

Abstract
While the molecular effectors of apoptotic cell death have been largely annotated over the past 30 years, leading to a strong biologic understanding of this process and its importance in cell biology, cell death through necrosis has only recently been accepted as a similarly regulated process with definable molecular effectors. The mitochondria are important and central mediators of both apoptosis and regulated necrosis. In apoptosis the Bcl-2 family members Bax and Bak undergo oligomerization in the outer mitochondrial membrane resulting in the release of apoptosis inducing substrates and the activation of caspases and nucleases. In contrast, during necrosis the mitochondria become dysfunctional and maladaptive in conjunction with reactive oxygen species production and the loss of ATP production, in part through opening of the mitochondrial permeability transition pore. While regulated necrosis is caspase independent, recent evidence has shown that it still requires the apoptotic regulators Bax and Bak, which can regulate the permeability characteristics of the outer mitochondrial membrane in their non-oligomerized state. Here we review the non-apoptotic side of Bcl-2 family, specifically the role of Bax and Bak in regulated necrotic cell death. We will also discuss how these Bcl-2 family member effectors could be part of a larger integrated network that ultimately decides the fate of a given cell somewhere within a molecular continuum between apoptosis and regulated necrosis.
Keywords: Apoptosis, necrosis, Bcl-2, calcium, mitochondria
HISTORICAL CONTEXT OF CELL DEATH
Cell death has been a recognized physiological process for over 150 years,1, 2 although it was not until the 1970s when the terms apoptosis and necrosis were adopted based on distinct morphological features underlying discrete forms of cell death.3 The first molecular breakthrough in defining the genes involved in cell death came in 1983 with the identification of cell death 1 and 2 (CED-1,-2), as genes involved in cell death in Caenorhabditis elegans (C. elegans).4 In hindsight, C. elegans are a perfect model for defining the physiological and genetic underpinnings of cell death given the ability to account for all the cells in this organism during development, with 131 of 1090 cells being destined to undergo programmed cell death.4, 5 The first identified mammalian cell death gene was B-cell leukemia/lymphoma 2 (Bcl-2), which was cloned from a translocation hot spot in follicular lymphoma from hematopoietic cell lines.6 Bcl-2 was later shown to be an anti-apoptotic protein with a conserved ortholog in C. elegans, CED-9 (ref 7). Remarkably, expression of mammalian Bcl-2 in C. elegans protected cells against apoptosis.7 The discovery of Bcl-2 led to the identification of many other key cell death regulators, such as Bcl-2 like 1 (Bcl-x), Bcl-2 associated protein x (Bax) and Bcl-2 homologues antagonist/killer (Bak), among several others.8–10
With respect to defining the molecular effectors of apoptosis, the caspases were discovered in the early to mid-1990s as cell death effector proteins. Caspases are also conserved in C. elegans where CED-3 functions as an intracellular protease to degrade key proteins to facilitate immediate cell death.11, 12 Finally, also in the mid-1990s the mitochondrion was shown to underlie the apoptotic pathway by harboring and subsequently releasing apoptogenic factors such as cytochrome c (cyt-c), which then initiates caspase activation through formation of the apoptosome.13 The mitochondrion was also suspected as being involved in apoptosis due to the localization of various Bcl-2 family members to this organelle.14–16 The evolving data subsequently connected the Bcl-2 family of proteins to the permeability and rupture of mitochondria, followed by the activation of caspases in the initiation of apoptosis, thus providing a model whereby this energy producing organelle was capable of also rapidly killing a cell through a programmed process.
Apoptotic cell death appears to be specialized for the programmed clearing of cells during development, ongoing positive and negative selection of T and B cells in the immune system, culling of cells with inappropriate cell cycle activity (cancer) and for general cellular turnover that underlies tissue homeostasis in select organs. In contrast, cells also die in adult vertebrate organisms in response to ischemic injury largely through a morphologic type of cell death known as necrosis. Ischemia, such as after myocardial infarction injury, results in a wide-spread loss of adult cardiomyocytes through a necrotic process that is driven by the lack of oxygen, calcium overload, and the generation of aberrant reactive oxygen species (ROS).17 Calcium overload of the mitochondria during ischemic injury is perhaps the most central and damaging effect, which leads to swelling and rupture of this organelle 18 through a process that would become known as mitochondrial permeability transition pore (MPTP) formation and opening.19–21 In 1980, this increase of mitochondrial calcium during ischemic injury was already proposed to be causative in mitochondrial dysfunction and cell death, as treatment with ruthenium red, a pharmacological inhibitor of mitochondrial calcium uptake, preserved mitochondrial function and prevented tissue damage.22 This was the first evidence showing that mitochondrial preservation protected tissue after ischemic injury (necrotic cell death), suggesting that necrosis can be a regulated process and not simply a default pathway of cellular killing.
