus E2 plus TMS
Three groups were utilized including: (a) ovariectomy plus vehicle, (b) ovariectomy plus 130 pg/ml E2, and (c) ovariectomy plus 280 pg/ml of E2. Owing to variations in the level of aromatase expression in breast tissue in these transgenic animals, we stratified according to the pre-treatment degree of breast hyperplasia before randomization. To accomplish this, we removed the left fourth mammary gland in 3-week-old mice, prepared whole mounts, and staged them into four groups based on degree of proliferation. The treatment protocol involved a 3-month period. Month 1: ovaries were removed at the start of the month and the fifth mammary gland on the right side obtained at the end of the month. Month 2: vehicle or estrogens were started and at the end of the month, the third mammary gland on the right side of each mouse was removed, whole mounted and assessed to quantitate ductal branching. Month 3: E2 or vehicle was continued and 30 mg TMS administered to groups b and c. At the end of the month, mice were sacrificed and the third mammary gland on the left side was removed and whole mounted for quantitation with Metamorph analysis.
TMS versus letrozole
The left fourth mammary glands were removed from 3-week-old mice to serve as a control in each of three groups. Group 1 then received vehicle for 1 month, group 2 received 60 mg TMS, and group 3 received 1 mg/kg/letrozole/day. At the end of the month, mammary glands were excised for analysis.
TMS in older animals
Low dose: TMS was given as a 30 mg silastic implant that delivered 77 μg TMS/kg/mouse/day. High dose: TMS was given daily at 50 mg/kg via subcutaneous injection.
Cyp1b1 (mouse) PCR
Was performed as reported earlier [16].
Measurement of 4-OHE2 and E2-adenine adducts
Analysis of estrogen metabolites was determined by UPLC coupled to a tandem mass spectrometer as previously described [17]. The metabolites that were analyzed are shown in Supplemental Table 1c, d.
Whole mount preparations
Whole mounts were prepared as described previously [18]. Whole mount photographs were taken using an Olympus SZX12 low magnification microscope with a 0.5× objective. Photographs were analyzed for tertiary ductal structures and alveolar buds (TDSABs) which are analogous to TDLUs in humans and for whole ductal structures (whole duct = primary and secondary ducts + TDSABs). A transparency film was placed on top of the whole mount picture and then the ductal branches excluding the lymph node, blood vessels, and squamous nodule were traced by hand. Transparencies were scanned and imported into Metamorph analysis software. Metamorph (Molecular devices, Sunnyvale, CA) was used to evaluate TDSABs and whole ductal structures. In addition, TDSABs were analyzed by drawing and counting the number of TDSABs to determine the % change in TDSABs (see Fig. 5d).
Fig. 5.

TMS increased alveolar buds when administered to older mice at high concentrations. From left to right the panels represent: 1 intact control, 2 TMS treatment of intact mice >5 months of age, 3 ovariectomized mice >5 months of age; top panels are before treatment and bottom panels are after treatment. a Whole mount of No. 4 mammary gland demonstrating increased alveolar buds and tertiary lateral branches with 50 mg/kg/day TMS treatment, but not in the intact control or ovariectomy group. b Metamorph analysis was used to determine the change in whole duct structure (primary and secondary ducts + TDSABs) in mice older than 6 months of age. c Metamorph analysis of TDSABs (tertiary ductal structures and alveolar buds). d Visual analysis of TDSABs. The graph indicates the mean ± SE for the percentage change (P = 0.04 between TMS group and control, P < 0.01 between TMS and ovariectomy, and P = 0.008 between control and ovariectomy)
Statistical analysis
Box plot analysis was used to examine the differences between the control, 30 mg TMS-treated, and 60 mg TMS-treated animals. Analyses compared all 2-OH metabolites in the three groups and all 4-OH metabolites (see Supplemental Table 1a, b for specific metabolites measured). The Kruskal–Wallis test was used to determine the statistical significance of the data, n = 57 for comparing 2-OHE2 data and n = 42 for comparing 4-OHE2 data. Mammary gland whole mount results were expressed as means ± SEM for % ductal branching after treatment and % change of TDSABs between before and after treatment. Differences of TDSABs between before and after treatment were analyzed by one-way ANOVA test by using SPSS12.0 for Windows.
Results
Effect of TMS on CYP1B1 activity
As shown in Fig. 1a., TMS decreased the conversion of E2 to 4-OHE2 in a dose responsive fashion (Fig. 1a). We demonstrated an even greater effect on 2-OHE2 formation, indicating that TMS blocks CYP1A1 as well as CYP1B1. Previous reports from in vitro studies had suggested that TMS inhibits CYP1B1 by a reduction of CYP1B1 transcription as well as by blockade of its enzyme activity [19]. Such effects had never been confirmed with in vivo experiments; accordingly, we designed experiments to examine TMS function in an animal model. In vivo studies were conducted to examine the effects of TMS on mouse Cyp1b1 and Cyp1a1 in vivo, using measurements of 2- and 4-hydroxy metabolites of E2 as biochemical endpoints. To enhance the sensitivity of detection of metabolites, we utilized aromatase-transfected mice, which synthesize large amounts of E2 in breast tissue. Mice were randomized into a control group, a 30 mg TMS group, and a 60 mg TMS group, and treatment was given via silastic implants for 8 weeks. At the end of this time, mice were sacrificed and the mammary glands were removed and analyzed for catecholestrogens, quinone conjugates, and depurinating DNA adducts [17]. TMS inhibited the formation of both the 2-hydroxylated (n = 57, P = 0.003) (Fig. 1b, Supplemental Table 1a) and 4-hydroxylated (n = 42, P = 0.03) (Fig. 1c, Supplemental Table 1b) metabolites. Taken together, these studies demonstrated that TMS blocks human CYP1B1, CYP1A1 (Fig. 1a) and mouse Cyp1b1, Cyp1a1 (Fig. 1b, c).
Fig. 1.

Increasing concentrations of TMS block the activity of CYP1B1 and CYP1A1. a MCF-7 cells were incubated with increasing concentrations of TMS in the presence of [3H]E2 and [3H]2-OHE2 (black circles) or [3H]4-OHE2 (black triangles) were measured as a reflection of enzyme activity, b, c 2-OHE1,2 and 4-OHE1,2 and their downstream metabolites were measured in the mammary glands of aromatase over-expressing mice treated with vehicle, 30 or 60 mg TMS. The box plots illustrate log plots of the median values (heavy line), the third quartile (area above the median), the 90th percentile (the line above the 3rd quartile), and the maximum value (circle). All values below the medians in the treated animals are undetectable. Precise values are provided in Supplemental Table 1 a and b. Asterisks indicate the individual TMS data points achieved statistical significance when compared to the intact control as determined using the Wilcoxon signed-ranked test, 30 mg TMS (P = 0.001) and 60 mg TMS (P = 0.001). The mean percentage variation between groups was compared using the Kruskal–Wallis non-parametric test and 2-OHE1,2 (P = 0.003) and 4-OHE1,2 (P = 0.033). d qRT-PCR of Cyp1b1 (mouse) from aromatase-transfected mouse breast tissue in animals treated with vehicle or 50 mg/kg TMS. Results have been normalized to β-actin. The control mice are shown on the left (striped bars) and the mice treated with TMS are on the right (black bars)
TMS does not regulate Cyp1b1 at the level of transcription
Previous reports from in vitro studies had suggested that TMS inhibits CYP1B1 by a reduction of CYP1B1 transcription. Therefore, we sought to confirm this observation in vivo. Mice were given either 0.3% HPC vehicle control or 50 mg/kg TMS subcutaneously daily for 8 weeks and breast tissue was obtained for PCR analysis. Surprisingly, TMS did not decrease mouse Cyp1b1 message as shown in three separate experiments (see Fig. 1d, compare control, leftmost striped bars, to TMS-treated, rightmost solid black bars).
Effects of TMS on cellular proliferation
Our prior studies had demonstrated that TMS reduces cell number in MCF-7 cells. As this agent blocks the conversion of E2 to its catechol metabolites, we questioned whether some of the effects of TMS might be due to reduced catecholestrogen levels. Safe et al. had previously reported that 2- and 4-OHE2 can exert estrogenic actions [20]. If correct, adding back of the catecholestrogens might partially rescue the effects of TMS on cell number. To examine this issue, we initially sought to confirm that the catecholestrogens could stimulate cell number in our experimental system. Both 2-OHE2 and 4-OHE2 enhanced cell proliferation in a dose responsive fashion (Fig. 2a) with peak effects between 10−7 and 10−6 M. These effects were mediated by estrogen receptors since fulvestrant (ICI) blocked the proliferative effects of the catecholestrogens (Fig. 2b). We then determined whether adding back of the catecholestrogens would partially rescue the TMS effect on cell number. Addition of 10−7 M 4-OHE2 or a combination of 2-OHE2 and 4-OH E2 at 10−7 M did not alter the results (see Fig. 2c). These observations allowed us to conclude that the inhibitory effects of TMS occur independently of the blockade of E2 to the catecholestrogens.