The first inhibitor of mitochondria permeability transition was identified in 1987–1988 when cyclosporin A (CsA), an immunosuppressant agent, was shown to desensitize calcium-induced mitochondrial swelling without affecting calcium uptake.23, 24 In addition, CsA was shown to protect against calcium- and oxidative stress-induced cell death in hepatocytes,25 providing yet another piece of evidence that necrotic cell death can be regulated. The target of CsA was determined to be cyclophilin D (CypD), a peptidyl-prolyl isomerase that resides in the matrix of the mitochondria where it regulates opening of the MPTP. 26 In the late 1990s a number of investigators reported data whereby the apoptosis regulating Bcl-2 family members could interact with presumed components of the MPTP and also regulate the opening of this pore, suggesting an intersection between apoptotic and necrotic cell death (more on this later).27–30 More recently, additional evidence has emerged whereby the Bcl-2 family members Bax and Bak are heavily involved in mediating multiple forms of regulated necrosis.31
MITOCHONDRIAL EFFECTS OF APOPTOSIS VS NECROSIS
An apoptotic cell is morphologically defined by chromatin condensation, cell shrinkage, membrane blebbing, and the removal of packaged cellular compartments by phagocytes in a non-inflammatory process. Necrotic cell death is morphologically defined by cell and organelle swelling, early plasma membrane rupture, and the spilling of cellular material into the tissue with subsequent inflammation.2, 32 Due to this, necrosis is considered a more harmful way for a cell to die compared to apoptosis. While mitochondria and a shared set of molecular effectors (Bax and Bak) are at the center of both forms of cell death, activation of caspases appears to be a uniquely apoptotic process.12 Caspase activation and function during apoptosis is ATP dependent and typically requires some degree of mitochondrial function, while necrosis is an ATP independent process where it progresses in conjunction with a complete loss of mitochondrial function.33 Indeed, a hallmark of necrotic cell death is mitochondrial dysfunction itself, such that if mitochondria structure-function are preserved during a necrotic insult, the cells affected typically no longer die.34 This observation further supports the hypothesis that necrosis is not purely an accidental form of cell death and that it can be regulated and prevented.
Mitochondrial dysfunction occurs during necrosis typically due to prolonged opening of the MPTP, which leads to inner membrane depolarization and loss of ATP production, swelling of the organelle and the production of ROS.35 However, MPTP opening and mitochondrial swelling and rupture also cause the release of apoptogenic factors such as cyt c, although in the absence of sufficient ATP caspase activation and the formation of the apoptosome is blocked so that the cell still perishes through a necrotic process.33, 35 While the outer membrane of the mitochondria is altered and engaged during MPTP opening, the inner mitochondrial membrane opening and loss of membrane potential is the first regulated and driving aspect of MPTP-mediated necrotic cell death. However, if only a small percentage of mitochondria undergo cyt c release with MPTP opening the remaining functioning mitochondria might still provide enough high energy phosphate for apoptotic cell death to ensue.
During apoptosis, the regulated release of cyt c from the mitochondria is due to mitochondrial outer membrane permeability (MOMP).36 MOMP is an event initiated directly by the Bcl-2 family members Bax and Bak.36 When Bax/Bak become activated by apoptotic signaling cues they form expansive hetero-homo oligomers within the outer membrane of the mitochondria to directly generate large pores leading to the release of cyt c.36 Importantly, during MOMP the mitochondria are still functional and produce ATP as the inner mitochondrial membrane is typically intact and able to maintain respiration for prolonged periods of time.36 Hence, while the mitochondria are intimately involved in both apoptotic and necrotic cell death, MOMP is a specific event for apoptosis while inner membrane opening of the MPTP is uniquely geared toward necrosis. However, as we will discuss later, Bax and Bak appear to integrate both forms of cell death at the level of the mitochondria by not only affecting MOMP, but also the ability of MPTP opening to culminate in the swelling and rupture of this organelle.37–39
THE BCL-2 FAMILY
Bcl-2 family members are characterized by containing at least one Bcl-2 homology (BH) domain (Figure 1).40 There are a total of four different BH domains (BH1-BH4), each of which can impart functional effects to Bcl-2 family members in impacting survival or cell death.40 The other characteristic of the Bcl-2 family is that many members contain a trans-membrane (TM) domain to allow their insertion into various organelle membranes, mainly the mitochondria but also the endoplasmic reticulum, lysosomes and even the nuclear envelop (Figure 1).40–42 There are a total of 25 human Bcl-2 family members that are divided into two main groups, the anti-apoptotic and the pro-apoptotic members (Figure 1).43 The pro-apoptotic Bcl-2 family members are further divided into two additional groups. The first is the BH3-only subfamily that functions to integrate signals from various pro-death stimuli and signaling pathways that either subsequently alter the activity of Bax and Bak or directly bind to and activate Bax and Bak.44 The second is the multidomain proapoptotic effectors Bax and Bak that contain BH1-3 domains along with a TM domain.43 The Bcl-2 anti-apoptotic family members, such as Bcl-2, Bcl-xl, MCL-1, and others, contain four BH domains and all but one contains a TM domain.43 The BH4 domain, which is largely exclusive to this subset of family members, is indispensable for their anti-apoptotic activities.45–47 The interplay between all the Bcl-2 family members controls the equilibrium of apoptotic versus necrotic cell death in response to various stimuli.
Figure 1. The Bcl-2 family members and their domains.

Schematic showing the 25 human Bcl-2 family members separated by their anti- vs pro- apoptotic activities and the domains they contain. Each family member is characterized by containing at least one Bcl-2 homology (BH) domain. Over half of the family members contain a transmembrane (TM) domain which allows for the insertion into various organelles. Unique to Bcl-rambo is an additional domain (BHNo). Bcl2L12 contains a unique domain with 1 proline rich (PR) region and 6 PxxP motifs.