Fig. 2.

The effect of estrogen metabolites, 2-OHE2 and 4-OHE2, on the growth of MCF-7 breast cancer cells. a 2-OHE2 (black bars) and 4-OHE2 (gray striped bars) were titrated from 10−9 to 10−6 M. A vehicle control, ethanol (white bar), is shown on the left. b The anti-estrogen, ICI 182,780 (fulvestrant), blocked 2-OHE2- and 4-OHE2-stimulated growth. MCF-7 cells were grown as indicated in the figure and the concentrations of anti-estrogen, estrogen, and metabolites used were ICI at 10−8 M, E2 10−10 M, and 2-OHE2 and 4-OHE2 at 10−7 M. c MCF-7 cells were treated with increasing concentrations of TMS and then either 10−10 M E2 (black circles), 10−10 M E2 + 10−7 M 4-OHE2 (open circles) or 10−10 M E2 + 10−7 M 4-OHE2 + 10−7 M 2-OHE2 (black triangles) were added back to the TMS-treated cells
Effects of TMS on developing breast structures
Prior published studies demonstrated that TMS could bring about apoptosis on breast cancer cells by invoking the intrinsic cell death pathway [11, 21]. TMS was also effective for reducing tumor burden in vivo [11]. Our current data indicate that TMS blocks growth in vitro independently of its catecholestrogen blocking effects. Therefore, it was of interest to determine whether TMS blocked the processes of breast development in young animals or maintenance in older ones. For initial experiments, we chose to examine effects on developing breast in a murine model with rapidly proliferating breast tissue. As ovariectomy reduces breast ductal development, we compared the effects of TMS to those of ovariectomy (oophorectomy) with and without added E2 as biologic yardsticks in all experiments. This experimental design obviates any effects potentially mediated through pituitary negative feedback. Figure 3a demonstrates the effect of ovariectomy to reduce ductal development over a 3-month period. Estrogen implants (130, Fig. 3b and 280 pg/ml, Fig. 3c) increased ductal branching in ovariectomized animals in a dose responsive fashion. During month 3, TMS given along with estrogen reduced ductal structure formation (see Fig. 3 compare 3b, c with 4b, c). Visnal assessments were confirmed by quantitative Metamorph analysis (refer to Fig. 3d). The 30 mg TMS implants given during month 3 reduced ductal branching in both the 130 pg/ml (black bars, P = 0.01) and 280 pg/ml (gray bars, P = 0.018) groups of mice. To further confirm the in vitro data (see Fig. 2c), TMS exerted a direct effect rather than by a reduction of 4-OHE2 formation since addition of 4-OHE2 in combination with E2 and TMS did not rescue the reduction in ductal branching (see Supplemental Fig. 2). These data, taken together, demonstrate a direct TMS effect to reduce ductal development in oophorectomized animals replaced with E2.
Fig. 3.

30 mg of TMS caused reductions of ductal structures in oophorectomized young mice treated with E2. The four whole mount panels from left to right represent: 1 before ovariectomy, 2 1 month after ovariectomy, 3 1 month treatment with vehicle or E2, 4 1 month treatment with vehicle or TMS plus E2. The three panels from top to bottom represent a vehicle treated, b 130 pg/ml E2 treated, and c 280 pg/ml E2 treated. d Metamorph quantitation of the reduction of ductal branching in young mice by 30 mg TMS
Fig. 4.