Generally, the prosurvival Bcl-2 subfamily members inhibit the activation of Bax and Bak, while the BH3-only subfamily members activate the Bax subfamily members. The Bcl-2 subfamily members inhibit Bax and Bak by either direct binding that results in their sequestration, or by binding to and sequestering specific activators such as the BH3-only effectors.48, 49 As an example, Bid binds Bax to activate it but at the same time can bind and nullify Bcl-2 resulting in even greater Bax activity.50, 51 A simplified model for the Bcl-2 proteins is that of a rheostat, with all of the prosurvival Bcl-2 subfamily members affecting the cell death equilibrium by sequestering Bax/Bak activity versus the BH3-only subfamily members that would otherwise free Bax/Bak to initiate MOMP and cell death.52
REGULATED NECROSIS
Historically, necrosis has been viewed as an uncontrollable form of cell death with default status, and there are certainly examples of stimuli that result in uncontrolled and unregulated necrotic cell death, such as with freeze-thaw injury. However, more recent studies have shown that under certain cellular contexts necrosis can be a highly regulated form of cell death in adult vertebrate organisms. Regulated necrosis is simply defined as caspase independent cell death that has all the morphological hallmarks of classical necrosis but that can be inhibited or accelerated by affecting one or more key molecular components. As stated previously, the first identified molecular effector of regulated necrosis was CypD, which was shown to control the opening probability of the MPTP, such that inhibitors of this opening preserved cells after ischemic injury. 53 In addition to ischemic injuries, inhibition of the MPTP and regulated necrosis has also been shown to mitigate muscular dystrophy, neurological degenerative disorders, and liver toxicity models.54–58
Another type of regulated necrosis is one in which cell death is induced by caspase inhibition in conjunction with apoptotic death ligand stimulation, which is referred to as necroptosis. This type of necrosis is mediated by apoptotic death receptors and the receptor interacting protein kinases 1 and 3 (RIP1 and RIP3).59 This form of necrosis has been implicated in ischemic injuries, pancreatitis, and viral infections. An additional form of regulated necrosis involves the hyper-activation of Poly (ADP-ribose) polymerase (PARP) induced by DNA damage, which was first suggested as a mechanism whereby cancer cells are killed with DNA alkylating agents.60
Taken together, these three independently defined forms of regulated necrosis provide strong evidence that necrosis can be a controlled form of cell death that is not purely accidental as once thought. Additionally, present studies have showed that these three independent forms of necrosis may be more integrated than first thought, as cells lacking Bax and Bak are resistant to all three forms of necrosis.38, 39
MPTP-DEPENDENT NECROSIS
The MPTP forms within the inner membrane of the mitochondrion where it permits diffusion of molecules up to 1.5 kDa. 53 MPTP opening, or something similar to it, has been shown to occur in yeast, plants, fish, amphibians, and mammals.61 The necrosis that is most prevalent in ischemic injuries is dependent on the opening of the MPTP and is due to calcium overload in the cell along with ROS production. 53 MPTP opening with ischemic injury results in the diminution of cellular energy production and loss of ATP levels that secondarily prevents caspase activation and induction of apoptosis despite mitochondrial rupture and the accumulation of apoptogenic substrates like cyt c.33
The classical model of the MPTP was that of one contiguous pore spanning the outer and inner mitochondrial membranes, consisting of the voltage-dependent anion channel (VDAC, outer membrane) and adenine nucleotide translocator (ANT, inner membrane), regulated by CypD within the mitochondrial matrix.31 Unfortunately, this model has not held up to genetic scrutiny as deletion of the genes encoding the ANT and VDAC isoforms in mice or mammalian cells showed that neither were essential components of the pore, mitochondrial swelling, or necrotic cell death.62, 63 However, deletion of the gene encoding CypD (Ppif) did reveal a role for this protein in regulating the opening of the MPTP, making it the first genetically defined component of this regulated necrotic process.54, 56 More recently, evidence has emerged suggesting that the mitochondrial F1F0 ATP synthase serves as the core component of the MPTP within the inner membrane, such that purified components of the F1F0 ATP synthase reconstituted into lipid bilayers could recapitulate pore activity similar to that of the MPTP (Figure 2).64–67
Figure 2. MPTP-dependent necrotic pathway.

When a cell receives a stress that leads to increased levels of intracellular calcium, the mitochondrial calcium uniporter (MCU) takes up the calcium into the matrix of the mitochondria where it can trigger MPTP opening through CypD. The MPTP is thought to be composed of the F1F0 ATP synthase regulated by ANT and the mitochondrial phosphate carrier (PiC). Upon prolonged opening of the MPTP there is an osmotic alteration and mitochondrial swelling and dysfunction occurs with loss of ATP production and ROS generation. MPTP-dependent mitochondrial dysfunction requires the presence of Bax or Bak on the outer mitochondrial membrane. Therefore, proteins that affect the content of Bax/Bak on the outer mitochondrial membrane, such as the pro-survival the Bcl-2 family members, can secondarily affect MPTP-dependent mitochondrial dysfunction. Abbreviations: IMM, inner mitochondrial membrane; IMS, intra-mitochondrial membrane space; OMM, outer mitochondrial membrane; ROS, reactive oxygen species; ANT, adenine nucleotide translocator.
While the components that constitute the MPTP are still debated today, a number of older publications in the literature from the late 1990s and early 2000s showed that Bcl-2 family members could directly interact with either VDAC or ANT,68–72 implying, at that time, that apoptotic regulators might affect necrotic cell death through the MPTP. While we now believe that ANT and VDAC are not direct components of the MPTP, these previous observations might still hold biologically relevant insights that indeed suggest an intersection between apoptosis and necrosis through Bcl-2 family members and the MPTP. For example, Tsujimoto and colleagues demonstrated that Bcl-2 over-expression could prevent MPTP dependent mitochondrial swelling,73 and Kroemer and colleagues showed that Bax was required to permit MPTP formation and the permeabilization of the mitochondrial membrane.71 Similarly, Bax over-expression in Jurkat cells resulted in cell death that was inhibited by CsA but not by caspase inhibitors.27 Finally, Bax can induce mitochondrial swelling in the presence of calcium.74 One opposing study concluding that Bax was not required for MPTP opening showed that mitochondria isolated from Bax−/− HTC116 cell lines were as susceptible to CypD dependent mitochondrial swelling as wild type mitochondria.75 Furthermore, mitochondria isolated from Bax and Bak1 (encodes Bak protein) null baby mouse kidney (BMK) cells were also shown to undergo mitochondrial swelling in response to calcium.75 Importantly, the caveat of this result is that the swelling shown in the null BMK cells was not inhibited by CsA; therefore, it can be argued that this was not MPTP-dependent mitochondrial swelling.75 However, the overall hypothesis from the older literature is that the Bcl-2 family member of proteins, including Bax and Bak, appear to regulate the MPTP.