TMS reduces ductal hyperplasia from 5-month-old aromatase-transgenic mice. a From left to tight the panels represent: 1 before treatment whole mount, 2 after treatment whole mount, 3 after treatment with H and E sections shown. From top to bottom, the panels represent intact control, TMS given as 60 mg silastic implants, or Letrozole (CGS20267) given at 20 μg/mouse. b From left to right includes: 1 intact control, 2 TMS treatment in intact mice <5 months of age, 3 ovariectomized mice <5 months of age. c Metamorph analysis of whole duct structures (primary and secondary ducts + TDSABs). d Analysis restricted to TDSABs (tertiary ductal structures and alveolar buds). The TDSABs were graphed as the mean ± SE for % change of TDS. One-way ANOVA test showed a statistically significant difference between the TMS and control groups (P = 0.016)
TMS versus letrozole and terminal end bud formation
Ovariectomy (Oophorectomy) abrogates the ovarian release of progesterone and estrogen. Blockade of estrogen production with an aromatase inhibitor such as letrozole specifically lowers E2. With this as a rationale, we sought to examine the effect of TMS in animals with intact ovaries, but negligible E2 secretion and compared the effect of TMS and letrozole on the whole mount appearance of mammary tissue. Three-week-old intact aromatase-transgenic mice had the left fourth mammary gland removed and whole mounted. At the end of the 4 weeks of treatment, the right mammary glands were removed (Fig. 4a, compare panels 1 and 2). As assessed visually, the major effect appeared to be a reduction of terminal ductal structure formation, and this occurred in both the letrozole and TMS-treated mice.
Breast effects in young versus older mice
Using animals available to us at that time, we entered animals into a study in which we gave 30 mg TMS by silastic implant for 8 weeks in mice that ranged in age from 4 to 10 months. The percentage ductal branching (mean ± SEM) was found to be 6.48 ± 0.28% in the control group (n = 16), 5.45 ± 0.3% in the ovariectomy group (n = 16), and 6.05 ± 0.4% in the TMS group (n = 16). Thus, it initially appeared that development of the mammary glands was similar in the TMS and control groups, but a reduction was observed in the ovariectomy group. A closer examination of the data uncovered that only the younger animals with developing ducts were susceptible to TMS treatment (see Fig. 4b–d). The mean ± SEM for percentage change of TDS in mice younger than 6 months were +78.8 ± 31.7% for control group, +2.7 ± 6.7% for TMS group, and −26.9 ± 18.5% for the ovariectomy group (see Fig. 4d).
TMS given to mice older than 5 months
To determine whether there was any effect of TMS on the mammary gland in the older animals, we utilized the highest dose that we had previously utilized, 50 mg/kg, given by subcutaneous injection over an 8-week period. This high dose of TMS was chosen because we expected a degree of resistance in the developed breast. Clearly, no inhibition was observed in these older animals treated with TMS, since stimulation was observed (Fig. 5a2 compare before with after). We used the Metamorph program to analyze the change in whole ductal structures, which we defined as consisting of the large duct as well as TDSABs (see “Methods” and Fig. 5b). In addition, Metamorph analysis of the whole duct (Whole Duct Structure, see Fig. 5b, c) as well as a visual analysis of TSABs (Fig. 5d) confirmed the stimulatory properties of TMS. The mean ± SEM of percentage total ductal lobular units (TDLUs) was 4.99 ± 0.43 in control group (n = 11) and 6.26 ± 0.36 in TMS group (n = 12) with the Metamorph program (Fig. 5b). Changes in alveolar buds examined also indicated a stimulatory effect of TMS (Fig. 5c). The mean ± SEM of percent change of alveolar buds were −10.1 ± 6 for the control group (n = 11), 14.2 ± 8.5 for the TMS group (n = 12), and −43 ± 5.2 for the ovariectomy group (n = 12) (P = 0.04 between control and TMS, P < 0.01 between TMS and ovariectomy, and P = 0.008 between control and ovariectomy see Fig. 5b).
In vivo toxicity of TMS
The toxicity of TMS at three different doses was tested: low dose (30 mg via silastic implants for 8 weeks), mid-dose (60 mg via silastic implants for 8 weeks), and high dose (50 mg/kg via subcutaneous injection for 4 weeks). No changes in body or uterine weight were observed (see Supplemental Table IIa, b). However, H&E staining of the organs from the high 50 mg/kg TMS treatment group showed significant toxicity to the liver on histological sections and an increase in megakaryoctes in the spleen (but without thrombocytosis in blood, see Fig. 6). Whole cell blood counts were also taken (see Supplemental Table IIc). Interestingly, we observed estrogenic effects with endometrial stimulation (see Fig. 6), but without change in total uterine weight (see Supplemental Table IIa, b).
Fig. 6.