Recently, our lab and others have shown that isolated mitochondria from mouse embryonic fibroblasts (MEFs), hepatocytes, and cardiomyocytes lacking Bax and Bak (DKO, double knock-outs) do not undergo MPTP-dependent swelling and that they have an increased calcium holding capacity compared to wild type mitochondria.39, 76 However, in the absence of Bax and Bak the inner membrane portion of the MPTP was still functional and able to induce opening that resulted in loss of membrane potential.39 It was simply that in the absence of Bax and Bak the outer mitochondrial membrane remained intact, thus allowing the mitochondria to persist without rupture. Importantly, this requirement of Bax and Bak to permit outer membrane rupture was independent of their activation and oligomerization that normally underlies MOMP and apoptosis, as oligomeric dead mutant versions of Bax restored necrotic killing in the DKO MEFs.39 Indeed, the inactive/passive state of Bax and Bak permitted necrosis by simply changing the physical properties of the outer mitochondrial membrane and making it more permeable and amenable to rupturing following inner mitochondrial membrane opening of the MPTP.39, 77 Hence, a small increase in permeability created by inactive/passive Bax/Bak as they reside in the outer mitochondrial membrane was required for mitochondrial rupture post MPTP opening to ultimately mediate necrosis (Figure 2).39 Other notable findings were that the activity of Bax and Bak in the outer mitochondrial membrane were not functionally coupled to the inner membrane, suggesting that the MPTP is not a coordinated activity that directly spans the outer and inner mitochondrial membranes simultaneously.39 Finally, Whelan et al showed that Bax may also function in necrotic cell death by supporting or driving fusion of mitochondria.76
NECROPTOSIS
Necroptosis was originally identified as a type of cell death that occurs when one of the 7 different apoptotic death receptors (extrinsic pathway) was stimulated with ligand and at the same time caspases were inhibited with pharmacologic agents. For example, tumor necrosis factor-α (TNFα)-induced cell death in L929 cells was dramatically enhanced when caspase inhibitors were used, producing a morphological type of necrotic cell death (Figure 3).78 While necroptotic cell death might appear to be fairly arbitrary and of unknown physiologic relevance, subsequent work in gene-targeted mice lacking select caspase encoding genes or other genetic components of the extrinsic apoptotic death receptor have suggested a role for this type of cell death in vivo.79–81 The RIP1 and RIP3 kinases are part of a complex that is initiated in conjunction with death receptor stimulation where they then facilitate the necroptotic killing of cells. Indeed, cells lacking RIP3 protein are protected against cell death after viral infection that might normally cause necroptosis, while gene-targeted mice lacking RIP3 protein show reduced survival compared to wild type mice after viral infection,82 but are protected from cerulein-induced necrosis of the pancreas.83 RIP-mediated necrosis has also been implicated in killing cells during ischemic injuries across multiple tissues, as the use of a RIP1 inhibitor, such as necrostatin-1,84 is protective suggesting that the this kinase might also be involved in other forms of regulated necrosis.85–87
Figure 3. Necroptotic pathway.

The combinatorial treatment with an apoptotic death receptor ligand and a caspase inhibitor leads to necroptosis with RIP1 activation. Without the caspase inhibitor present, caspase 8 would normally cleave and inactivate RIP1. When RIP1 is left unchecked in the presence of a caspase inhibitor, it complexes with RIP3 and together they lead to the phosphorylation and activation of MLKL. MLKL is a required protein for necroptosis.
During necroptosis death receptor stimulation results in caspase 8 activation where it then cleaves and inactivates RIP1 (Figure 3).88 It is only in the context of death receptor stimulation and caspase inactivation that RIP1 has pro-death activities in vivo. Indeed, when caspase 8 is inhibited or deleted in mice, this cleavage event does not occur and RIP1 is able to interact with RIP3 to initiate cell death, which is prevented in cells lacking RIP3 protein.82, 89, 90 These studies also determined that necrostatin-1 blocks necroptosis by inhibiting the interaction between RIP1 and RIP3.82, 90 Finally, directly downstream of the RIPs is mixed lineage kinase like (MLKL), a pseudokinase that when phosphorylated directly induces cellular necrosis, and while the mechanism of killing remains under investigation, this protein has recently been shown to directly generate pores in membranes that could directly mediate cell death.91 Indeed, cells and mice lacking MLKL are resistant to necroptosis.92, 93 MLKL has also been shown to translocate to the plasma membrane where it induces calcium entry through interaction with transient receptor potential melastatin 7 during a necroptotic stimuli, suggesting another potential mechanism for cell death through enhanced calcium entry. 94
Given that necroptosis was originally thought to be a highly unique form of cell death, in conjunction with a more recent report demonstrating that mitochondria are supposedly dispensable for necroptotic killing,95 it came as a surprise when 2 groups independently showed that Bax and Bak are required for necroptotic cell death.38, 96 However, it is still not clear why Bax and Bak deficient cells are resistant to necroptosis and to what extent mitochondria are involved as a mechanism for necroptotic killing (see below).