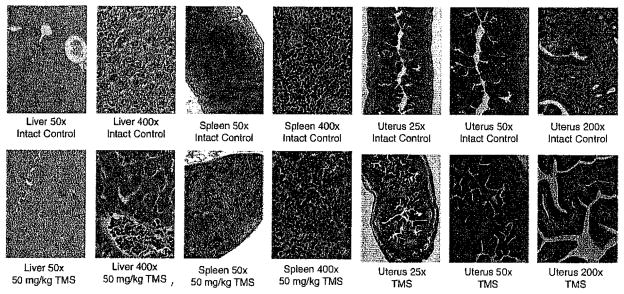
Evaluation of organs after treatment with high dose, 50 mg/kg TMS. Photomicrographs of hematoxylin and eosin stained formalin fixed, paraffin embedded slides of aromatase mouse organs following control injection or injection with 50 mg/kg TMS for 8 weeks
Discussion
2,3′,4′,5′-TMS was developed as a selective inhibitor of CYP1B1, based on in vitro studies of potency and selectivity. CYP1B1 catalyzes the conversion of E2 to 4-OHE2, a catechol estrogen reported to exert direct carcinogenic effects on breast [5, 22–24]. Our prior studies unexpectedly demonstrated a direct, pro-apoptotic effect of TMS on human breast cancer cells. These findings, taken together, suggested that TMS might be a promising agent to prevent breast cancer. For this reason, we conducted systematic studies on the effects of TMS on human cells in vitro and on breast structures in vivo. We report that TMS non-selectively blocks both 2-OHE2 and 4-OHE2 formation via inhibition of cytochrome P450 activity, but does not reduce message levels (see Fig. 1), a finding previously reported from in vitro studies [19]. We discovered that TMS prevents cellular proliferation (see Fig. 2c) by a different pathway than it uses to inhibit the cytochrome P450s.
It is of interest that our data demonstrate that catecholestrogens can stimulate breast cancer cells (see Fig. 2a). Previous study by Safe and coworkers had also shown the potential for catecholestrogens to stimulate the growth of MCF-7 and T47D, ER + breast cancer cells [25]. As these agents can bind to the ER, albeit with weak affinity, it is likely that these estrogenic effects are mediated through classical estrogen-related proliferation pathways.
As estrogen and progesterone are known to influence terminal duct structures in the breast, we utilized ovariectomy as a benchmark with which to compare the effects of TMS. Loss of ductal structures in response to ovariectomy in mice reflects the lack of estrogens and progesterone. Estrogens are considered to play a major role in promoting the proliferation of normal breast epithelium [26]. Progesterone is required for alveolar growth, but not ductal formation [27, 28]. When compared with ovariectomy, low dose TMS given to young mice with developing breasts produced reductions both in whole duct structures and in TDSABs (Figs. 3, 4). However, when TMS was given to mice older than 5 months of age, it was found to increase TDSABs (see Fig. 5).
Our findings with respect to TMS and terminal duct structures could be important with respect to the genesis of breast cancer. Wellings and Jensen concluded that TDLUs are the principal sites of origin of dysplastic, metaplastic, hyperplastic, anaplastic, and neoplastic lesions of the human breast [29]. Parks also concluded that breast cancer arises in the TDLU [30]. Compounds such as TMS, which block TDSABs in young mice, might then be useful in the prevention of breast cancer.
When TMS was administered at high doses, we found it exerted estrogenic properties, stimulating the endometrium and formation of total duct structures as well as larger ducts in mice older than 5 months (see Fig. 5). Other stilbenes such as diethylstilbestrol (DES) and resveratrol have estrogenic activity [31–33]. DES contains hydroxyl groups in positions analogous to the 3 and 17 positions of E2. In contrast, all hydroxyl groups on TMS are converted to methyl groups, and therefore, TMS would not be expected to bind to the estrogen receptor nor act as an estrogen. Although Phase I CYP enzymes are best known for their oxidation reactions, the CYPs can catalyze a variety of reactions including O-dealkylation [34]. Dawling and coworkers have shown that CYP1B1 is capable of demethylating methoxyestrogens [35]. Thus, if demethylation were to occur in vivo, metabolites of TMS might be estrogenic. Chun and coworker studied the metabolism of TMS and demonstrated a metabolite formed through the enzymatic activity of CYP1B1 which they thought to be an O-demethylated product [19]. We are in agreement with Chun et al. that TMS might not only be an inhibitor of CYP1B1, but could potentially be a substrate for CYP1B1. If true, the O-demethylated TMS could then exert estrogen action similar to other stilbenes. Interestingly, TMS also stimulated the endometrium (see Fig. 6), but without increasing uterine size (see Supplemental Table IIa, b). This effect might also represent an estrogenic effect on a target tissue, perhaps also through local demethylation.