The MPTP has been implicated in necroptosis as Ppif (CypD encoding gene) null cells are partially resistant to the treatment of TNFα with caspase inhibitors.83 In contrast, necroptosis has been shown to be CypD independent in vivo as the lethality normally observed in Casp8 null mice that is completely rescued with Ripk3 deletion (encodes the RIP3 gene), is not rescued with Ppif deletion.95 This suggests that the type of cell death that occurs in Casp8 null mice that leads to their embryonic lethality does not utilize the MPTP-type of regulated necrosis. Moreover, Ppif null mice subjected to kidney ischemia showed increased protection when also given necrostatins, suggesting that these 2 types of cell death are separate pathways.97 However, in the heart necrostatins provided no additional protection in Ppif null animals during cardiac ischemia-reperfusion (I/R)-injury, suggesting that they could be within the same linear pathway.98 One point that must be remembered here is that cells lacking CypD protein are still susceptible to apoptosis, and that in vivo, unlike in vitro with pan caspase inhibitors, additional caspases are still active in Casp8 null mice and hence could mediate an apoptotic pathway to effectively kill Ppif null cells.54, 56
PARP-DEPENDENT NECROSIS
PARP-mediated necrosis was first discovered through the observation that DNA alkylating agents induce very efficient necrotic cell death in cancer cells. The over activation of PARP by DNA damage results in the depletion of the NAD+ pool and the inhibition of glycolysis, which more specifically sensitizes cancer cells to death given their almost exclusive reliance on glycolysis for ATP production (Figure 4).60 However, cells relying on oxidative phosphorylation and not glycolysis were resistant to NAD+ depletion and death with DNA alkylating agents and PARP over-activation,60 suggesting that maintenance of mitochondrial function could be a protective variable to this type of necrosis. Another way NAD+ depletion affects cell viability is by inhibition of the sirtuin family. Sirtuins are NAD+ dependent deacetylases that have been shown to play a role in cell viability and depending on the injury and cell type, sirtuin inhibition can be either maladaptive or protective.99 As discussed earlier, Bax/Bak DKO cells are also resistant to DNA alkylating agent-induced necrotic cell death. An attractive hypothesis is that such resistance is due to a greater level of mitochondrial protection that is observed in the absence of Bax and Bak, which preserves the ATP status of the cell more effectively with PARP over activation.39, 100, 101 Notably, when Bax/Bak DKO cells were forced to utilize glycolysis for energy they became susceptible to DNA alkylation-induced necrosis.100 In addition to showing that Bax is required for DNA alkylation-induced necrosis by MNNG, Bcl-2 overexpression protected against this form of necrosis.101 PARP mediated necrosis has also been implicated in ischemic injuries in various tissues, as PARP inhibition or genetic deletion is protective.102–104
Figure 4. PARP-dependent necrotic pathway.
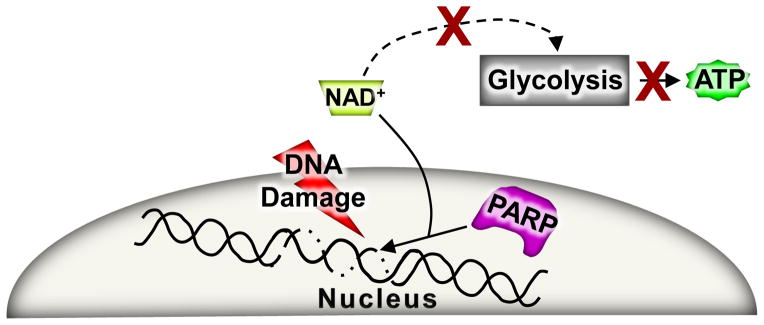
Upon an extensive amount of DNA damage PARP becomes over activated as a repair response, but in the process NAD+ levels can plummet. For a glycolytic-dependent cell this type of NAD+ depletion stress leads to a complete inhibition of ATP production and eventual necrotic cell death.
A UNIFIED MODEL OF REGULATED NECROSIS
Although the three types of necrosis discussed above were identified independently, it is possible that each represents part of an inter-related process. One possible integration point is the mitochondria where each form of cell death appears to require the collapse of the mitochondria at some level. This is somewhat the opposite of apoptosis as mitochondrial viability is required for the maintenance of ATP generation so that cell death can occur in an orderly process to package cellular contents for removal without tissue inflammation. However, during regulated necrosis the mitochondria swell, lose their ability to generate ATP and subsequently rupture. As discussed throughout this review Bax/Bak underlie both processes as DKO cells are resistant to necrotic and apoptotic cell death, and hence are likely a central integration point for potentially all forms of regulated cell death (Figure 5).
Figure 5. The unified model of regulated necrosis.

A schematic in which all three forms of regulated necrotic cell death are depicted together. Since energy depletion is a common feature for all forms of necrotic cell death, mitochondria dysfunction through MPTP opening and Bax and Bak are placed at the center of the pathway. Prior to mitochondrial dysfunction the extrinsic arm of the necrotic pathway can be engaged and it includes the RIPs and MLKL stimulated by death receptor activation with caspase inhibition. The intrinsic arm of the necrotic pathway includes stressors that increase intracellular calcium and/or induce DNA damage. Increases of calcium lead to mitochondrial dysfunction through the MPTP, while DNA damage leads to the over activation of PARP. Both of these later mechanisms result in dysfunctional mitochondria and a further exacerbation of the injury events and culmination in necrotic cell death. Due to the rapid decline of ATP generation and the rupture of mitochondria the cell loses the ability to regulate plasma membrane integrity and necrosis fully ensues.