Stilbenes other than TMS have been studied extensively and exert some of the effects reported here. For example, resveratrol, a stilbene family member, can also inhibit the formation of depurinating estrogen-DNA adducts [36–38]. It should be noted, however, that TMS was found to be more potent than resveratrol for blocking CYP1B1 activation in vitro. In addition, high dose TMS was found to be relatively non-toxic.
Supplementary Material
Acknowledgments
This study was supported by grants from the Susan G. Koinen Breast cancer foundation KG080267 (S. E. Aiyar), and the UVa. Cancer Center through The Women’s Oncology Research Fund and the NCI Cancer Center support grant P30 CA44579 (R.J. Santen). Core support at the Eppley Institute was provided by grant P30 CA36727 from the National Cancer Institute.
Abbreviations
- IMS
2,4,3′,5′-Tetramethoxystilbene
- CYP1A1
Cytochrome P450 1A1
- Cyp1a1
Mouse cytochrome P450 1B1
- CYP1B1
Human cytochrome P450 1B1
- Cyp1b1
Mouse cytochrome P450 1B1
- E2
Estradiol
- 4-OHE2
4-Hydroxyestradiol
- 2-OHE2
2-Hydroxyestradiol
- TDLU
Terminal duct lobular units
- TDSABs
Tertiary ductal structures and alveolar buds
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10549-010-1301-5) contains supplementary material, which is available to authorized users.
Contributor Information
Taehyun Kim, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908, USA. Department of Surgery, Pusan Paik Hospital, Inje University, Busan, South Korea.
Hoyong Park, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908, USA. Department of Surgery, Kyungpook National University Hospital, Daegu, South Korea.
Wei Yue, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908, USA.
Ji-Ping Wang, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908, USA.
Kristen A. Atkins, Department of Pathology, University of Virginia, Charlottesville, VA 22908, USA
Zhenguo Zhang, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908, USA.
Eleanor G. Rogan, Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 68198-6805, USA
Ercole L. Cavalieri, Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 68198-6805, USA
Khalid S. Mohammad, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN 46202, USA
Sanghee Kim, College of Pharmacy, Natural Products Research Institute, Seoul National University, Seoul, South Korea.
Richard J. Santen, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908, USA
Sarah E. Aiyar, Email: sa3p@virginia.edu, Division of Endocrinology and Metabolism, Department of Internal Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908, USA
References
- 1.Lareef MH, Garber J, Russo PA, Russo IH, Heulings R, Russo J. The estrogen antagonist ICI-182-780 does not inhibit the transformation phenotypes induced by 17-beta-estradiol and 4-OH estradiol in human breast epithelial cells. Int J Oncol. 2005;26:423–429. [PubMed] [Google Scholar]
- 2.Jefcoate CR, Liehr JG, Santen RJ, Sutter TR, Yager JD, Yue W, Santner SJ, Tekmal R, Demers L, Pauley R, Naftolin F, Mor G, Berstein L. Tissue-specific synthesis and oxidative metabolism of estrogens. J Natl Cancer Inst Monogr. 2000;27:95–112. doi: 10.1093/oxfordjournals.jncimonographs.a024248. [DOI] [PubMed] [Google Scholar]
- 3.Liehr JG, Ricci MJ. 4-Hydroxylation of estrogens as marker of human mammary tumors. Proc Natl Acad Sci USA. 1996;93:3294–3296. doi: 10.1073/pnas.93.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liehr JG. Dual role of oestrogens as hormones and pro-carcinogens: tumour initiation by metabolic activation of oestrogens. Eur J Cancer Prev. 1997;6:3–10. doi: 10.1097/00008469-199702000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, Muti P, Rogan E, Russo J, Santen R, Sutter T. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochem Biophys Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Guengerich FP, Chun YJ, Kim D, Gillam EM, Shimada T. Cytochrome P450 1B1: a target for inhibition in anticarcino-genesis strategies. Mutat Res. 2003;523–524:173–182. doi: 10.1016/s0027-5107(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 7.McKay JA, Melvin WT, Ah-See AK, Ewen SW, Greenlee WF, Marcus CB, Burke MD, Murray GI. Expression of cytochrome P450 CYP1B1 in breast cancer. FEBS Lett. 1995;374:270–272. doi: 10.1016/0014-5793(95)01126-y. [DOI] [PubMed] [Google Scholar]
- 8.Murray GI, Taylor MC, McFadyen MC, McKay JA, Greenlee WF, Burke MD, Melvin WT. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997;57:3026–3031. [PubMed] [Google Scholar]
- 9.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–124. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Guengerich FP, Chun YJ, Kim D, Gillam EM, Shimada T. Cytochrome P450 1B1: a target for inhibition in anticarcino-genesis strategies. Mutat Res. 2003;523–524:173–182. doi: 10.1016/s0027-5107(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 11.Park H, Aiyar SE, Fan P, Wang J, Yue W, Okouneva T, Cox C, Jordan MA, Demers L, Cho H, Kim S, Song RX, Santen RJ. Effects of tetramethoxystilbene on hormone-resistant breast cancer cells: biological and biochemical mechanisms of action. Cancer Res. 2007;67:5717–5726. doi: 10.1158/0008-5472.CAN-07-0056. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Ko H, Park JE, Jung S, Lee SK, Chun YJ. Design, synthesis, and discovery of novel trans-stilbene analogues as potent and selective human cytochrome P450 1B1 inhibitors. J Med Chem. 2002;45:160–164. doi: 10.1021/jm010298j. [DOI] [PubMed] [Google Scholar]
- 13.Jeng MH, Yue W, Eischeid A, Wang JP, Santen RJ. Role of MAP kinase in the enhanced cell proliferation of long term estrogen deprived human breast cancer cells. Breast Cancer Res Treat. 2000;62:167–175. doi: 10.1023/a:1006406030612. [DOI] [PubMed] [Google Scholar]
- 14.Spink DC, Hayes CL, Young NR, Christou M, Sutter TR, Jefcoate CR, Gierthy JF. The effects of 2,3,7,8-tetra-chlorodibenzo-p-dioxin on estrogen metabolism in MCF-7 breast cancer cells: evidence for induction of a novel 17 beta-estradiol 4-hydroxylase. J Steroid Biochem Mol Biol. 1994;51:251–258. doi: 10.1016/0960-0760(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 15.Tekmal RR, Ramachandra N, Gubba S, Durgam VR, Mantione J, Toda K, Shizuta Y, Dillehay DL. Overexpression of int-5/aromatase in mammary glands of transgenic mice results in the induction of hyperplasia and nuclear abnormalities. Cancer Res. 1996;56:3180–3185. [PubMed] [Google Scholar]
- 16.Xu M, Miller MS. Determination of murine fetal Cyp1a1 and 1b1 expression by real-time fluorescence reverse transcription-polymerase chain reaction. Toxicol Appl Pharmacol. 2004;201:295–302. doi: 10.1016/j.taap.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 17.Gaikwad NW, Yang L, Muti P, Meza JL, Pruthi S, Ingle JN, Rogan EG, Cavaheri EL. The molecular etiology of breast cancer: evidence from biomarkers of risk. Int J Cancer. 2008;122:1949–1957. doi: 10.1002/ijc.23329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moral R, Wang R, Russo IH, Mailo DA, Lamartiniere CA, Russo J. The plasticizer butyl benzyl phthalate induces genomic changes in rat mammary gland after neonatal/prepubertal exposure. BMC Genomics. 2007;8:453. doi: 10.1186/1471-2164-8-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun YJ, Lee SK, Kim MY. Modulation of human cytochrome P450 1B1 expression by 2,4,3′,5′-tetramethoxystilbene. Drug Metab Dispos. 2005;33:1771–1776. doi: 10.1124/dmd.105.006502. [DOI] [PubMed] [Google Scholar]
- 20.Wu F, Safe S. Differential activation of wild-type estrogen receptor alpha and C-terminal deletion mutants by estrogens, antiestrogens and xenoestrogens in breast cancer cells. J Steroid Biochem Mol Biol. 2007;103:1–9. doi: 10.1016/j.jsbmb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Aiyar SE, Park H, Aldo PB, Mor G, Gildea JJ, Miller AL, Thompson EB, Castle JD, Kim S, Santen RJ. TMS, a chemically modified herbal derivative of Resveratrol, induces cell death by targeting Bax. Breast Cancer Res Treat. 2010;124(1):265–277. doi: 10.1007/s10549-010-0903-2. [DOI] [PubMed] [Google Scholar]