Another consideration for a unified model of regulated necrosis is that separate inhibition of PARP mediated necrosis, necroptosis, or MPTP-dependent necrosis appears to protect tissues from cell death following ischemic injuries in vivo. Increased ROS production and calcium overload underlie ischemic injury-induced cell death in vivo, each of which impacts the mitochondria and causes MPTP opening. Indeed, the RIP1 inhibitor necrostatin-1 can delay MPTP opening in rat cardiomyocytes exposed to oxidative stress, suggesting a role for RIPs with necrotic inducers other than just death ligands and caspase inhibitors.105 Also, as stated earlier, there is no additive effect of necrostatin-1 treatment in Ppif null mice during heart I/R injury.86 In addition, Ppif null MEFs are partially resistant to RIP-mediated necrosis.90 These results suggest that RIP-mediated necrosis can function upstream of MPTP-mediated necrosis. However, other data whereby mitochondria appear dispensable for necroptosis95 might suggest that 2 distinct pathways underlie regulated necrosis, such as an extrinsic necrosis induced by death ligands and caspase inhibitors versus an intrinsic necrosis induced by calcium and/or ROS overload that induces MPTP opening.
ROS can also lead to DNA damage that can trigger PARP activation.106 However, for PARP activation to be deleterious and induce a form or regulated necrosis, mitochondrial ATP production has to cease.60 If mitochondrial dysfunction does not occur, the cell is able to maintain energy demands associated with PARP activation to facilitate DNA repair. This places PARP-dependent necrosis downstream of the MPTP-dependent mitochondrial dysfunction. Moreover, maintenance of mitochondrial integrity and ATP production associated with deletion of Bax/Bak protein reduces or inhibits regulated necrosis due to each of the inducers discussed above, again pointing to the convergence of Bcl-2 family members in supporting regulated necrosis (Figure 5).
Regulated necrosis and apoptosis were previously considered as 2 separate forms of cell death controlled by unique pathways although recent work has shown that the Bcl-2 family serves as a functional convergence point between both. The defining moment between apoptosis and necrosis appears to be at the level of the mitochondria and if MOMP occurs to drive apoptosis or if only the MPTP opens to drive a form of regulated necrosis, although both events clearly require Bax and Bak and are influenced by the activity of other Bcl-2 family members. In addition to mitochondrial permeability, Bcl-2 family members also regulate endoplasmic reticulum calcium levels, which can further impact the mitochondria and MPTP formation and necrosis through direct communication between these 2 organelles and the ultimate content of calcium within the mitochondria. Finally, Bax can also regulate lysosomal permeability adding even further levels of regulation of the necrotic pathway by the Bcl-2 family.42
FUTURE DIRECTIONS
One important question that needs to be addressed in moving forward is if regulated necrosis underlies cellular attrition outside of injury events, such as in response to development or physiologic stimuli. While this clearly appears to be the case for necroptosis, which mediates select forms of cell death in vivo, it is less clear if the MPTP-mediated mechanism of cellular necrosis might also occur outside of ischemic injury in vivo. Another issue is defining exactly how necroptosis results in cell death, as the ultimate effectors that bring about cell death downstream of death receptor activation with caspase inhibition remains mechanistically illusive. While there are data that mitochondria are completely dispensable for necroptotic cell death,95 we and others have observed a requisite role for the MPTP and Bax/Bak as potential downstream effectors of necroptosis. Finally, it will be important to continue to discover other mechanistic effectors of regulated necrotic cell killing, as this form of cell death appears to be the primary way that cells die in adult vertebrate organisms after an injury event or ischemic damage, as well as in response to long-term degenerative disorders, which together underlies most forms of disease with aging in adult humans.
Supplementary Material
Acknowledgments
Sources of funding
This work was supported by grants from the NIH (to J.D.M). J.D.M was also supported by the Howard Hughes Medical Institute.
Non-standard abbreviations
- ANT
adenine nucleotide translocator
- Bak
Bcl-2 homologues antagonist/killer
- Bax
Bcl-2 associated protein x
- Bcl-2
B-cell leukemia/lymphoma 2
- BH
Bcl-2 homology
- CED
cell death
- cyt-c
cytochrome c
- CsA
cyclosporin A
- CypD
cyclophilin D
- DKO
Bax/Bak1 double knock-out
- MEFs
mouse embryonic fibroblasts
- MLKL
mixed lineage kinase like
- MOMP
mitochondrial outer membrane permeability
- MPTP
mitochondrial permeability transition pore
- PARP
Poly (ADP-ribose) polymerase
- RIP
receptor interacting protein kinase
- ROS
reactive oxygen species
- TNFα
tumor necrosis factor-α
- VDAC
voltage-dependent anion channel
Footnotes
Disclosures:
None.
Conflict of interest of competing financial interest:
None
References
- 1.Clarke PG, Clarke S. Nineteenth century research on naturally occurring cell death and related phenomena. Anat Embryol (Berl) 1996;193:81–99. doi: 10.1007/BF00214700. [DOI] [PubMed] [Google Scholar]
- 2.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3. [PMC free article] [PubMed] [Google Scholar]
- 3.Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hedgecock EM, Sulston JE, Thomson JN. Mutations affecting programmed cell deaths in the nematode caenorhabditis elegans. Science. 1983;220:1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- 5.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 6.Tsujimoto Y, Croce CM. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci USA. 1986;83:5214–5218. doi: 10.1073/pnas.83.14.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaux DL, Weissman IL, Kim SK. Prevention of programmed cell death in caenorhabditis elegans by human bcl-2. Science. 1992;258:1955–1957. doi: 10.1126/science.1470921. [DOI] [PubMed] [Google Scholar]
- 8.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 9.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 10.Farrow SN, White JH, Martinou I, Raven T, Pun KT, Grinham CJ, Martinou JC, Brown R. Cloning of a bcl-2 homologue by interaction with adenovirus e1b 19k. Nature. 1995;374:731–733. doi: 10.1038/374731a0. [DOI] [PubMed] [Google Scholar]
- 11.Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction of apoptosis in fibroblasts by il-1 beta-converting enzyme, a mammalian homolog of the c. Elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 12.Cohen G. Caspases: The executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: Requirement for datp and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 14.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 15.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl xL and Bcl-2, displaces bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 16.Krajewski S, Krajewska M, Reed JC. Immunohistochemical analysis of in vivo patterns of bak expression, a proapoptotic member of the bcl-2 protein family. Cancer Res. 1996;56:2849–2855. [PubMed] [Google Scholar]
- 17.Kristián T, Siesjö BK. Calcium in ischemic cell death. Stroke. 1998;29:705–718. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- 18.Henry PD, Shuchleib R, Davis J, Weiss ES, Sobel BE. Myocardial contracture and accumulation of mitochondrial calcium in ischemic rabbit heart. Am J Physiol. 1977;233:H677–H684. doi: 10.1152/ajpheart.1977.233.6.H677. [DOI] [PubMed] [Google Scholar]
- 19.Raaflaub J. Swelling of isolated mitochondria of the liver and their susceptibility to physicochemical influences. Helv Physiol Pharmacol Acta. 1953;11:142–156. [PubMed] [Google Scholar]
- 20.Hunter FE, Jr, Ford L. Inactivation of oxidative and phosphorylative systems in mitochondria by preincubation with phosphate and other ions. J Biol Chem. 1955;216:357–369. [PubMed] [Google Scholar]
- 21.Tapley DF. The effect of thyroxine and other substances on the swelling of isolated rat liver mitochondria. J Biol Chem. 1956;222:325–339. [PubMed] [Google Scholar]
- 22.Peng C-F, Kane JJ, Straub KD, Murphy ML. Improvement of mitochondrial energy production in ischemic myocardium by in vivo infusion of ruthenium red. J Cardiovasc Pharmacol. 1980;2:45–54. doi: 10.1097/00005344-198001000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- 24.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 25.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin a is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–7830. [PubMed] [Google Scholar]
- 26.Woodfield KY, Price NT, Halestrap AP. Cdna cloning of rat mitochondrial cyclophilin. Biochim Biophys Acta. 1997;1351:27–30. doi: 10.1016/s0167-4781(97)00017-1. [DOI] [PubMed] [Google Scholar]
- 27.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 29.Pastorino JG, Chen ST, Tafani M, Snyder JW, Farber JL. The overexpression of bax produces cell death upon induction of the mitochondrial permeability transition. J Biol Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- 30.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 31.Karch J, Molkentin JD. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci USA. 2014;111:10396–10397. doi: 10.1073/pnas.1410104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. BBA Bioenergetics. 2006;1757:1371–1387. doi: 10.1016/j.bbabio.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 33.Skulachev V. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006;11:473–485. doi: 10.1007/s10495-006-5881-9. [DOI] [PubMed] [Google Scholar]
- 34.Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin a. J Mol Cell Cardiol. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- 35.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 36.Chipuk J, Bouchier-Hayes L, Green D. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 37.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic bax and bak: A requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, Balachandran S, Madesh M. Requirement of fadd, nemo, and bax/bak for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol Cell Biol. 2011;31:3745–3758. doi: 10.1128/MCB.05303-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EHY, Robbins J, Kinnally KW, Molkentin JD. Bax and bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife. 2013:2. doi: 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cory S, Adams JM. The bcl2 family: Regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 41.Szegezdi E, MacDonald DC, Chonghaile TN, Gupta S, Samali A. Bcl-2 family on guard at the er. AM J Physiol-Cell PH. 2009;296:C941–C953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 42.Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. AM J Physiol Gastr L. 2006;290:G1339–G1346. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Youle RJ, Strasser A. The bcl-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 44.Dewson G, Kluck RM. Mechanisms by which bak and bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009;122:2801–2808. doi: 10.1242/jcs.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugioka R, Shimizu S, Funatsu T, Tamagawa H, Sawa Y, Kawakami T, Tsujimoto Y. Bh4-domain peptide from bcl-xl exerts anti-apoptotic activity in vivo. Oncogene. 2003;22:8432–8440. doi: 10.1038/sj.onc.1207180. [DOI] [PubMed] [Google Scholar]
- 46.Huang D, Adams JM, Cory S. The conserved n-terminal bh4 domain of bcl-2 homologues is essential for inhibition of apoptosis and interaction with ced-4. The EMBO J. 1998;17:1029–1039. doi: 10.1093/emboj/17.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimizu S, Konishi A, Kodama T, Tsujimoto Y. Bh4 domain of antiapoptotic bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci USA. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for bcl-xl and bcl-2, displaces bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 49.Zha H, Aime-Sempe C, Sato T, Reed JC. Proapoptotic protein bax heterodimerizes with bcl-2 and homodimerizes with bax via a novel domain (bh3) distinct from bh1 and bh2. J Biol Chem. 1996;271:7440–7444. doi: 10.1074/jbc.271.13.7440. [DOI] [PubMed] [Google Scholar]
- 50.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruffolo SC, Shore GC. Bcl-2 selectively interacts with the bid-induced open conformer of bak, inhibiting bak auto-oligomerization. J Biol Chem. 2003;278:25039–25045. doi: 10.1074/jbc.M302930200. [DOI] [PubMed] [Google Scholar]