- 22.Russo J, Russo IH. Genotoxicity of steroidal estrogens. Trends Endocrinol Metah. 2004;15:211–214. doi: 10.1016/j.tem.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 23.Russo J, Russo IH. Breast development, hormones and cancer 247. Adv Exp Med Biol. 2008;630:52–56. doi: 10.1007/978-0-387-78818-0_4. [DOI] [PubMed] [Google Scholar]
- 24.Santen R, Cavalieri E, Rogan E, Russo J, Guttenplan J, Ingle J, Yue W. Estrogen mediation of breast tumor formation involves estrogen receptor-dependent, as well as independent, genotoxic effects. Ann N Y Acad Sci. 2009;1155:132–140. doi: 10.1111/j.1749-6632.2008.03685.x. [DOI] [PubMed] [Google Scholar]
- 25.Gupta M, McDougal A, Safe S. Estrogenic and antiestrogenic activities of 16alpha- and 2-hydroxy metabolites of 17beta-estradiol in MCF-7 and T47D human breast cancer cells. J Steroid Biochem Mol Biol. 1998;67:413–419. doi: 10.1016/s0960-0760(98)00135-6. [DOI] [PubMed] [Google Scholar]
- 26.Russo J, Santen RJ, Russo IH. Hormonal control of breast development. In: DeGroot LJ, Jameson JL, editors. Endocrinology. 5. W.B. Saunders; Philadelphia: 2005. pp. 3045–3055. [Google Scholar]
- 27.Conneely OM, Mulac-Jericevic B, Arnett-Mansfield R. Progesterone signaling in mammary gland development. Ernst Schering Found Symp Proc. 2007;1:45–54. [PubMed] [Google Scholar]
- 28.Topper YJ, Freeman CS. Multiple hormone interactions in the developmental biology of the mammary gland. Physiol Rev. 1980;60:1049–1106. doi: 10.1152/physrev.1980.60.4.1049. [DOI] [PubMed] [Google Scholar]
- 29.Wellings SR, Jensen HM. On the origin and progression of ductal carcinoma in the human breast. J Natl Cancer Inst. 1973;50:1111–1118. doi: 10.1093/jnci/50.5.1111. [DOI] [PubMed] [Google Scholar]
- 30.Parks AG. The micro-anatomy of the breast. Ann R Coll Surg Engl. 1959;25:235–251. [PMC free article] [PubMed] [Google Scholar]
- 31.Basly JP, Marre-Fournier F, Le Bail JC, Habrioux G, Chulia AJ. Estrogenic/antiestrogenic and scavenging properties of (E)- and (Z)-resveratrol. Life Sci. 2000;66:769–777. doi: 10.1016/s0024-3205(99)00650-5. [DOI] [PubMed] [Google Scholar]
- 32.Gehm BD, Levenson AS, Liu H, Lee EJ, Amundsen BM, Cushman M, Jordan VC, Jameson JL. Estrogenic effects of resveratrol in breast cancer cells expressing mutant and wild-type estrogen receptors: role of AF-1 and AF-2. J Steroid Biochem Mol Biol. 2004;88:223–234. doi: 10.1016/j.jsbmb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Wu F, Safe S. Differential activation of wild-type estrogen receptor alpha and C-terminal deletion mutants by estrogens, antiestrogens and xenoestrogens in breast cancer cells. J Steroid Biochem Mol Biol. 2007;103:1–9. doi: 10.1016/j.jsbmb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 35.Dawling S, Roodi N, Parl FF. Methoxyestrogens exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res. 2003;63:3127–3132. [PubMed] [Google Scholar]
- 36.Lu F, Zahid M, Wang C, Saeed M, Cavalieri EL, Rogan EG. Resveratrol prevents estrogen-DNA adduct formation and neoplastic transformation in MCF-10F cells. Cancer Prev Res (Phila) 2008;1:135–145. doi: 10.1158/1940-6207.CAPR-08-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zahid M, Gaikwad NW, Rogan EG, Cavalieri EL. Inhibition of depurinating estrogen-DNA adduct formation by natural compounds. Chem Res Toxicol. 2007;20:1947–1953. doi: 10.1021/tx700269s. [DOI] [PubMed] [Google Scholar]
- 38.Zahid M, Gaikwad NW, Ali MF, Lu F, Saeed M, Yang L, Rogan EG, Cavalieri EL. Prevention of estrogen-DNA adduct formation in MCF-10F cells by resveratrol. Free Radic Biol Med. 2008;45:136–145. doi: 10.1016/j.freeradbiomed.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.