- 52.Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. Bcl-2/bax: A rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol. 1993;4:327–332. [PubMed] [Google Scholar]
- 53.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 54.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin d reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 55.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin d is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin d-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 57.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramachandran A, Lebofsky M, Baines CP, Lemasters JJ, Jaeschke H. Cyclophilin d deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic Res. 2011;45:156–164. doi: 10.3109/10715762.2010.520319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galluzzi L, Kepp O, Kroemer G. Rip kinases initiate programmed necrosis. J Mol Cell Biol. 2009;1:8–10. doi: 10.1093/jmcb/mjp007. [DOI] [PubMed] [Google Scholar]
- 60.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, Bernardi P. The mitochondrial permeability transition from yeast to mammals. FEBS Lett. 2010;584:2504–2509. doi: 10.1016/j.febslet.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The adp/atp translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin d modulates mitochondrial f0f1-atp synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial atp synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the fo atp synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park H-A, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimizu S, Narita M, Tsujimoto Y, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel vdac. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 69.Shimizu S, Shinohara Y, Tsujimoto Y. Bax and bcl-xl independently regulate apoptotic changes of yeast mitochondria that require vdac but not adenine nucleotide translocator. Oncogene. 2000;19:4309–4318. doi: 10.1038/sj.onc.1203788. [DOI] [PubMed] [Google Scholar]
- 70.Chandra D, Choy G, Daniel PT, Tang DG. Bax-dependent regulation of bak by voltage-dependent anion channel 2. J Biol Chem. 2005;280:19051–19061. doi: 10.1074/jbc.M501391200. [DOI] [PubMed] [Google Scholar]
- 71.Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HL, Prévost M-C, Xie Z, Matsuyama S, Reed JC. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 72.Brenner C, Cadiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, Leber B, Andrews D, Duclohier H, Reed JC. Bcl-2 and bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- 73.Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H, Tsujimoto Y. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci USA. 1998;95:1455–1459. doi: 10.1073/pnas.95.4.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pastorino JG, Tafani M, Rothman RJ, Marcinkeviciute A, Hoek JB, Farber JL. Functional consequences of the sustained or transient activation by bax of the mitochondrial permeability transition pore. J Biol Chem. 1999;274:31734–31739. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- 75.De Marchi U, Campello S, Szabo I, Tombola F, Martinou JC, Zoratti M. Bax does not directly participate in the Ca2+-induced permeability transition of isolated mitochondria. J Biol Chem. 2004;279:37415–37422. doi: 10.1074/jbc.M314093200. [DOI] [PubMed] [Google Scholar]
- 76.Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, Crow MT, Gavathiotis E, Dorn GW, II, O’Rourke B, Kitsis RN. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci USA. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin SH, Perera MN, Nguyen T, Datskovskiy D, Miles M, Colombini M. Bax forms two types of channels, one of which is voltage-gated. Biophys J. 2011;101:2163–2169. doi: 10.1016/j.bpj.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of l929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 80.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. Rip3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang T-B, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-caspase-8-cFLIPL complex. Cell reports. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the rip1-rip3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 84.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of rip1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 86.Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC. The cardioprotective effect of necrostatin requires the cyclophilin-d component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21:467–469. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M. Cleavage of rip3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 89.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. Rip3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 90.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 91.Galluzzi L, Kepp O, Kroemer G. Mlkl regulates necrotic plasma membrane permeabilization. Cell Res. 2014;24:139–140. doi: 10.1038/cr.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X. Mlkl knockout mice demonstrate the indispensable role of mlkl in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of rip3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 94.Cai Z, Jitkaew S, Zhao J, Chiang H-C, Choksi S, Liu J, Ward Y, Wu L-g, Liu Z-G. Plasma membrane translocation of trimerized mlkl protein is required for tnf-induced necroptosis. Nat Cell biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell reports. 2013;5:878–885. doi: 10.1016/j.celrep.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tischner D, Manzl C, Soratroi C, Villunger A, Krumschnabel G. Necrosis-like death can engage multiple pro-apoptotic bcl-2 protein family members. Apoptosis. 2012;17:1197–1209. doi: 10.1007/s10495-012-0756-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller J-O, De Zen F, Weinlich R, Ortiz A, Walczak H. Two independent pathways of regulated necrosis mediate ischemia–reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lim S, Davidson S, Mocanu M, Yellon D, Smith C. The cardioprotective effect of necrostatin requires the cyclophilin-d component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21:467–469. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ying W. Roles of nad(+), parp-1, and sirtuins in cell death, ischemic brain injury, and synchrotron radiation x-ray-induced tissue injury. Scientifica. 2013;2013:691251. doi: 10.1155/2013/691251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zong W-X, Ditsworth D, Bauer DE, Wang Z-Q, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, Susin SA. Sequential activation of poly (adp-ribose) polymerase 1, calpains, and bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(adp-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 103.Halmosi R, Berente Z, Osz E, Toth K, Literati-Nagy P, Sumegi B. Effect of poly(adp-ribose) polymerase inhibitors on the ischemia-reperfusion-induced oxidative cell damage and mitochondrial metabolism in langendorff heart perfusion system. Mol Pharmacol. 2001;59:1497–1505. doi: 10.1124/mol.59.6.1497. [DOI] [PubMed] [Google Scholar]
- 104.Mota-Filipe H, Sepodes B, McDonald MC, Cuzzocrea S, Pinto R, Thiemermann C. The novel parp inhibitor 5-aminoisoquinolinone reduces the liver injury caused by ischemia and reperfusion in the rat. Med Sci Monit. 2002;8:BR444–453. [PubMed] [Google Scholar]
- 105.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: A potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 106.Jeggo PA. DNA repair: Parp - another guardian angel? Curr Biol. 1998;8:R49–51. doi: 10.1016/s0960-9822(98)70032-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.